The Regulation of JNK Signaling Pathways in Cell Death through the Interplay with Mitochondrial SAB and Upstream Post-Translational Effects

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
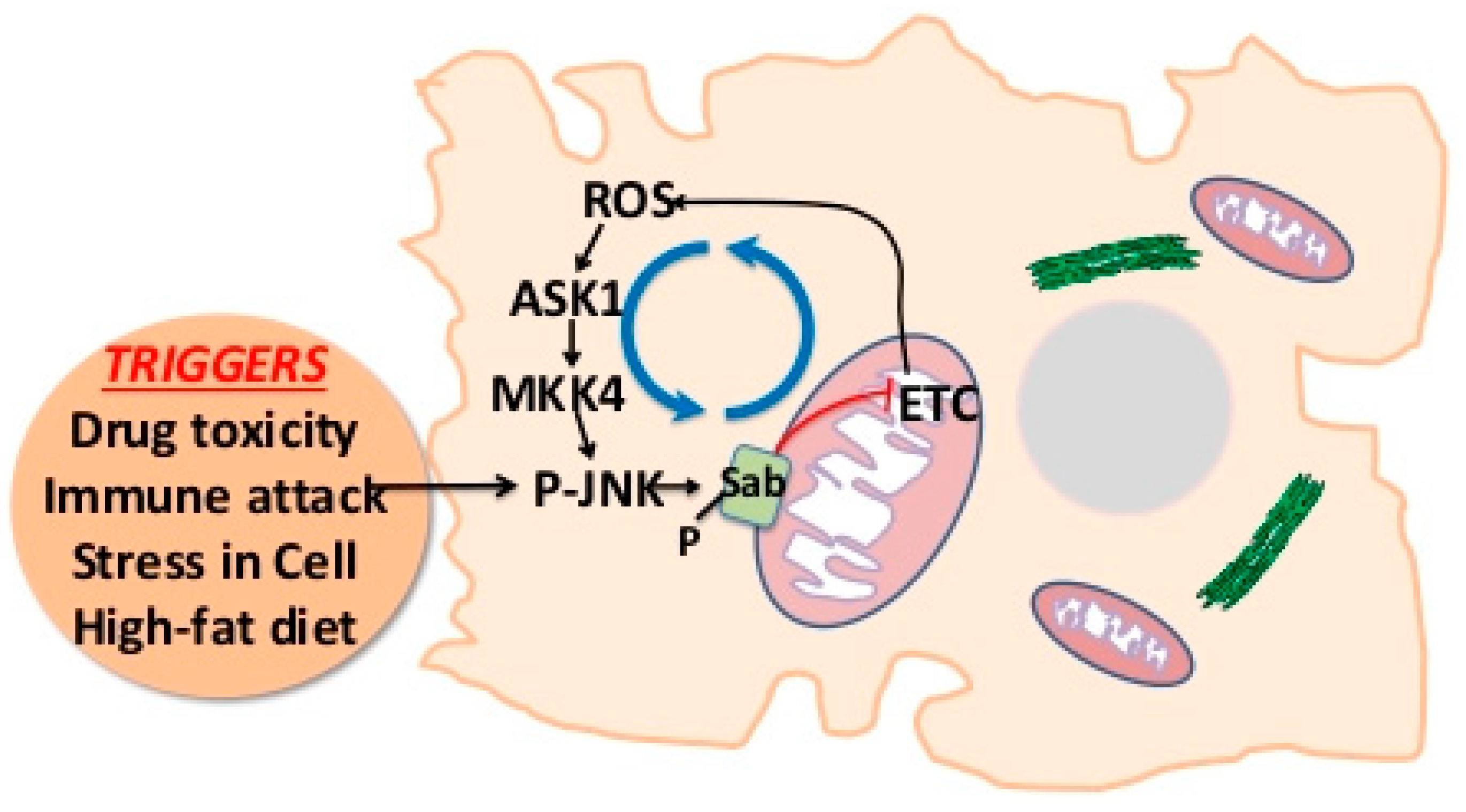
2. JNK-SAB-ROS Activation Loop
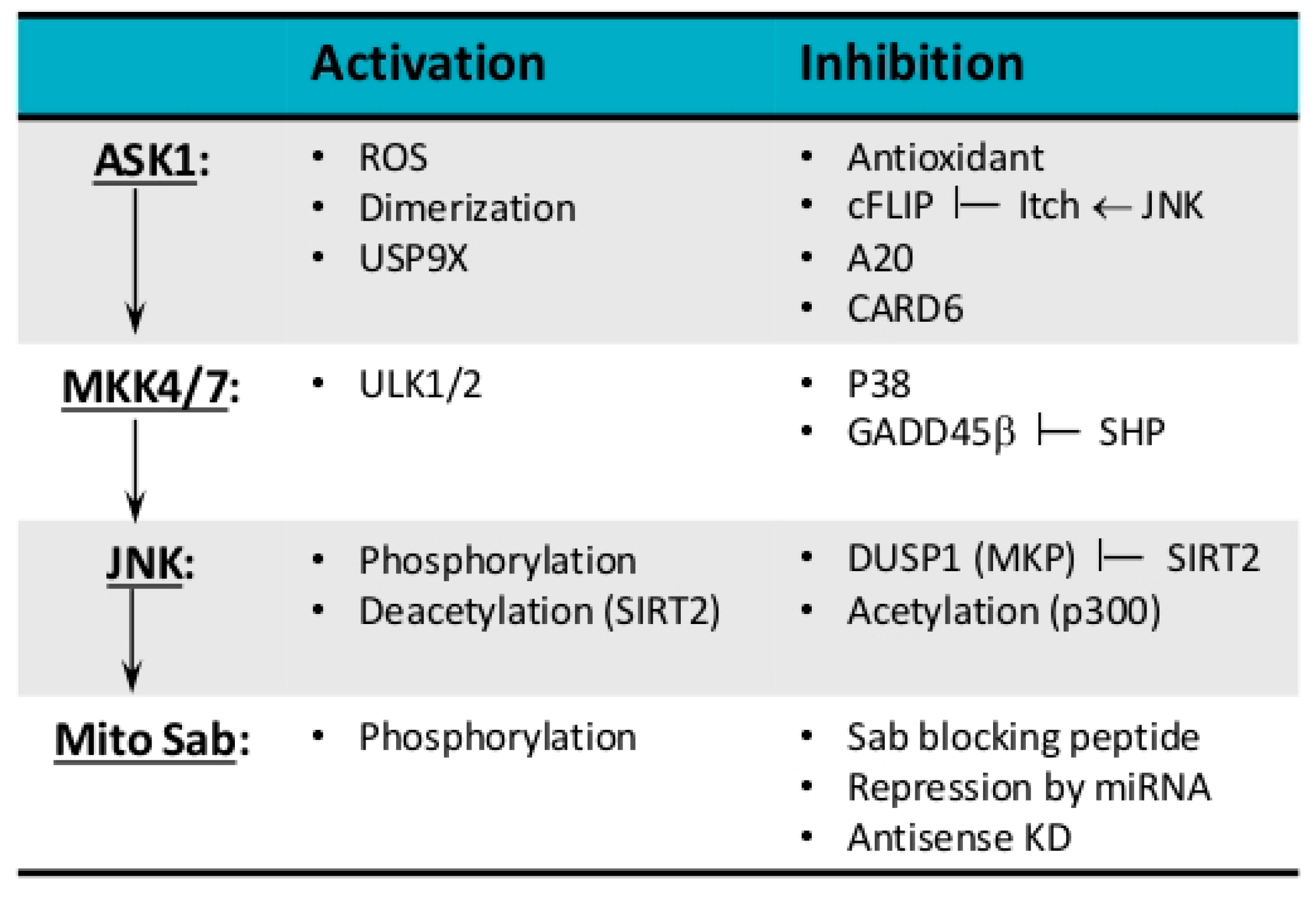
3. Modulation of JNK Activation Loop
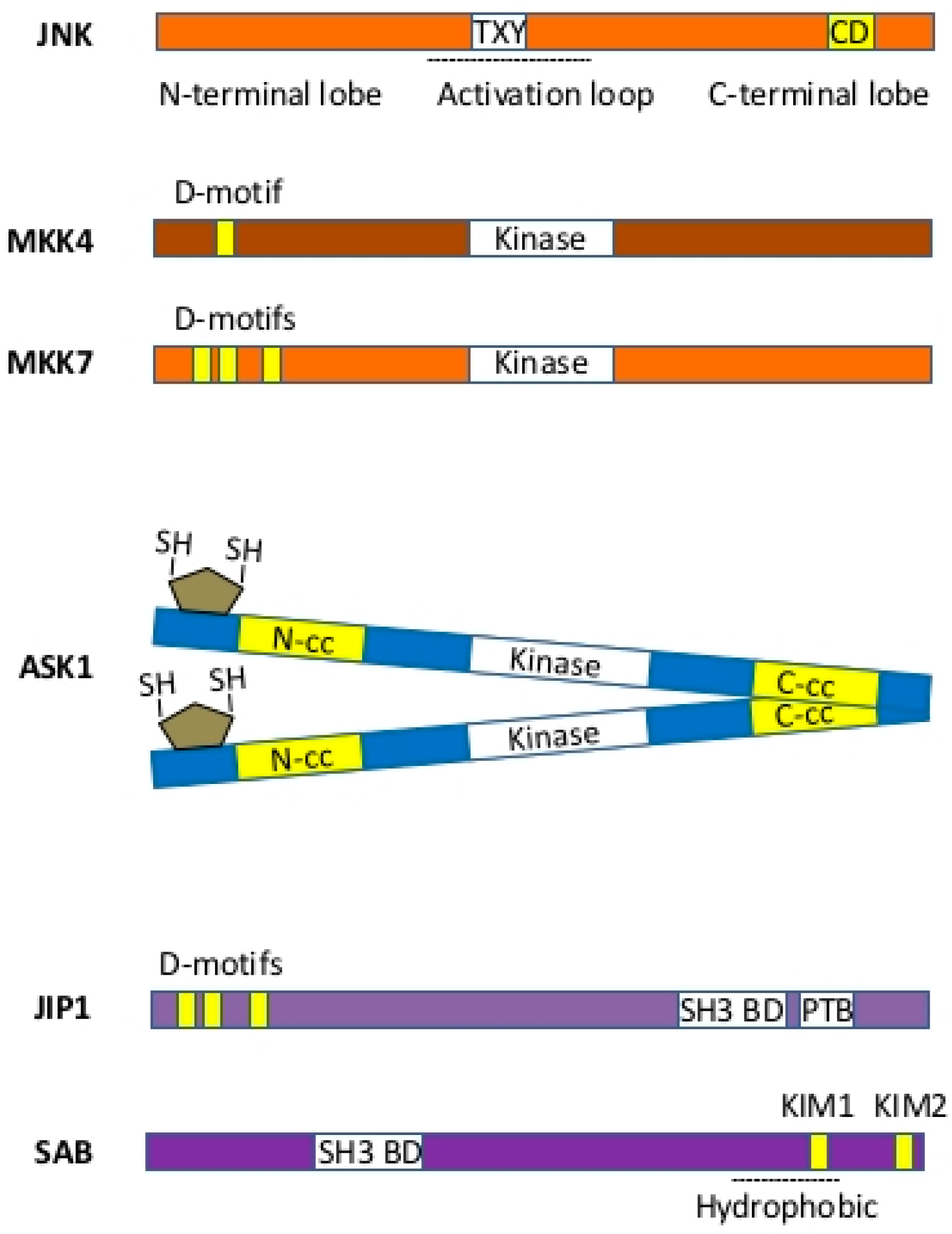
3.1. MAP Kinases—JNK and p38
3.2. MKKs—MKK4 and MKK7
3.3. MAP3K—MLK2/3, ASK1, TAK1
3.4. Scaffold Protein—SAB
3.5. Regulation of SAB Expression
4. Perspectives on the Intervention of the JNK Activation Loop
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKT | AKT serine/threonine kinase 1 |
| APAP | acetaminophen |
| ASK1 | apoptosis signal-regulating kinase 1 |
| ATF2 | activating transcription factor 2 |
| ATP | adenosine triphosphate |
| CDC42 | cell division cycle 42 |
| cFLIP | CASP8 and FADD like apoptosis regulator |
| c-JUN | Jun proto-oncogene |
| DOK4 | downstream of tyrosine kinase/docking protein 4 |
| DUSP | dual-specificity phosphatase |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| GalN | N-acetyl-galactosamine |
| ITCH | Itchy E3 ubiquitin protein ligase |
| JIP | JNK-interacting protein |
| JNK | c-Jun N-terminal kinases |
| KIM | kinase interaction motif |
| KO | knock out |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MAP2K | mitogen-activated protein kinase kinase |
| MAP3K | mitogen-activated protein kinase kinase kinase |
| MKK4 | MAPK kinase 4 |
| MKP | mitogen-activated protein kinase phosphatase |
| MLK | mixed lineage kinase |
| MPT | mitochondrial permeability transition |
| NASH | nonalcoholic steatohepatitis |
| NCOR | nuclear receptor corepressor |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| P-JNK | phosphoactivated JNK |
| RAC | Rac family small GTPase 1 |
| ROS | reactive oxygen species |
| SAB (Sh3bp5) | SH3-domain binding protein 5 |
| SH3 | SRC Homology 3 Domain |
| SHP1 | SH2 phosphatase 1 |
| SRC | SRC proto-oncogene non-receptor tyrosine kinase |
| TAK1 | TGF-beta activated kinase 1 |
| TNF | tumor necrosis factor |
References
