Prenatal Programming of Neuroendocrine System Development by Lipopolysaccharide: Long-Term Effects


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. LPS Structure and Induction of Cytokine Synthesis
3. LPS Effect on Permeability of Blood–Brain, Blood–Testis, and Blood–Placenta Barriers
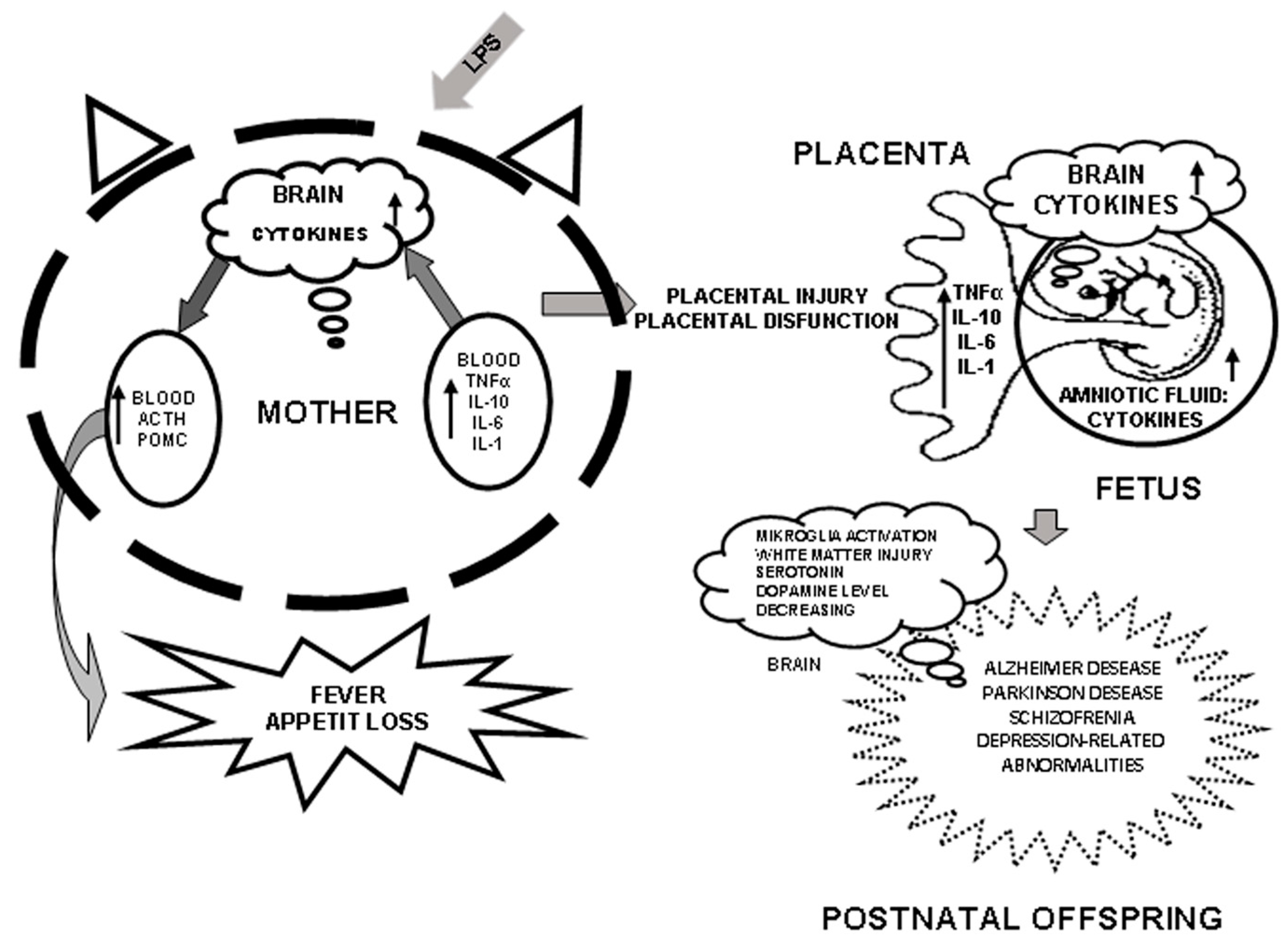
4. Role of Cytokines in Mother–Fetus Interaction and Induction of Their Synthesis by LPS
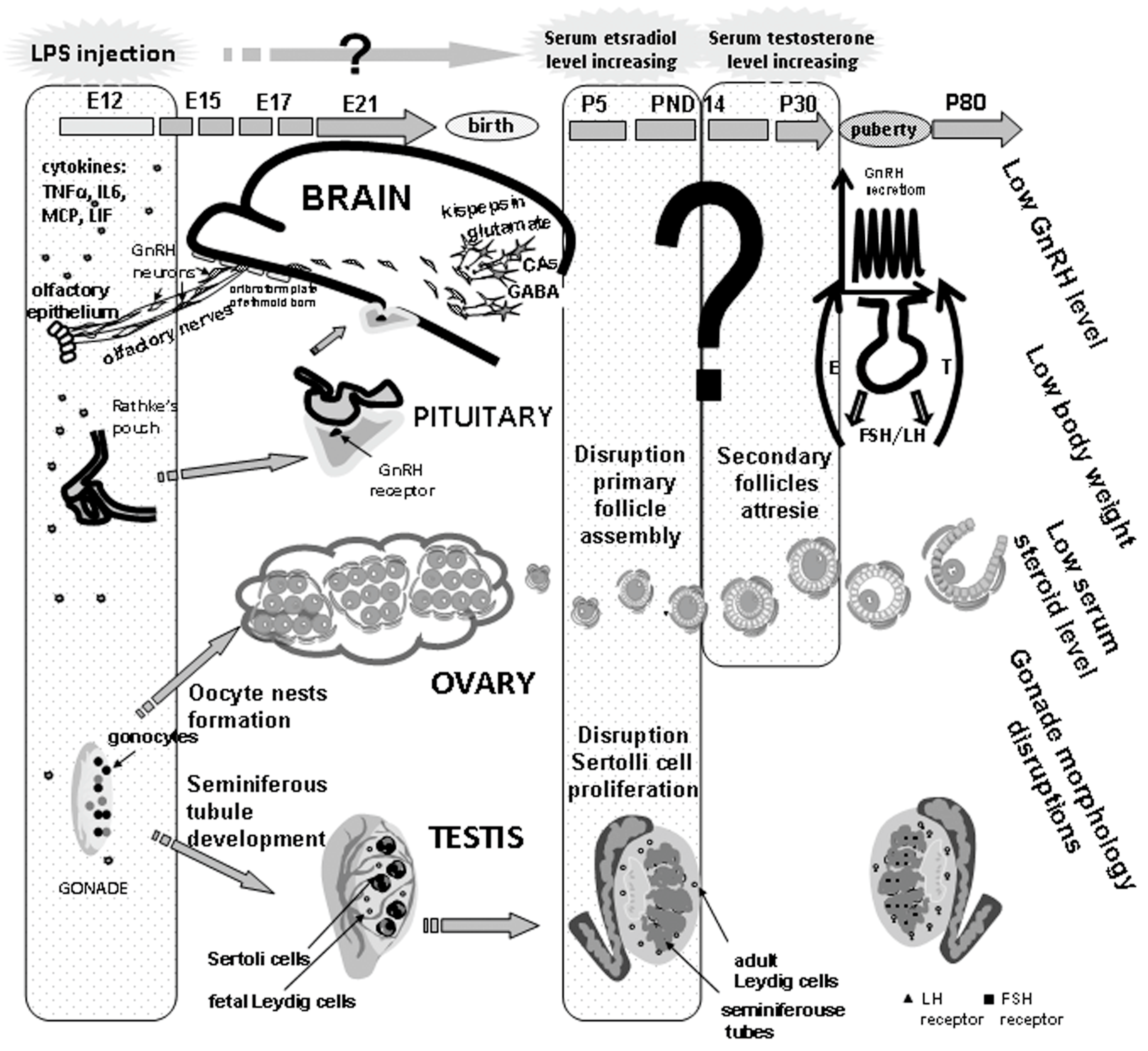
5. Long-Term Effects of Perinatal Administration of LPS during Critical Periods of Development
6. Conclusions
Funding
Conflicts of Interest
References
- Kohmura, Y.; Kirikae, T.; Kirikae, F.; Nakano, M.; Sato, I. Lipopolysaccharide (LPS)-induced intra-uterine fetal death (IUFD) in mice is principally due to maternal cause but not fetal sensitivity to LPS. Microbiol. Immunol. 2000, 44, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yan, J.Y.; Lo, Y.K.; Carvey, P.M.; Ling, Z. Dopaminergic and serotoninergic deficiencies in young adult rats prenatally exposed to the bacterial lipopolysaccharide. Brain Res. 2009, 1265, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, L.A. Plasticity of neuroendocrine-immune interactions during ontogeny: Role of perinatal programming in pathogenesis of inflammation and stress-related diseases in adults. Recent Patents Endocr. Metab. Immune Drug Discov. 2009, 3, 11–27. [Google Scholar] [CrossRef]
- Zakharova, L. Perinatal stress in brain programming and pathogenesis of psychoneurological Disorders. Biol. Bull. 2015, 42, 12–20. [Google Scholar] [CrossRef]
- Zambrano, E.; Guzman, C.; Rodríguez-González, G.L.; Durand-Carbajal, M.; Nathanielsz, P.W. Fetal programming of sexual development and reproductive function. Mol. Cell. Endocrinol. 2014, 382, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Li, X.F.; Kinsey Jones, J.S.; Knox, A.M.; Wu, X.Q.; Tahsinsoy, D.; Brain, S.D.; Lightman, S.L.; O’Byrne, K.T. Neonatal lypopolysaccharide exposure exacerbates stressinduced suppression of luteinizing hormone pulse frequency in adulthood. Endocrinology 2007, 148, 5984–5990. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, E.C.; Seckl, J.R. Prenatal stress, glucocorticoids and the programming of adult disease. Front. Behav. Neurosci. 2009, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C.R. Biochemistry of endotoxins. Annu. Rev. Biochem. 1990, 59, 129–170. [Google Scholar] [CrossRef] [PubMed]
- Kastowsky, M.; Gutberlet, T.; Bradaczek, H. Molecular Modelling of the Three-Dimensional Structure and Conformational Flexibility of Bacterial Lipopolysaccharide. J. Bacterol. 1992, 174, 4798–4806. [Google Scholar] [CrossRef]
