A Nationwide Survey on Danon Disease in Japan

 ,
,
Abstract
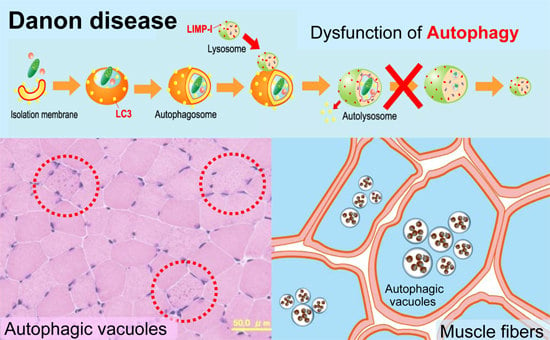
:
1. Introduction
2. Results
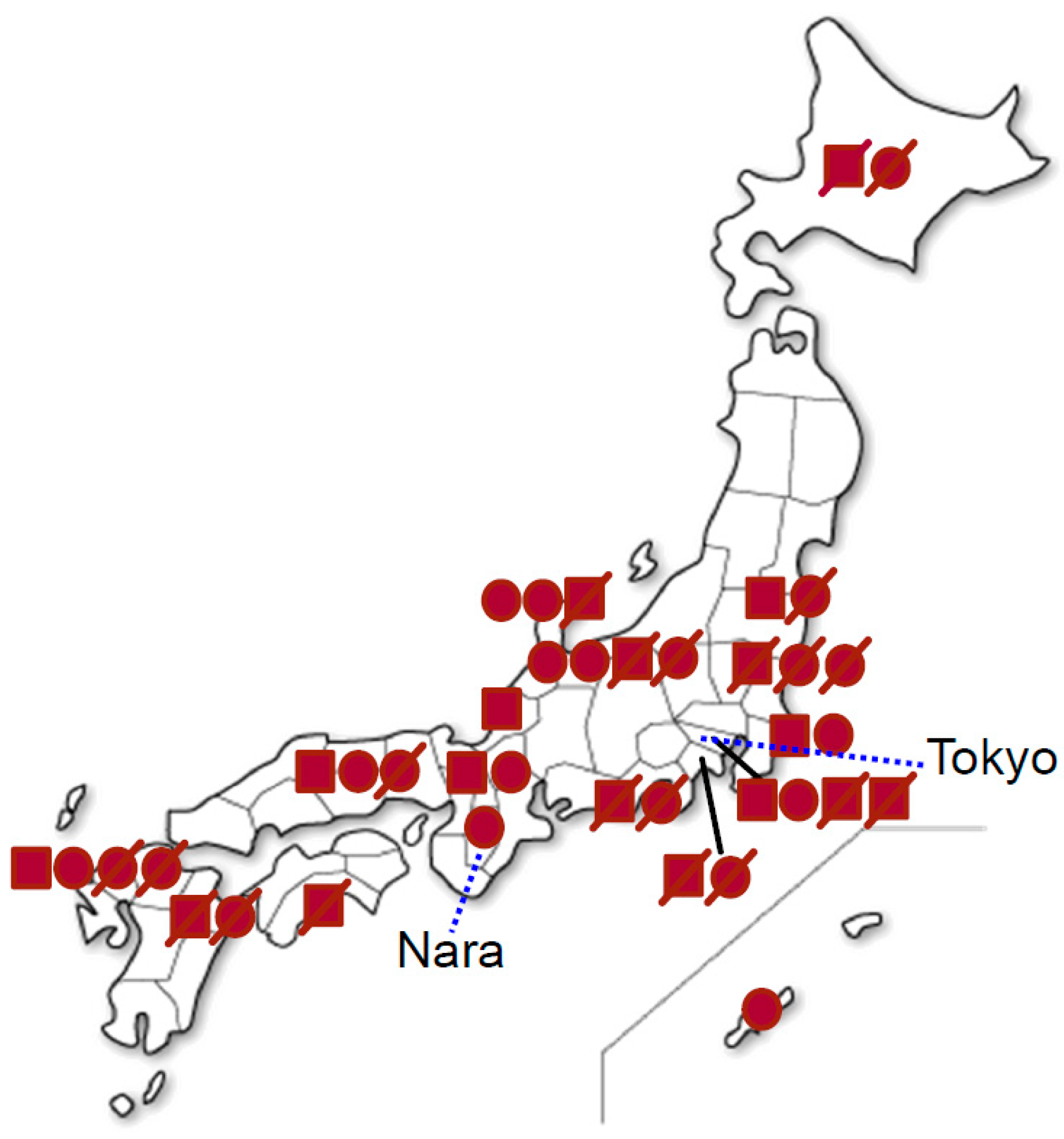
2.1. Patients
2.2. Clinical Features
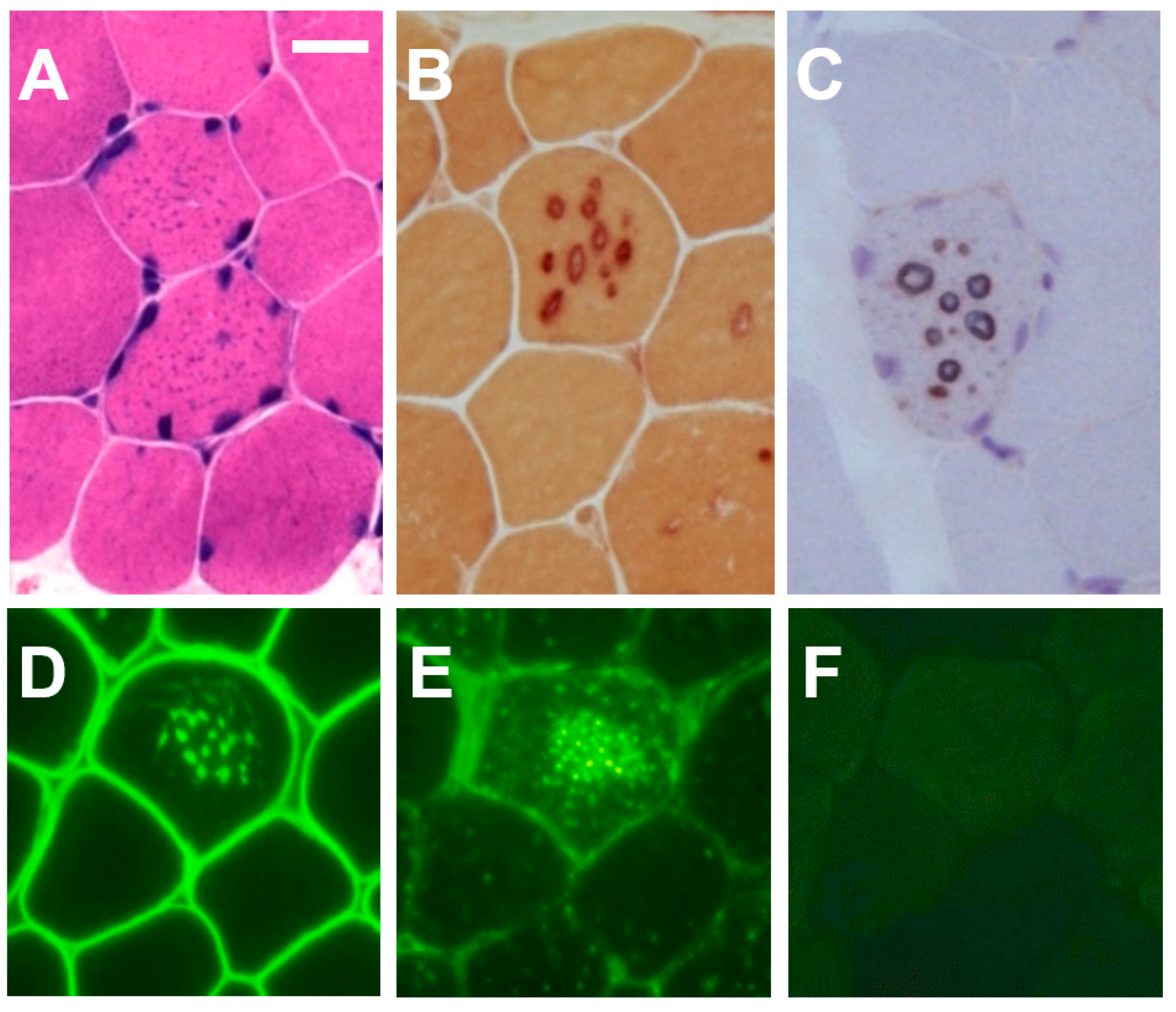
2.3. Histochemistry and Immunohistochemistry
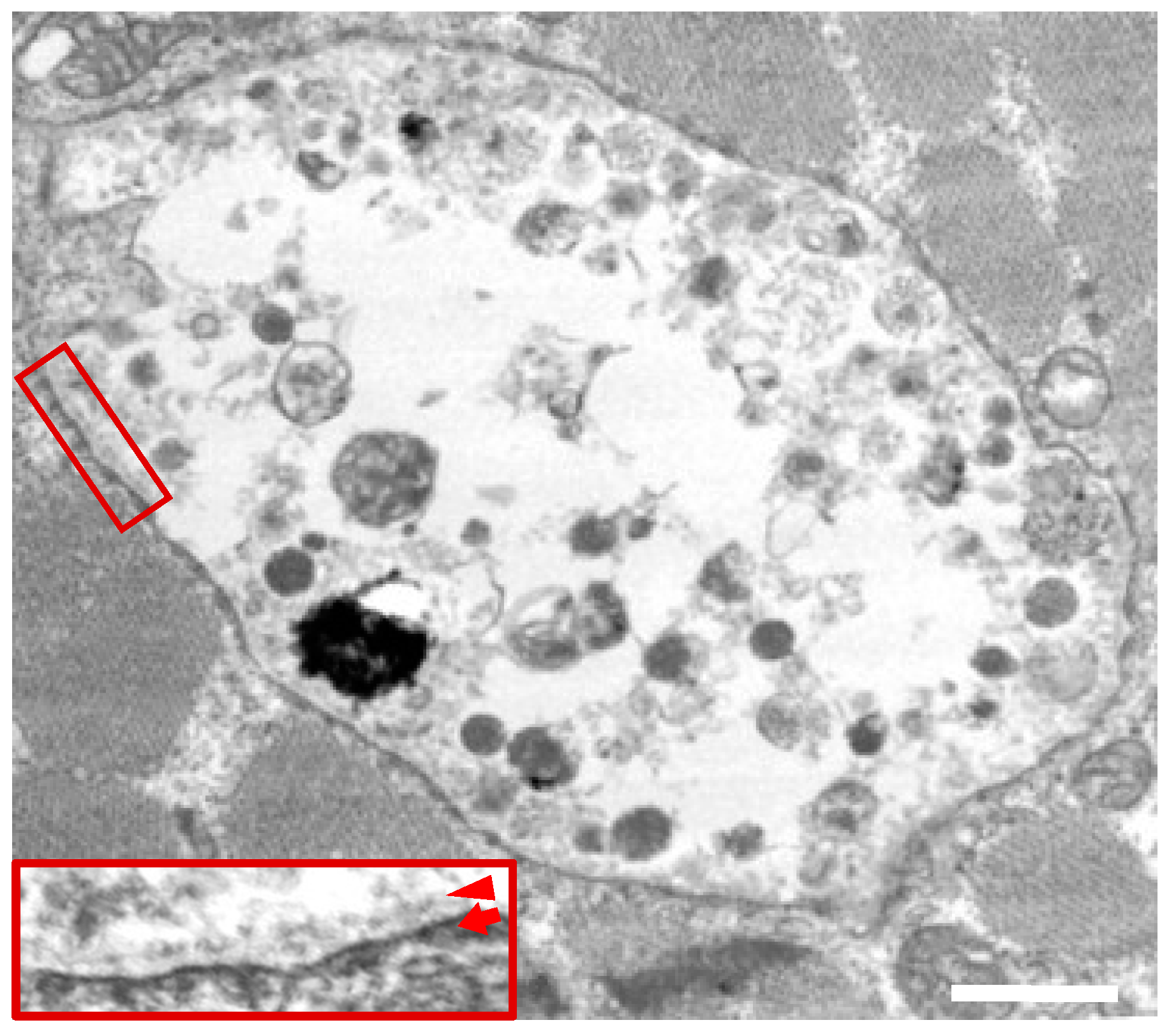
2.4. Electron Micsroscopy
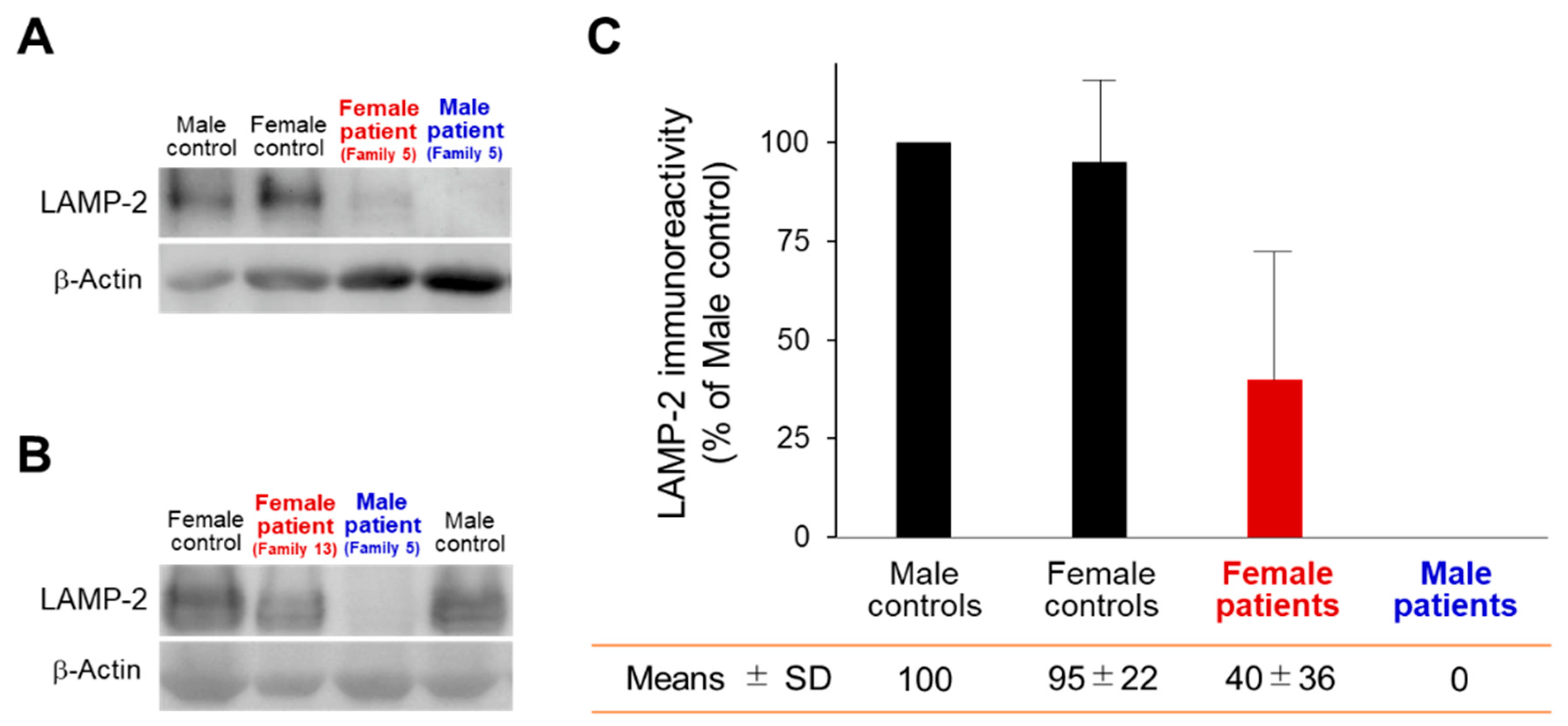
2.5. Western Blot Analysis
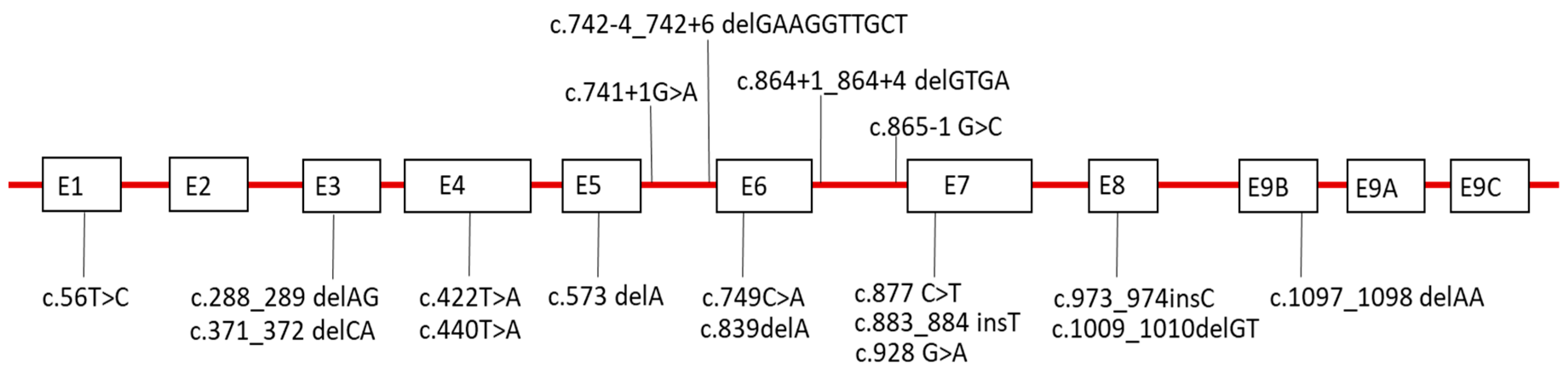
2.6. Sequence Analysis of LAMP-2
3. Discussion
4. Materials and Methods
4.1. Nationwide, Questionnaire-Based Survey
4.2. Histochemistry and Immunohistochemistry
4.3. Electron Microscopy
4.4. Western Blot Analysis of Muscle Specimens
4.5. Sequence Analysis of LAMP-2
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AVSF | autophagic vacuoles with sarcolemmal features |
| CK | creative kinase |
| CNS | central nervous system |
| EEG | electroencephalography |
| ECG | electrocardiogram |
| LAMP-2 | lysosome-associated membrane protein-2 |
| LIMP-1 | lysosomal integral membrane protein-1 |
| LVAD | left ventricular assist devices |
| WPW | Wolff–Parkinson–White |
References
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Sugie, K.; Yamamoto, A.; Murayama, K.; Oh, S.J.; Takahashi, M.; Mora, M.; Riggs, J.E.; Colomer, J.; Iturriaga, C.; Meloni, A.; et al. Clinicopathological features of genetically confirmed Danon disease. Neurology 2002, 58, 1773–1778. [Google Scholar] [CrossRef] [PubMed]
