The Role of TGF-β Signaling in Lung Cancer Associated with Idiopathic Pulmonary Fibrosis


Abstract
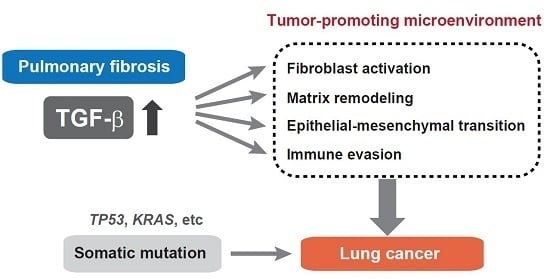
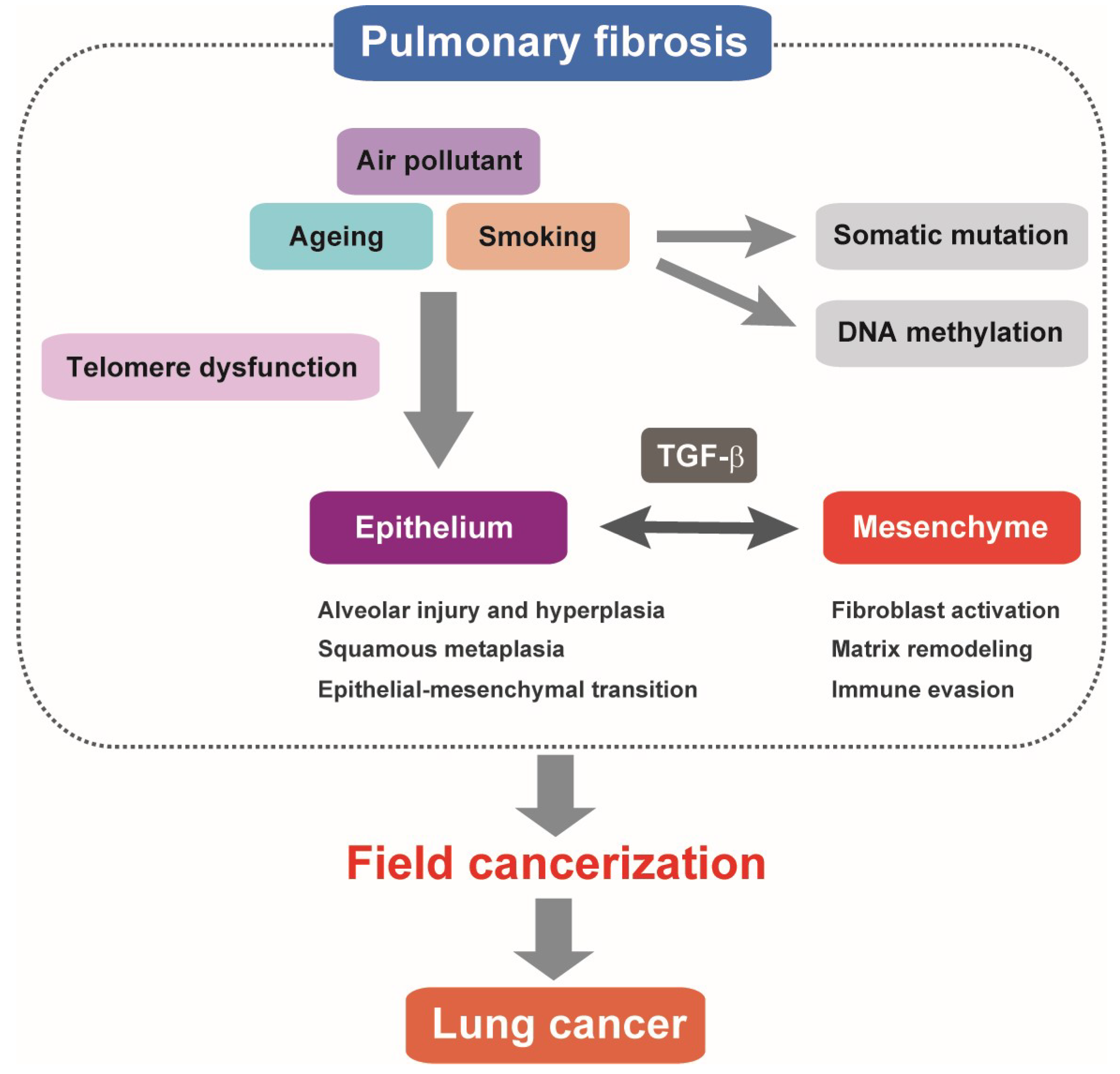
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Genetic Predispositions to Pulmonary Fibrosis
3. Genomic Features of Lung Cancer That Occurs in Pulmonary Fibrosis
4. Genome-Wide Methylation Profiles of Pulmonary Fibrosis and Lung Cancer
5. Roles of TGF-β in Alveolar Epithelial Cells
6. TGF-β-Mediated Fibroproliferative Reactions in Pulmonary Fibrosis and Lung Cancer
7. Clinical Implications and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.; Venn, A.; Lewis, S.; Britton, J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am. J. Respir. Crit. Care Med. 2000, 161, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Amatto, V.C.; Behr, J.; Stowasser, S. Comorbidities in idiopathic pulmonary fibrosis patients: A systematic literature review. Eur. Respir. J. 2015, 46, 1113–1130. [Google Scholar] [CrossRef] [PubMed]
- Le Jeune, I.; Gribbin, J.; West, J.; Smith, C.; Cullinan, P.; Hubbard, R. The incidence of cancer in patients with idiopathic pulmonary fibrosis and sarcoidosis in the UK. Respir. Med. 2007, 101, 2534–2540. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Suda, T.; Naito, T.; Enomoto, N.; Hashimoto, D.; Fujisawa, T.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology 2009, 14, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Tomassetti, S.; Gurioli, C.; Ryu, J.H.; Decker, P.A.; Ravaglia, C.; Tantalocco, P.; Buccioli, M.; Piciucchi, S.; Sverzellati, N.; Dubini, A.; et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest 2015, 147, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Ostman, A. Tumour-stroma interaction: Cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 2004, 45, S163–S175. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Nagase, T. Hippo and TGF-β interplay in the lung field. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L756–L767. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Kaiser, R.; Mellemgaard, A.; Douillard, J.Y.; Orlov, S.; Krzakowski, M.; von Pawel, J.; Gottfried, M.; Bondarenko, I.; Liao, M.; et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014, 15, 143–155. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.O.; Dellaripa, P.F.; Lederer, D.J.; Khanna, D.; Young, L.R.; Martinez, F.J. Interstitial lung disease: NHLBI Workshop on the primary prevention of chronic lung diseases. Ann. Am. Thorac. Soc. 2014, 11, S169–S177. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.O.; Ren, P.; Avila, N.A.; Chow, C.K.; Franks, T.J.; Travis, W.D.; McCoy, J.P.J.; May, R.M.; Wu, H.P.; Nguyen, D.M.; et al. Early interstitial lung disease in familial pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.; Malvezzi, M.; Negri, E.; La Vecchia, C.; Boffetta, P. Risk factors for lung cancer worldwide. Eur. Respir. J. 2016, 48, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Mushiroda, T.; Wattanapokayakit, S.; Takahashi, A.; Nukiwa, T.; Kudoh, S.; Ogura, T.; Taniguchi, H.; Kubo, M.; Kamatani, N.; Nakamura, Y. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J. Med. Genet. 2008, 45, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armanios, M.; Blackburn, E.H. The telomere syndromes. Nat. Rev. Genet. 