- Rietschel, E.T.; Kirikae, T.; Schade, F.U.; Mamat, U.; Schmidt, G.; Loppnow, H.; Ulmer, A.Z.; Zahringer, U.; Seydel, U.; Di Padova, F. Bacterial endotoxin: Molecular relationship of structure to activity and function. FASEB. J. 1994, 8, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Kim, H.S.; Moon, S.C.; Lee, Y.R.; Yu, K.Y.; Lee, B.K.; Youn, H.Z.; Jeong, Y.J.; Kim, B.S.; Lee, S.H.; et al. Effects of protein concentration and detergent on endotoxin reduction by ulfiltration. BMB Rep. 2009, 42, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Akashi, S.; Nagafuku, M.; Ogata, M.; Ywakura, Y.; Akira, S.; Kitamura, T. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat. Immunol. 2002, 3, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L. Toll receptors, CD14, MD-2 and NOD2: their role in health and acute and chronic infectious diseases. Curr. Opin. Infect. Dis. 2002, 15, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, A.; Hedger, M.P. Immunological, paracrine and endocrine aspects of testicular immune privilege. Mol. Cell. Endocrinol. 2011, 335, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Zaga, V.; Estrada-Gutierrez, G.; Beltran-Montoya, J.; Maida-Claros, R.; Lopez-Vancell, R.; Vadillo-Ortega, F. Secretions of interleukin-1beta and tumor necrosis factor alpha by whole fetal membranes depend on initial interactions of amnion or choriodecidua with lipopolysaccharides or group B. streptococci. Biol. Reprod. 2004, 71, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Ivey, N.S.; Martin, E.N., Jr.; Scheld, W.M.; Nahtan, B.R. A new method for measuring blood-brain barrier permeability demonstrated with Europium-bound albumin during experimental lipopolysaccharide (LPS) induced meningitis in the rat. J. Neurosci. Methods 2005, 142, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Jiang, Y. How does peripheral lipopolysaccharide induce gene expression in the brain of rats? Toxicology 2004, 201, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Nakaoke, R.; Dohgu, S.; Banks, W.A. Release of cytokines by brain endothelial cells: A polarized response to lipopolysaccharide. Brain Behav. Immun. 2006, 20, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Yu, C.; Hsuchou, H.; Zhang, Y.; Kastin, A.J. Neuroinflammation facilitates LIF entry into brain: Role of TNF. Am. J. Physiol. Cell Physiol. 2008, 294, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, S.; Herkenham, M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflamation during endotoxemia, independent of systemic cytokines. J. Neurosci. 2005, 25, 1788–1796. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewska-Zaremba, D.; Herman, A. The role of immunological system in the regulation of gonadoliberin and gonadotropin secretion. Reprod. Biol. 2009, 9, 11–23. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, Y.; Wang, L.; Xue, F.; Hu, Y.; Hu, R.; Xu, C. MKP-1 attenuates LPS-induced blood-testis barrier dysfunction and inflammatory response through p38 and IκBα pathways. Oncotarget 2016, 7, 84907–84923. [Google Scholar] [CrossRef] [PubMed]