- Danon, M.J.; Oh, S.J.; DiMauro, S.; Manaligod, J.R.; Eastwood, A.; Naidu, S.; Schliselfeld, L.H. Lysosomal glycogen storage disease with normal acid maltase. Neurology 1981, 31, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Sugie, K.; Nishino, I. Lysosomal Membrane Disorders: LAMP-2 Deficiency. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 5th ed.; Rosenberg, R.N., Pascual, J.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 411–417. [Google Scholar]
- Nishino, I.; Yamamoto, A.; Sugie, K.; Hirano, M.; Nonaka, I. Danon disease and related disorders. Acta Myologica 2001, 20, 120–124. [Google Scholar]
- Sugie, K.; Noguchi, S.; Kozuka, Y.; Arikawa-Hirasawa, E.; Tanaka, M.; Yan, C.; Saftig, P.; von Figura, K.; Hirano, M.; Ueno, S.; et al. Autophagic vacuoles with sarcolemmal features delineate Danon disease and related myopathies. J. Neuropathol. Exp. Neurol. 2005, 64, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Furuta, A.; Nishino, I. Danon disease: A phenotypic expression of LAMP-2 deficiency. Acta Neuropathol. 2015, 129, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Sugie, K.; Koori, T.; Yamamoto, A.; Ogawa, M.; Hirano, M.; Inoue, K.; Nonaka, I.; Nishino, I. Characterization of Danon disease in a male patient and his affected mother. Neuromuscul. Disord. 2003, 13, 708–711. [Google Scholar] [CrossRef]
- Sugie, K.; Yoshizawa, H.; Onoue, K.; Nakanishi, Y.; Eura, N.; Ogawa, M.; Nakano, T.; Sakaguchi, Y.; Hayashi, Y.K.; Kishimoto, T.; et al. Early onset of cardiomyopathy and intellectual disability in a girl with Danon disease associated with a de novo novel mutation of the LAMP-2 gene. Neuropathology 2016, 36, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Yamamoto, A.; Takano, K.; Sudo, A.; Wada, T.; Goto, Y.; Nishino, I.; Saitoh, S. Germline mosaicism of a novel mutation in lysosome-associated membrane protein-2 deficiency (Danon disease). Ann. Neurol. 