2012, 13, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, R.J.; Porte, J.; Braybrooke, R.; Flores, C.; Fingerlin, T.E.; Oldham, J.M.; Guillen-Guio, B.; Ma, S.F.; Okamoto, T.; John, A.E.; et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome-wide association study. Lancet Respir. Med. 2017, 5, 869–880. [Google Scholar] [CrossRef]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunninghake, G.M.; Hatabu, H.; Okajima, Y.; Gao, W.; Dupuis, J.; Latourelle, J.C.; Nishino, M.; Araki, T.; Zazueta, O.E.; Kurugol, S.; et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N. Engl. J. Med. 2013, 368, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Oldham, J.M.; Ma, S.F.; Martinez, F.J.; Anstrom, K.J.; Raghu, G.; Schwartz, D.A.; Valenzi, E.; Witt, L.; Lee, C.; Vij, R.; et al. TOLLIP, MUC5B, and the response to n-acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.F.; Garcia, J.G.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.E.; et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef]
- Evans, C.M.; Fingerlin, T.E.; Schwarz, M.I.; Lynch, D.; Kurche, J.; Warg, L.; Yang, I.V.; Schwartz, D.A. Idiopathic pulmonary fibrosis: A genetic disease that involves mucociliary dysfunction of the peripheral airways. Physiol. Rev. 2016, 96, 1567–1591. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.G.; Livraghi-Butrico, A.; Fletcher, A.A.; McElwee, M.M.; Evans, S.E.; Boerner, R.M.; Alexander, S.N.; Bellinghausen, L.K.; Song, A.S.; Petrova, Y.M.; et al. Muc5b is required for airway defence. Nature 2014, 505, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Van Moorsel, C.H.; Van Oosterhout, M.F.; Barlo, N.P.; de Jong, P.A.; Van der Vis, J.J.; Ruven, H.J.; Van Es, H.W.; Van den Bosch, J.M.; Grutters, J.C. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med. 2010, 182, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S.; Loyd, J.E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1717–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogan, J.D.; Kropski, J.A.; Zhao, M.; Mitchell, D.B.; Rives, L.; Markin, C.; Garnett, E.T.; Montgomery, K.H.; Mason, W.R.; McKean, D.F.; et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2015, 191, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Bertuch, A.A. The molecular genetics of the telomere biology disorders. RNA Biol. 2016, 13, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.H.; Segal, M.; Boyraz, B.; Guinan, E.; Hofmann, I.; Cahan, P.; Tai, A.K.; Agarwal, S. Poly(A)-specific ribonuclease (PARN) mediates 3’-end maturation of the telomerase RNA component. Nat. Genet. 2015, 47, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Masutomi, K.; Possemato, R.; Wong, J.M.; Currier, J.L.; Tothova, Z.; Manola, J.B.; Ganesan, S.; Lansdorp, P.M.; Collins, K.; Hahn, W.C. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc. Natl. Acad. Sci. USA 2005, 102, 8222–8227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannier, J.B.; Sandhu, S.; Petalcorin, M.I.; Wu, X.; Nabi, Z.; Ding, H.; Boulton, S.J. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 2013, 342, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.Y.; Cramb, S.M.; Baade, P.D.; Youlden, D.R.; Nwogu, C.; Reid, M.E. The international epidemiology of lung cancer: Latest trends, disparities, and tumor characteristics. J. Thorac. Oncol. 2016, 11, 1653–1671. [Google Scholar] [CrossRef] [PubMed]
- Karampitsakos, T.; Tzilas, V.; Tringidou, R.; Steiropoulos, P.; Aidinis, V.; Papiris, S.A.; Bouros, D.; Tzouvelekis, A. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2017, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Incze, J.; Vaughan, C.W.J.; Lui, P.; Strong, M.S.; Kulapaditharom, B. Premalignant changes in normal appearing epithelium in patients with squamous cell carcinoma of the upper aerodigestive tract. Am. J. Surg. 1982, 144, 401–405. [Google Scholar] [CrossRef]