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Cell. Biol. 2007, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Zhang, J.; Oo, C.; Min, R.W.M.; Kaplowitz, N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 2018, 67, 2013–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunawan, B.K.; Liu, Z.X.; Han, D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 2006, 131, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Han, D.; Petrovic, L.M.; Kaplowitz, N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J. Biol. Chem. 2011, 286, 35071–35078. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Fernandez-Checa, J.C.; Kaplowitz, N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014, 5, e989. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Le, B.H.; García-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Wiltshire, C.; Matsushita, M.; Tsukada, S.; Gillespie, D.A.; May, G.H. A new c-Jun N-terminal kinase (JNK)-interacting protein, Sab (SH3BP5), associates with mitochondria. Biochem. J. 2002, 367, 577–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, S.; Than, T.A.; Min, R.W.; Aghajan, M.; Kaplowitz, N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Rehman, H.; Krishnasamy, Y.; Schnellmann, R.G.; Lemasters, J.J.; Zhong, Z. Improvement of liver injury and survival by JNK2 and iNOS deficiency in liver transplants from cardiac death mice. J. Hepatol. 2015, 63, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, Y.; Win, S.; Than, T.A.; Yin, S.; Ye, M.; Hu, H.; Kaplowitz, N. Antcin H Protects Against Acute Liver Injury Through Disruption of the Interaction of c-Jun-N-Terminal Kinase with Mitochondria. Antioxid. Redox Signal. 2017, 26, 207–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, S.; Lemay, S.; Osawa, M.; Che, W.; Duan, Y.; Tompkins, A.; Brookes, P.S.; Sheu, S.S.; Abe, J. Mitochondrial Dok-4 recruits Src kinase and regulates NF-kappaB activation in endothelial cells. J. Biol. Chem. 2005, 280, 26383–26396. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Günther, S. New insights into the structural dynamics of the kinase JNK3. Sci. Rep. 2018, 8, 9435. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, J.D.; Nwachukwu, J.C.; Figuera-Losada, M.; Cherry, L.; Nettles, K.W.; LoGrasso, P.V. Structural mechanisms of allostery and autoinhibition in JNK family kinases. Structure 2012, 20, 2174–2184. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, A.; Czech, M.P.; Davis, R.J. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes Dev. 2004, 18, 1976–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, C.; Standen, C.L.; Jung, D.Y.; Gray, S.; Ong, H.; Flavell, R.A.; Kim, J.K.; Davis, R.J. Requirement of JIP1-mediated c-Jun N-terminal kinase activation for obesity-induced insulin resistance. Mol. Cell. Biol. 2010, 30, 4616–4625. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Standen, C.L.; Morel, C.; Jung, D.Y.; Kim, J.K.; Swat, W.; Flavell, R.A.; Davis, R.J. A Protein Scaffold Coordinates SRC-Mediated JNK Activation in Response to Metabolic Stress. Cell Rep. 2017, 20, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; LoGrasso, P.V. Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 2011, 286, 16052–16062. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.Y.; Adler, V.; Pincus, M.R.; Ronai, Z. MEKK1/JNK signaling stabilizes and activates p53. Proc. Natl. Acad. Sci. USA 1998, 95, 10541–10546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signaling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Pérez, S.; Rius-Pérez, S.; Tormos, A.M.; Finamor, I.; Nebreda, Á.R.; Taléns-Visconti, R.; Sastre, J. Age-dependent regulation of antioxidant genes by p38α MAPK in the liver. Redox Biol. 2018, 16, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.O.; Ludwig, R.L.; Morrison, D.; Kotlyarov, A.; Gaestel, M.; Vousden, K.H. HDM2 phosphorylation by MAPKAP kinase 2. Oncogene 2005, 24, 1965–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmlöf, M.; Roudier, E.; Högberg, J.; Stenius, U. MEK-ERK-mediated phosphorylation of Mdm2 at Ser-166 in hepatocytes. Mdm2 is activated in response to inhibited Akt signaling. J. Biol. Chem. 2007, 282, 2288–2296. [Google Scholar] [CrossRef] [PubMed]
- Alarcon-Vargas, D.; Ronai, Z. p53-Mdm2--the affair that never ends. Carcinogenesis 2002, 23, 541–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, M.I.; Jones, S.N. Regulation of the Mdm2-p53 signaling axis in the DNA damage response and tumorigenesis. Transl. Cancer Res. 2016, 5, 707–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, Y.; Yin, S.; Yan, M.; Win, S.; Aung Than, T.; Aghajan, M.; Hu, H.; Kaplowitz, N. Protective role of p53 in acetaminophen hepatotoxicity. Free Radic Biol. Med. 2017, 106, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Shu, L.; Dilling, M.B.; Easton, J.; Harwood, F.C.; Ichijo, H.; Houghton, P.J. Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21(Cip1). Mol. Cell 2003, 11, 1491–1501. [Google Scholar] [CrossRef]