2002, 52, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Harimura, Y.; Yamasaki, H.; Sekiguchi, Y.; Ishizu, T.; Seo, Y.; Kawano, S.; Aonuma, K. Utility of real-time 3-dimensional echocardiography and magnetic resonance imaging for evaluation of Danon disease. Circulation 2010, 121, e390–e392. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H.; Yamagishi, T.; Dobashi, T.; Nakazawa, M.; Hayashi, T.; Furumichi, K. A case of Danon disease with paroxysmal supraventricular tachycardia (translation). Pediatr. Cardiol. Card. Surg. 2004, 20, 417. (In Japanese) [Google Scholar]
- Dougu, N.; Joho, S.; Shan, L.; Shida, T.; Matsuki, A.; Uese, K.; Hirono, K.; Ichida, F.; Tanaka, K.; Nishino, I.; et al. Novel LAMP-2 mutation in a family with Danon disease presenting with hypertrophic cardiomyopathy. Circ. J. 2009, 73, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Awaya, T.; Yoshida, T.; Shibata, M.; Kato, T. Flowcytometric diagnosis of Danon disease (LAMP-2 Deficiency) (translation). No to Hattatsu 2012, 44, S183. (In Japanese) [Google Scholar]
- Tamura, T.; Nishida, K. A case of Danon disease diagnosed by hypertrophic cardiomyopathy complicated with eye lesions (translation). Pediatr. Cardiol. Card. Surg. 2010, 26, s255. (In Japanese) [Google Scholar]
- Hashida, Y.; Wada, T.; Saito, T.; Ohta, K.; Kasahara, Y.; Yachie, A. Early diagnosis of Danon disease: Flow cytometric detection of lysosome-associated membrane protein-2-negative leukocytes. J. Cardiol. 2015, 66, 168–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitahara, H.; Nawata, K.; Kinoshita, O.; Itoda, Y.; Shintani, Y.; Fukayama, M.; Ono, M. Implantation of a Left Ventricular Assist Device for Danon Cardiomyopathy. Ann. Thorac. Surg. 2017, 103, e39–e41. [Google Scholar] [CrossRef] [PubMed]