- Georgiades, P.; Ferguson-Smith, A.C.; Burton, G.J. Comparative developmental anatomy of the murine and human definitive placentae. Placenta 2002, 23, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Ashdown, H.; Dumont, Y.; Ng, M.; Poole, S.; Boksa, P.; Luheshi, G.N. The role of cytokines in mediating effects of prenatal infection on the fetus: Implications for schizophrenia. Mol. Psychiatry 2006, 11, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Simamura, E.; Shimada, H.; Higashi, N.; Uchishiba, M.; Otani, H.; Hatta, T. Maternal leukemia inhibitory factor (LIF) promotes fetal neurogenesis via LIF-ACTH-LIF signaling relay pathway. Endocrinology 2010, 151, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, T.; Simamura, E.; Shimada, H.; Arai, T.; Higashi, N.; Akai, T.; Iizuka, H.; Hatta, T. The suppression of maternal-fetal leukemia inhibitory factor signal relay pathway by maternal immune activation impairs brain development in mice. PLoS ONE. 2015, 10, e0129011. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, M.V.; Alexander, J.M.; Byrd, W.; Bawdon, R.E. Transfer of inflammatory cytokines across the placenta. Obstet. Gynecol. 2004, 103, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, R.; Heikkinen, T.; Hakala, K.; Laine, K.; Alanen, A. Transfer of proinflammatory cytokines across term placenta. Obstet. Gynecol. 2005, 106, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.J. Mother-offspring dialogue in early pregnancy: Impact of adverse environment on pregnancy maintenance and neurobiology. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Gayle, D.A.; Beloosesky, R.; Desai, M.; Amidi, F.; Nunez, S.E.; Ross, M.G. Maternal LPS induces cytokines in the amniotic fluid and corticotropin releasing hormone in the fetal rat brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R1024–R1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togher, K.L.; O’Keeffe, M.M.; Khashan, A.S.; Gutierrez, H.; Kenny, L.C.; O’Keeffe, G.W. Epigenetic regulation of the placental HSD11B2 barrier and its role as a critical regulator of fetal development. Epigenetics 2014, 9, 816–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charmandari, E.; Kino, T.; Souvatzoglou, E.; Chrousos, G.P. Pediatric stress: Hormonal mediators and human development. Horm. Res. 2003, 59, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.T. The link between perinatal glucocorticoids exposure and psychiatric disorders. Pediatr. Res. 2011, 69, 19R–25R. [Google Scholar] [CrossRef] [PubMed]
- Straley, M.E.; Togher, K.L.; Nolan, A.M.; Kenny, L.C.; O’Keeffe, G.W. LPS alters placental inflammatory and endocrine mediators and inhibits fetal neurite growth in affected offspring during late gestation. Placenta 2014, 35, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Holmlund, U.; Cebers, G.; Dahlfors, A.R. Expression and regulation of the pattern recognition receptors toll-like receptor-2 and Toll-like receptor-4 in the human placenta. Immunology 2002, 107, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Beijar, E.C.; Mallard, C.; Powell, T.L. Expression and subcellular localization of TLR-4 in term and first trimester human placenta. Placenta 2006, 27, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Liverman, C.S.; Kaftan, H.A.; Cui, L.; Hersperger, S.G.; Taboada, E.; Klein, R.M.; Berman, N.E. Altered expression of pro-inflammatory and developmental genes in the fetal brain in a mouse model of maternal infection. Neurosci. Lett. 2006, 399, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Pan, Z.L.; Pang, Y.; Evans, O.B.; Rhodes, P.G. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr. Res. 2000, 47, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Sharova, V.S.; Izvolskaia, M.S.; Zakharova, L.A. Lipopolysaccharide-induced maternal inflammation affects the gonadotropin-releasing hormone neuron development in fetal mice. Neuroimmunomodulation 2015, 22, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Murray, P.J.; Urwyler, A.; Yee, B.K.; Schedlowski, M.; Feldon, J. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol. Psychiatry 2008, 13, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.J.; Hallenbeck, J.M.; Gallo, V. Determining the fetal inflammatory response in an experimental model of intrauterine inflammation in rats. Pediatr. Res. 2004, 56, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.X.; Wang, H.; Ning, H. Maternally administered melatonin differentially regulates lipopolysaccharide-induced proinflammatory and anti-inflammatory cytokines in maternal serum, amniotic fluid, fetal liver, and fetal brain. J. Pineal Res. 2007, 43, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J. Neuroinflamm. 2009, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, J.H.; Jarskog, L.F.; Vadlamudi, S. Maternal infection regulates BDNF and NGF expression in fetal and neonatal brain and maternal-fetal unit of the rat. Mol. Psychiatry 2003, 11, 47–55. [Google Scholar] [CrossRef]
- Yu, H.M.; Yuan, T.M.; Gu, W.Z.; Li, J.P. Expression of glial fibrillary acidic protein in developing rat brain after intrauterine infection. Neuropathology 2004, 24, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Rousset, C.I.; Chalon, S.; Cantagrel, S.; Bodard, S.; Andres, C.; Gressens, P.; Saliba, E. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr. Res. 2006, 59, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Girgis, R.R.; Kumar, S.S.; Brown, A.S. The cytokine model of schizophrenia: Emerging therapeutic strategies. Biol. Psychiatry 2014, 75, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Baharnoori, M.; Brake, W.G.; Srivastava, L.K. Prenatal immune challenge induces developmental changes in the morphology of pyramid DAl neurons of the prefrontal cortex and hippocampus in rats. Schizophr. Res. 2009, 107, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginsberg, Y.; Khatib, N.; Weiner, Z.; Beloosesky, R. Maternal Inflammation, Fetal Brain Implications and Suggested Neuroprotection: A Summary of 10 Years of Research in Animal Models. Rambam Maimonides Med. J. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Carvey, P.M.; Ling, Z. Altered glutathione homeostasis in animals prenatally exposed to lipopolysaccharide. Neurochem. Int. 2007, 50, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvey, P.M.; Chang, Q.; Lipton, J.W.; Ling, Z. Prenatal exposure to the bacteriotoxin lipopolysaccharide leads to long-term losses of dopamine neurons in offspring: A potential, new model of Parkinson’s disease. Front. Biosci. 2003, 8, s826–s837. [Google Scholar] [CrossRef] [PubMed]