- Curtius, K.; Wright, N.A.; Graham, T.A. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 2018, 18, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.A.; Vachani, A.; Whitney, D.; Elashoff, M.; Porta Smith, K.; Ferguson, J.S.; Parsons, E.; Mitra, N.; Brody, J.; Lenburg, M.E.; et al. A bronchial genomic classifier for the diagnostic evaluation of lung cancer. N. Engl. J. Med. 2015, 373, 243–251. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hironaka, M.; Fukayama, M. Pulmonary fibrosis and lung carcinoma: A comparative study of metaplastic epithelia in honeycombed areas of usual interstitial pneumonia with or without lung carcinoma. Pathol. Int. 1999, 49, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Ogura, T.; Yokose, T.; Nagai, K.; Nishiwaki, Y.; Esumi, H. p53 gene alteration in atypical epithelial lesions and carcinoma in patients with idiopathic pulmonary fibrosis. Hum. Pathol. 2001, 32, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Munakata, M.; Ohtsuka, Y.; Nisihara, H.; Nasuhara, Y.; Kamachi-Satoh, A.; Dosaka-Akita, H.; Homma, Y.; Kawakami, Y. Expression and alteration of ras and p53 proteins in patients with lung carcinoma accompanied by idiopathic pulmonary fibrosis. Cancer 2002, 95, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masai, K.; Tsuta, K.; Motoi, N.; Shiraishi, K.; Furuta, K.; Suzuki, S.; Asakura, K.; Nakagawa, K.; Sakurai, H.; Watanabe, S.I.; et al. Clinicopathological, immunohistochemical, and genetic features of primary lung adenocarcinoma occurring in the setting of usual interstitial pneumonia pattern. J. Thorac. Oncol. 2016, 11, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Nakaoku, T.; Tsuta, K.; Tsuchihara, K.; Matsumoto, S.; Yoh, K.; Goto, K. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl. Lung Cancer Res. 2015, 4, 156–164. [Google Scholar] [PubMed]
- Duruisseaux, M.; Antoine, M.; Rabbe, N.; Rodenas, A.; Mc Leer-Florin, A.; Lacave, R.; Poulot, V.; Duchêne, B.; Van Seuningen, I.; Cadranel, J.; et al. Lepidic predominant adenocarcinoma and invasive mucinous adenocarcinoma of the lung exhibit specific mucin expression in relation with oncogenic drivers. Lung Cancer 2017, 109, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Shin, D.H.; Kim, K.B.; Shin, N.; Park, W.Y.; Lee, J.H.; Choi, K.U.; Kim, J.Y.; Lee, C.H.; Sol, M.Y.; et al. MUC5AC and MUC5B enhance the characterization of mucinous adenocarcinomas of the lung and predict poor prognosis. Histopathology 2015, 67, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.A.; Kim, D.; Chun, S.M.; Bae, S.; Song, J.S.; Kim, M.Y.; Koo, H.J.; Song, J.W.; Kim, W.S.; Lee, J.C.; et al. Genomic profiles of lung cancer associated with idiopathic pulmonary fibrosis. J. Pathol. 2018, 244, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Kaczkowski, B.; Ohshima, M.; Matsuzaki, H.; Noguchi, S.; Mikami, Y.; Lizio, M.; Itoh, M.; Kawaji, H.; Lassmann, T.; et al. Integrative CAGE and DNA methylation profiling identify epigenetically regulated genes in NSCLC. Mol. Cancer Res. 2017, 15, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chen, X.; Hong, Q.; Deng, Z.; Ma, H.; Xin, Y.; Fang, Y.; Ye, H.; Wang, R.; Zhang, C.; et al. Meta-analyses of gene methylation and smoking behavior in non-small cell lung cancer patients. Sci. Rep. 2015, 5, 8897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djureinovic, D.; Hallström, B.M.; Horie, M.; Mattsson, J.S.; La Fleur, L.; Fagerberg, L.; Brunnström, H.; Lindskog, C.; Madjar, K.; Rahnenführer, J.; et al. Profiling cancer testis antigens in non-small-cell lung cancer. JCI Insight 2016, 1, e86837. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Ambalavanan, N.; Halloran, B.; Zhang, X.; Liu, H.; Crossman, D.K.; Bray, M.; Zhang, K.; Thannickal, V.J.; Hagood, J.S. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Pedersen, B.S.; Rabinovich, E.; Hennessy, C.E.; Davidson, E.J.; Murphy, E.; Guardela, B.J.; Tedrow, J.R.; Zhang, Y.; Singh, M.K.; et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.V.; Yu, G.; Yakhini, Z.; Kaminski, N. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e33770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, W.E.; Cheng, D.S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Ibarra, G.H.; Kaminski, N. Fibrosis: Lessons from OMICS analyses of the human lung. Matrix Biol. 2018, 68, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Kolahian, S.; Fernandez, I.E.; Eickelberg, O.; Hartl, D. Immune mechanisms in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Broekelmann, T.J.; Limper, A.H.; Colby, T.V.; McDonald, J.A. Transforming growth factor β1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 1991, 88, 6642–6646. [Google Scholar] [CrossRef] [PubMed]
- Coker, R.K.; Laurent, G.J.; Jeffery, P.K.; du Bois, R.M.; Black, C.M.; McAnulty, R.J. Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax 2001, 56, 549–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.G.; Cho, S.J.; Kang, M.J.; Chapoval, S.P.; Lee, P.J.; Noble, P.W.; Yehualaeshet, T.; Lu, B.; Flavell, R.A.; Milbrandt, J.; et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J. Exp. Med. 2004, 200, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Investig. 1997, 100, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.M.; Mineo-Kuhn, M.M.; Kramer, C.M.; Finkelstein, J.N. Growth factors alter neonatal type II alveolar epithelial cell proliferation. Am. J. Physiol. 1994, 266, L17–L22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Kage, H.; Borok, Z. EMT and interstitial lung disease: A mysterious relationship. Curr. Opin. Pulm. Med. 2012, 18, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, M.; Saito, A.; Mikami, Y.; Ohshima, M.; Morishita, Y.; Nakajima, J.; Kohyama, T.; Nagase, T. Characterization of human lung cancer-associated fibroblasts in three-dimensional in vitro co-culture model. Biochem. Biophys. Res. Commun. 2012, 423, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Suzuki, H.I.; Horie, M.; Ohshima, M.; Morishita, Y.; Abiko, Y.; Nagase, T. An integrated expression profiling reveals target genes of TGF-β and TNF-α possibly mediated by microRNAs in lung cancer cells. PLoS ONE 2013, 8, e56587. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Ostman, A. Exploring the tumour environment: Cancer-associated fibroblasts as targets in cancer therapy. Expert. Opin. Ther. Targets 2005, 9, 1217–1233. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Saito, A.; Noguchi, S.; Yamaguchi, Y.; Ohshima, M.; Morishita, Y.; Suzuki, H.I.; Kohyama, T.; Nagase, T. Differential knockdown of TGF-β ligands in a three-dimensional co-culture tumor-stromal interaction model of lung cancer. BMC Cancer 2014, 14, 580. [Google Scholar] [CrossRef] [PubMed]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef] [PubMed]
- Yamanouchi, H.; Fujita, J.; Yoshinouchi, T.; Hojo, S.; Kamei, T.; Yamadori, I.; Ohtsuki, Y.; Ueda, N.; Takahara, J. Measurement of hepatocyte growth factor in serum and bronchoalveolar lavage fluid in patients with pulmonary fibrosis. Respir. Med. 1998, 92, 273–278. [Google Scholar] [CrossRef]