- Namatame, S.; Segawa, M.; Matsuda, N.; Hoshi, A.; Ugawa, Y. A case of Danon disease (translation). Rinsho Shinkeigaku 2015, 55, 62. (In Japanese) [Google Scholar]
- Nguyen, H.T.; Noguchi, S.; Sugie, K.; Matsuo, Y.; Nguyen, C.T.H.; Koito, H.; Shiojima, I.; Nishino, I.; Tsukaguchi, H. Small-Vessel Vasculopathy Due to Aberrant Autophagy in LAMP-2 Deficiency. Sci. Rep. 2018, 8, 3326. [Google Scholar] [CrossRef] [PubMed]
- Takumi, Y.; Akagi, T.; Hiratsuka, T.; Shibata, T.; Ueda, T.; Tojigamori, M.; Shiroshita, H.; Etoh, T.; Inomata, M.; Noguchi, T.; et al. Laparoscopic transverse colon resection in a patient of juvenile transverse colon cancer with hypertrophic cardiomyopathy due to Danon disease (translation). J. Jpn. Surg. Assoc. 2015, 76, 2852. (In Japanese) [Google Scholar]
- Nagatomo, Y.; Kuraoka, A.; Muneuchi, J.; Terashi, E.; Sugitani, Y.; Takenaka, S.; Watanabe, M.; Shiroo, K. A girl patient of Danon disease diagnosed because of school cardiac examination (translation). J. Jpn. Pediatr. Soc. 2014, 118, 1014. (In Japanese) [Google Scholar]
- Yamamoto, K.; Ootsuka, Y.; Miyake, K.; Takahashi, K.; Nakayashiro, M.; Takakuwa, H.; Fukushima, N.; Ichida, F.; Nishino, I. A girl patient of Danon disease with a novel LAMP-2 mutation diagnosed by severe heart failure (translation). J. Jpn. Pediatr. Soc. 2013, 117, 554. (In Japanese) [Google Scholar]