- Huang, C.Y.; Tan, T.H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarikhani, M.; Mishra, S.; Desingu, P.A.; Kotyada, C.; Wolfgeher, D.; Gupta, M.P.; Singh, M.; Sundaresan, N.R. SIRT2 regulates oxidative stress-induced cell death through deacetylation of c-Jun NH2-terminal kinase. Cell Death Differ. 2018, 25, 1638–1656. [Google Scholar] [CrossRef] [PubMed]
- Villalba, J.M.; Alcaín, F.J. Sirtuin activators and inhibitors. Biofactors 2012, 38, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Chen, Q.; Dong, Z.; Xu, L.; Lu, T.; Li, D.; Zhang, J.; Zhang, M.; Xia, Q. Inactivation of Sirt1 in mouse livers protects against endotoxemic liver injury by acetylating and activating NF-κB. Cell Death Dis. 2016, 7, e2403. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.T.; Bardwell, A.J.; Abdollahi, M.; Bardwell, L. A docking site in MKK4 mediates high affinity binding to JNK MAPKs and competes with similar docking sites in JNK substrates. J. Biol. Chem. 2003, 278, 32662–32672. [Google Scholar] [CrossRef] [PubMed]
- Kragelj, J.; Palencia, A.; Nanao, M.H.; Maurin, D.; Bouvignies, G.; Blackledge, M.; Jensen, M.R. Structure and dynamics of the MKK7-JNK signaling complex. Proc. Natl. Acad. Sci. USA 2015, 112, 3409–3414. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, T.Y.; Song, L.; Zhang, C.; Li, J.; Lin, Z.Z.; Lin, S.C.; Lin, S.Y. Liver-specific deficiency of unc-51 like kinase 1 and 2 protects mice from acetaminophen-induced liver injury. Hepatology 2018, 67, 2397–2413. [Google Scholar] [CrossRef] [PubMed]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrichsdorff, J.; Luedde, T.; Perdiguero, E.; Nebreda, A.R.; Pasparakis, M. p38 alpha MAPK inhibits JNK activation and collaborates with IkappaB kinase 2 to prevent endotoxin-induced liver failure. EMBO Rep. 2008, 9, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girnius, N.; Davis, R.J. TNFα-Mediated Cytotoxic Responses to IAP Inhibition Are Limited by the p38α MAPK Pathway. Cancer Cell 2016, 29, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Lalaoui, N.; Hänggi, K.; Brumatti, G.; Chau, D.; Nguyen, N.N.; Vasilikos, L.; Spilgies, L.M.; Heckmann, D.A.; Ma, C.; Ghisi, M.; et al. Targeting p38 or MK2 Enhances the Anti-Leukemic Activity of Smac-Mimetics. Cancer Cell 2016, 30, 499–500. [Google Scholar] [CrossRef] [PubMed]