- Cameron, N.M.; Shahrokh, D.; Del Corpo, A. Epigenetic programming of phenotypic variations in reproductive strategies in the rat through maternal care. J. Neuroendocrinol. 2008, 20, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, L.L.; Hu, Y.F.; Wang, B.W.; Huang, Y.Y.; Zhang, C.; Chen, Y.H.; Xu, D.X. Maternal LPS exposure during pregnancy impairs testicular development, steroidogenesis and spermatogenesis in male offspring. PLoS ONE. 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Loveland, K.L.; Klein, B.; Pueschl, D.; Indumathy, S.; Bergmann, M.; Loveland, B.E.; Hedger, M.P.; Schuppe, H.C. Cytokines in Male Fertility and Reproductive Pathologies: Immunoregulation and Beyond. Front. Endocrinol. (Lausanne) 2017, 8, 307. [Google Scholar] [CrossRef] [PubMed]
- Sharova, V.S.; Izvol’skaia, M.S.; Voronova, S.N.; Zakharova, LA. Effect of bacterial endotoxin on migration of gonadotropin-releasing, hormone producing neurons in rat embryogenesis. Ontogenez 2011, 42, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Izvolskaia, M.S.; Tillet, Y.; Sharova, V.S.; Voronova, S.N.; Zakharova, L.A. Disruptions in the hypothalamic-pituitary-gonadal axis in rat offspring following prenatal maternal exposure to lipopolysaccharide. Stress 2016, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ebling, F.J. The neuroendocrine timing of puberty. Reproduction 2005, 129, 675–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dozio, E.; Ruscica, M.; Galliera, E.; Corsi, M.M.; Magni, P. Leptin, ciliary neurotrophic factor, leukemia inhibitory factor and interleukin-6: Class-I cytokines involved in the neuroendocrine regulation of the reproductive function. Curr. Protein Pept. Sci. 2009, 10, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, SR.; Rutkowski, H.; Vrezas, I. Cytokines and steroidogenesis. Mol. Cell. Endocrinol. 2004, 215, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Skinner, M.K. Regulation of primordial follicle assembly and development. Hum. Reprod. Update 2005, 11, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Gearing, D.P.; White, L.S.; Compton, D.L.; Schooley, K.; Donovan, P.J. Role of leukemia inhibitory factor and its receptor in mouse primordial germ cell growth. Development 1994, 120, 3145–3153. [Google Scholar] [PubMed]