- Maron, B.J.; Roberts, W.C.; Arad, M.; Haas, T.S.; Spirito, P.; Wright, G.B.; Almquist, A.K.; Baffa, J.M.; Saul, J.P.; Ho, C.Y. Clinical outcome and phenotypic expression in LAMP-2 cardiomyopathy. JAMA 2009, 301, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; McMahon, C.J.; Smith, L.R.; Bersola, J.; Adesina, A.M.; Breinholt, J.P.; Kearney, D.L.; Dreyer, W.J.; Denfield, S.W.; Price, J.F.; et al. Danon disease as an underrecognized cause of hypertrophic cardiomyopathy in children. Circulation 2005, 112, 1612–1617. [Google Scholar] [CrossRef] [PubMed]
- Arad, M.; Maron, B.J.; Gorham, J.M.; Johnson, W.H., Jr.; Saul, J.P.; Perez-Atayde, A.R.; Spirito, P.; Wright, G.B.; Kanter, R.J.; Seidman, C.E.; et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med. 2005, 352, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Riggs, J.E.; Schochet, S.S., Jr.; Gutmann, L.; Shanske, S.; Neal, W.A.; DiMauro, S. Lysosomal glycogen storage disease without acid maltase deficiency. Neurology 1983, 33, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Mahdhaoui, A.; Bouraoui, H.; Tabarki, B.; Majdoub, M.; Trimeche, B.; Mahdhaoui, N.; Chabrak, S.; Ernez-Hajri, S.; Jeridi, G.; Ammar, H. Familial hypertrophic cardiomyopathy associated with Wolff-Parkinson-White syndrome. Acta. Clin. Belg. 2003, 58, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Marriott, H.T.L. Electrocardiographic abnormalities, conduction disorders and arrhythmias in primary myocardial disease. Prog. Cardiovasc. Dis. 1964, 7, 99–114. [Google Scholar] [CrossRef]