- Wang, W.; Li, Q.; Yamada, T.; Matsumoto, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Kayano, Y.; Nishioka, Y.; Sone, S.; et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin. Cancer Res. 2009, 15, 6630–6638. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T. cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, J.C.; Osterholzer, J.J.; Marazioti, A.; Stathopoulos, G.T. “Scar-cinoma”: Viewing the fibrotic lung mesenchymal cell in the context of cancer biology. Eur. Respir. J. 2016, 47, 1842–1854. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Kiura, K.; Takigawa, N.; Yoshioka, H.; Harita, S.; Kuyama, S.; Yonei, T.; Fujiwara, K.; Maeda, T.; Aoe, K.; et al. Comparison of the incidence and pattern of interstitial lung disease during erlotinib and gefitinib treatment in Japanese Patients with non-small cell lung cancer: The Okayama Lung Cancer Study Group experience. J. Thorac. Oncol. 2010, 5, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, A.; Kishi, K.; Yoshimura, K. A nationwide survey concerning lung surgery for lung cancer associated with idiopathic interstitial pneumonia. Nihon. Kokyuki. Gakkai. Zasshi. 2011, 49, 148–150. [Google Scholar] [PubMed]
- Sato, T.; Teramukai, S.; Kondo, H.; Watanabe, A.; Ebina, M.; Kishi, K.; Fujii, Y.; Mitsudomi, T.; Yoshimura, M.; Maniwa, T.; et al. Impact and predictors of acute exacerbation of interstitial lung diseases after pulmonary resection for lung cancer. J. Thorac. Cardiovasc. Surg. 2014, 147, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.J.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Kardatzke, D.R.; Daigl, M.; Kirchgaessler, K.U.; Lancaster, L.H.; et al. Effect of pirfenidone on mortality: Pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir. Med. 2017, 5, 33–41. [Google Scholar] [CrossRef]
- Richeldi, L.; Cottin, V.; du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS® trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Saito, A.; Ohshima, M.; Suzuki, H.I.; Nagase, T. YAP and TAZ modulate cell phenotype in a subset of small cell lung cancer. Cancer Sci. 2016, 107, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, N.; Horie, M.; Suzuki, H.I.; Yoshihara, M.; Djureinovic, D.; Persson, J.; runnström, H.; Lindskog, C.; Elfving, H.; Micke, P.; et al. An integrative analysis of transcriptome and epigenome features of ASCL1-positive lung adenocarcinomas. J. Thorac. Oncol. 2018, 13, 1676–1691. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tamiya, M.; Tamiya, A.; Nakahama, K.; Taniguchi, Y.; Shiroyama, T.; Isa, S.I.; Nishino, K.; Kumagai, T.; Kunimasa, K.; et al. Analysis of early death in japanese patients with advanced non-small-cell lung cancer treated with nivolumab. Clin. Lung Cancer 2018, 19, e171–e176. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, A.; Horie, M.; Micke, P.; Nagase, T. The Role of TGF-β Signaling in Lung Cancer Associated with Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 3611. https://doi.org/10.3390/ijms19113611
Saito A, Horie M, Micke P, Nagase T. The Role of TGF-β Signaling in Lung Cancer Associated with Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences. 2018; 19(11):3611. https://doi.org/10.3390/ijms19113611
Chicago/Turabian StyleSaito, Akira, Masafumi Horie, Patrick Micke, and Takahide Nagase. 2018. "The Role of TGF-β Signaling in Lung Cancer Associated with Idiopathic Pulmonary Fibrosis" International Journal of Molecular Sciences 19, no. 11: 3611. https://doi.org/10.3390/ijms19113611