- Van der Hoek, K.H.; Woodhouse, C.M.; Brännström, M.; Norman, R.J. Effects of interleukin (IL)-6 on luteinizing hormone- and IL-1beta-induced ovulation and steroidogenesis in the rat ovary. Biol. Reprod. 1998, 58, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Eddie, S.L.; Childs, A.J.; Jabbour, H.N.; Anderson, R.A. Developmentally regulated IL6-type cytokines signal to germ cells in the human fetal ovary. Mol. Hum. Reprod. 2012, 18, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Izvolskaia, M.S.; Sharova, V.S.; Ignatiuk, V.M.; Voronova, S.N.; Zakharova, L.A. Abolition of prenatal LPS-induced reproductive disorders in rat male offspring by fulvestrant. Andrologia 2018, in press. [Google Scholar]
- Ignatiuk, V.M.; Izvolskaya, M.S.; Sharova, V.S.; Voronova, S.N.; Zakharova, L.A. Disruptions in reproductive system in female rats after prenatal LPS-induced immunological stress: Role of sex steroids. Stress 2018. [Google Scholar] [CrossRef] [PubMed]
- Beloosesky, R.; Weiner, Z.; Khativ, N.; Maravi, N.; Mandel, R.; Boles, J.; Ross, M.G.; Itskovitz-Eldor, J. Prophylactic maternal n-acetylcysteine before lipopolysaccharide suppresses fetal inflammatory cytokine responses. Am. J. Obstet. Gynecol. 2009, 200, e1–e5. [Google Scholar] [CrossRef] [PubMed]
- Chua, J.S.; Cowley, C.J.; Manavis, J.; Rofe, A.M.; Coyle, P. Prenatal exposure to lipopolysaccharide results in neurodevelopmental damage that is ameliorated by zinc in mice. Brain Behav. Immun. 2012, 26, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Beloosesky, R.; Khatib, N.; Ginsberg, Y. Maternal magnesium sulfate fetal neuroprotective effects to the fetus: Inhibition of neuronal nitric oxide synthase and nuclear factor kappa-light-chain-enhancer of activated B cells activation inthom. Am. J. Obstet. Gynecol. 2016, 215, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Yu, Z.; Fu, L.; Xia, MZ.; Zhao, M.; Wang, H.; Zhang, C.; Hu, YF.; Tao, FB.; Xu, D.X. Supplementation with vitamin D3 during pregnancy protects against lipopolysaccharide-induced neural tube defects through improving placental folate transportation. Toxicol. Sci. 2015, 145, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Labrousse, V.F.; Leyrolle, Q.; Amadieu, C.; Aubert, A.; Sere, A.; Coutureau, E.; Grégoire, S.; Bretillon, L.; Pallet, V.; Gressens, P.; et al. Dietary omega-3 deficiency exacerbates inflammation and reveals spatial memory deficits in mice exposed to lipopolysaccharide during gestation. Brain Behav. Immun. 2018, 7, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izvolskaia, M.; Sharova, V.; Zakharova, L. Prenatal Programming of Neuroendocrine System Development by Lipopolysaccharide: Long-Term Effects. Int. J. Mol. Sci. 2018, 19, 3695. https://doi.org/10.3390/ijms19113695
Izvolskaia M, Sharova V, Zakharova L. Prenatal Programming of Neuroendocrine System Development by Lipopolysaccharide: Long-Term Effects. International Journal of Molecular Sciences. 2018; 19(11):3695. https://doi.org/10.3390/ijms19113695
Chicago/Turabian StyleIzvolskaia, Marina, Viktoria Sharova, and Liudmila Zakharova. 2018. "Prenatal Programming of Neuroendocrine System Development by Lipopolysaccharide: Long-Term Effects" International Journal of Molecular Sciences 19, no. 11: 3695. https://doi.org/10.3390/ijms19113695