- Roos, J.C.P.; Daniels, M.J.; Morris, E.; Hyry, H.I.; Cox, T.M. Heterogeneity in a large pedigree with Danon disease: Implications for pathogenesis and management. Mol. Genet. Metab. 2018, 123, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Boucek, D.; Jirikowic, J.; Taylor, M. Natural history of Danon disease. Genet. Med. 2011, 13, 563–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedberg Oldfors, C.; Máthé, G.; Thomson, K.; Tulinius, M.; Karason, K.; Östman-Smith, I.; Oldfors, A. Early onset cardiomyopathy in females with Danon disease. Neuromuscul. Disord. 2015, 25, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Cho, A.; Lim, B.C.; Kim, M.J.; Kim, K.J.; Nishino, I.; Hwang, Y.S.; Chae, J.H. A 13-year-old girl with proximal weakness and hypertrophic cardiomyopathy with Danon disease. Muscle Nerve 2010, 41, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Dworzak, F.; Casazza, F.; Mora, M.; De Maria, R.; Gronda, E.; Baroldi, G.; Rimoldi, M.; Morandi, L.; Cornelio, F. Lysosomal glycogen storage with normal acid maltase: A familial study with successful heart transplant. Neuromuscul. Disord. 1994, 4, 243–247. [Google Scholar] [CrossRef]
- Furuta, A.; Kikuchi, H.; Fujita, H.; Yamada, D.; Fujiwara, Y.; Kabuta, T.; Nishino, I.; Wada, K.; Uchiyama, Y. Property of lysosomal storage disease associated with midbrain pathology in the central nervous system of Lamp-2-deficient mice. Am. J. Pathol. 2015, 185, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Prall, F.R.; Drack, A.; Taylor, M.; Ku, L.; Olson, J.L.; Gregory, D.; Mestroni, L.; Mandava, N. Ophthalmic manifestations of Danon disease. Ophthalmology 2006, 113, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Musumeci, O.; Paleologo, G.; Cucinotta, M.; Migliorato, A.; Rodolico, C.; Toscano, A. Ischemic stroke due to hypoperfusion in a patient with a previously unrecognized Danon disease. Neuromuscul. Disord. 