- De Smaele, E.; Zazzeroni, F.; Papa, S.; Nguyen, D.U.; Jin, R.; Jones, J.; Cong, R.; Franzoso, G. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signaling. Nature 2001, 414, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Zazzeroni, F.; Bubici, C.; Jayawardena, S.; Alvarez, K.; Matsuda, S.; Nguyen, D.U.; Pham, C.G.; Nelsbach, A.H.; Melis, T.; et al. Gadd45 beta mediates the NF-kappa B suppression of JNK signaling by targeting MKK7/JNKK2. Nat. Cell Biol. 2004, 6, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [PubMed]
- Barthwal, M.K.; Sathyanarayana, P.; Kundu, C.N.; Rana, B.; Pradeep, A.; Sharma, C.; Woodgett, J.R.; Rana, A. Negative regulation of mixed lineage kinase 3 by protein kinase B/AKT leads to cell survival. J. Biol. Chem. 2003, 278, 3897–3902. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Liu, T.; Kamp, D.W.; Wang, Y.; He, H.; Zhou, X.; Li, D.; Yang, L.; Zhao, B.; Liu, G. AKT/mTOR and c-Jun N-terminal kinase signaling pathways are required for chrysotile asbestos-induced autophagy. Free Radic. Biol. Med. 2014, 72, 296–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.J.; Lee, Y.J. Dissociation of Akt1 from its negative regulator JIP1 is mediated through the ASK1-MEK-JNK signal transduction pathway during metabolic oxidative stress: A negative feedback loop. J. Cell Biol. 2005, 170, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Soga, M.; Matsuzawa, A.; Ichijo, H. Oxidative Stress-Induced Diseases via the ASK1 Signaling Pathway. Int. J. Cell Biol. 2012, 2012, 439587. [Google Scholar] [CrossRef] [PubMed]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, A.; Saegusa, K.; Noguchi, T.; Sadamitsu, C.; Nishitoh, H.; Nagai, S.; Koyasu, S.; Matsumoto, K.; Takeda, K.; Ichijo, H. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 2005, 6, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Weijman, J.F.; Kumar, A.; Jamieson, S.A.; King, C.M.; Caradoc-Davies, T.T.; Ledgerwood, E.C.; Murphy, J.M.; Mace, P.D. Structural basis of autoregulatory scaffolding by apoptosis signal-regulating kinase 1. Proc. Natl. Acad. Sci. USA 2017, 114, E2096–E2105. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.W.; Cho, J.H.; Lee, J.K.; Kang, Y.H.; Chae, J.S.; Kim, Y.M.; Kim, J.; Kim, E.K.; Kim, S.E.; Baik, J.H.; et al. CIB1 functions as a Ca(2+)-sensitive modulator of stress-induced signaling by targeting ASK1. Proc. Natl. Acad. Sci. USA 2009, 106, 17389–17394. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, T.; Li, X.; Yang, M.; Tang, S.; Zhu, X.; Gu, Y.; Su, X.; Xia, M.; Li, W.; et al. Lys29-linkage of ASK1 by Skp1-Cullin 1-Fbxo21 ubiquitin ligase complex is required for antiviral innate response. Elife 2016, 5, e14087. [Google Scholar] [CrossRef] [PubMed]
- Cockrell, L.M.; Puckett, M.C.; Goldman, E.H.; Khuri, F.R.; Fu, H. Dual engagement of 14-3-3 proteins controls signal relay from ASK2 to the ASK1 signalosome. Oncogene 2010, 29, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Kawarazaki, Y.; Ichijo, H.; Naguro, I. Apoptosis signal-regulating kinase 1 as a therapeutic target. Expert Opin. Ther. Targets 2014, 18, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Cebula, M.; Schmidt, E.E.; Arnér, E.S. TrxR1 as a potent regulator of the Nrf2-Keap1 response system. Antioxid. Redox Signal. 2015, 23, 823–853. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Noguchi, T.; Homma, K.; Katagiri, K.; Takeda, K.; Matsuzawa, A.; Ichijo, H. Ubiquitin-like sequence in ASK1 plays critical roles in the recognition and stabilization by USP9X and oxidative stress-induced cell death. Mol. Cell 2009, 36, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, P.X.; Zhao, L.P.; Zhang, X.; Ji, Y.X.; Zhang, X.J.; Fang, C.; Lu, Y.X.; Yang, X.; Gao, M.M.; et al. The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat. Med. 2018, 24, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Won, M.; Park, K.A.; Byun, H.S.; Sohn, K.C.; Kim, Y.R.; Jeon, J.; Hong, J.H.; Park, J.; Seok, J.H.; Kim, J.M.; et al. Novel anti-apoptotic mechanism of A20 through targeting ASK1 to suppress TNF-induced JNK activation. Cell Death Differ. 2010, 17, 1830–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.X.; Ji, Y.X.; Zhang, X.J.; Zhao, L.P.; Yan, Z.Z.; Zhang, P.; Shen, L.J.; Yang, X.; Fang, J.; Tian, S.; et al. Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat. Med. 2017, 23, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L.) turnover. Cell 2006, 124, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Min, R.W.M.; Le, K.; Zhou, S.; Aghajan, M.; Than, T.A.; Win, S.; Kaplowitz, N. The role of MAP2 kinases and p38 kinase in acute murine liver injury models. Cell Death Dis. 2017, 