2016, 26, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Bottillo, I.; Giordano, C.; Cerbelli, B.; D’Angelantonio, D.; Lipari, M.; Polidori, T.; Majore, S.; Bertini, E.; D’Amico, A.; Giannarelli, D.; et al. A novel LAMP2 mutation associated with severe cardiac hypertrophy and microvascular remodeling in a female with Danon disease: A case report and literature review. Cardiovasc. Pathol. 2016, 25, 423–431. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Site of LAMP-2 Mutation | LAMP-2 Mutation | Effect on mRNA | Affected Males | Affected Females | References |
|---|---|---|---|---|---|---|
| 1 | E9b | c.1097_1098 delAA | Frame shift | proband, 1 cousin | mother, 1 sister | [2] |
| 2 | E4 | c.440 T > A | Stop codon | proband | - | [2] |
| 3 | I6 | c.865-1 G > C | Splicing | proband | mother | [2] |
| 4 | I5 | c.741+1G>A | Stop codon | proband | - | [2] |
| 5 | I5 | c.742-4_742+6 delGAAGGTTGCT | Splicing | proband | mother | [2,8] |
| 6 | E7 | c.883_884 insT | Frame shift | proband | 1 sister | [2,10] |
| 7 | E3 | c.288_289 delAG | Stop codon | proband | mother, 1 aunt | [11] |
| 8 | E7 | c.928 G>A | Splicing | proband | - | [12] |
| 9 | E5 | c.573 delA | Frame shift | proband | mother, 2 sisters | [13] |
| 10 | E7 | c.877 C>T | Stop codon | proband | mother, 1 sister | [14] |
| 11 | E3 | c.371_372 delCA | Stop codon | proband | - | [15] |
| 12 | I6 | c.864+1_864+4 delGTGA | Splicing | proband | 2 sisters | [16] |
| 13 | E6 | c.749C>A | Stop codon | - | proband | [9] |