8, e2903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Al-Lamki, R.; Bai, L.; Streb, J.W.; Miano, J.M.; Bradley, J.; Min, W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ. Res. 2004, 94, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Mao, W.; Wang, X.; Sun, P.; Cheng, D.; Tian, S.; Zhu, X.Y.; Yang, L.; Huang, Z.; Li, H. Caspase recruitment domain 6 protects against hepatic ischemia/reperfusion injury by suppressing ASK1. J. Hepatol. 2018, 69, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Zeng, Q.; Cheng, D.; Zhang, K.; Zheng, J.; Liu, Y.; Yuan, Y.F.; Tang, Y.D. Caspase Recruitment Domain Protein 6 protects against hepatic steatosis and insulin resistance by suppressing Ask1. Hepatology 2018. [Google Scholar] [CrossRef]
- Takaesu, G.; Ninomiya-Tsuji, J.; Kishida, S.; Li, X.; Stark, G.R.; Matsumoto, K. Interleukin-1 (IL-1) receptor-associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL-1 signaling pathway. Mol. Cell. Biol. 2001, 21, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Malato, Y.; Willenbring, H. The MAP3K TAK1: A chock block to liver cancer formation. Hepatology 2010, 52, 1506–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Urano, F.; Jaeschke, A. Cdc42 and Rac1 are major contributors to the saturated fatty acid-stimulated JNK pathway in hepatocytes. J. Hepatol. 2012, 56, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelkar, N.; Standen, C.L.; Davis, R.J. Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Mol. Cell. Biol. 2005, 25, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Court, N.W.; Kuo, I.; Quigley, O.; Bogoyevitch, M.A. Phosphorylation of the mitochondrial protein Sab by stress-activated protein kinase 3. Biochem. Biophys. Res. Commun. 2004, 319, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, A.; Sato, M.; Sato, K.; Gengyo-Ando, K.; Yorimitsu, T.; Nakai, J.; Hara, T.; Sato, K.; Sato, K. REI-1 Is a Guanine Nucleotide Exchange Factor Regulating RAB-11 Localization and Function in C. elegans Embryos. Dev. Cell 2015, 35, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Paudel, I.; Hernandez, S.M.; Portalatin, G.M.; Chambers, T.P.; Chambers, J.W. Sab Concentrations Indicate Chemotherapeutic Susceptibility in Ovarian Cancer Cell Lines. Biochem. J. 2018, 475, 3471–3492. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.P.; Portalatin, G.M.; Paudel, I.; Robbins, C.J.; Chambers, J.W. Sub-chronic administration of LY294002 sensitizes cervical cancer cells to chemotherapy by enhancing mitochondrial JNK signaling. Biochem. Biophys. Res. Commun. 2015, 463, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; Pachori, A.; Howard, S.; Iqbal, S.; LoGrasso, P.V. Inhibition of JNK mitochondrial localization and signaling is protective against ischemia/reperfusion injury in rats. J. Biol. Chem. 2013, 288, 4000–4011. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.P.; Santiesteban, L.; Gomez, D.; Chambers, J.W. Sab mediates mitochondrial dysfunction involved in imatinib mesylate-induced cardiotoxicity. Toxicology 2017, 382, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; Howard, S.; LoGrasso, P.V. Blocking c-Jun N-terminal kinase (JNK) translocation to the mitochondria prevents 6-hydroxydopamine-induced toxicity in vitro and in vivo. J. Biol. Chem. 2013, 288, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; Pachori, A.; Howard, S.; Ganno, M.; Hansen, D.J.; Kamenecka, T.; Song, X.; Duckett, D.; Chen, W.; Ling, Y.Y.; et al. Small Molecule c-jun-N-terminal Kinase (JNK) Inhibitors Protect Dopaminergic Neurons in a Model of Parkinson’s Disease. ACS Chem. Neurosci. 2011, 2, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; Cherry, L.; Laughlin, J.D.; Figuera-Losada, M.; Lograsso, P.V. Selective inhibition of mitochondrial JNK signaling achieved using peptide mimicry of the Sab kinase interacting motif-1 (KIM1). ACS Chem. Biol. 2011, 6, 808–818. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Win, S.; Than, T.A.; Kaplowitz, N. The Regulation of JNK Signaling Pathways in Cell Death through the Interplay with Mitochondrial SAB and Upstream Post-Translational Effects. Int. J. Mol. Sci. 2018, 19, 3657. https://doi.org/10.3390/ijms19113657
Win S, Than TA, Kaplowitz N. The Regulation of JNK Signaling Pathways in Cell Death through the Interplay with Mitochondrial SAB and Upstream Post-Translational Effects. International Journal of Molecular Sciences. 2018; 19(11):3657. https://doi.org/10.3390/ijms19113657
Chicago/Turabian StyleWin, Sanda, Tin Aung Than, and Neil Kaplowitz. 2018. "The Regulation of JNK Signaling Pathways in Cell Death through the Interplay with Mitochondrial SAB and Upstream Post-Translational Effects" International Journal of Molecular Sciences 19, no. 11: 3657. https://doi.org/10.3390/ijms19113657