| 14 | E7 | c.877C>T | Stop codon | proband | mother | [17] |
| 15 | E1 | c.56T>C | Frame shift | proband | mother | [18] |
| 16 | E8 | c.1009_1010delGT | Frame shift | proband | mother | [19] |
| 17 | NA | NA | NA | proband | mother | [20] |
| 18 | E8 | c.973_974insC | Frame shift | - | proband | [21] |
| 19 | E4 | c.422T>A | Stop codon | - | proband | [22] |
| 20 | E6 | c.839delA | Stop codon | - | proband | [17] |
| Characteristics | Male | Female |
|---|---|---|
| Subjects, n | 17 | 22 |
| Probands, n | 16 | 4 |
| Manifesting mothers of the probands, n (%) | 10/16 (63%) | 0/4 (0%) |
| Age at onset, n | ||
| Infantile | 4 | 0 |
| Childhood | 9 | 1 |
| Second decade | 4 | 5 |
| Adult | 0 | 1 |
| Unknown | 0 | 15 |
| Alive at the time of the survey, n | 7 | 11 |
| Dead at the time of the survey, n | 10 | 11 |
| Age at death, year-old, mean ± SD | 19 ± 5 (n = 7) | 37 ± 11 (n = 7) |
| Cause of death, n (%) | ||
| Cardiac failure | 9/10 (90%) | 11/11 (100%) |
| Cancer | 1/10 (10%) | 0/0 (0%) |
| Myopathy, n (%) | 17/17 (100%) | 2/22 (9%) |
| Muscle weakness | 13/17 (76%) | 2/22 (9%) |
| No weakness with myogenic change on EMG | 4/17 (24%) | 0/22 (0%) |
| Cardiomyopathy, n (%) | 17/17 (100%) | 21/22 (95%) |
| Hypertrophic | 15/17 (88%) | 11/22 (50%) |
| Dilated | 2/17 (12%) | 6/22 (27%) |
| Unknown | 0/17 (0%) | 4/22 (18%) |
| Mental retardation, n (%) | 8/17 (46%) | 2/22 (9%) |
| Retinopathy, n (%) | 1/17 (6%) | 1/22 (5%) |
| Pes cavus, n (%) | 1/17 (6%) | 1/22 (5%) |
| Cerebral infarction, n (%) | 0/17 (0%) | 2/22 (9%) |
| Heart transplantation, n (%) | 0/17 (0%) | 1/22 (5%) |
| Awaiting heart transplantation, n (%) | 2/17 (12%) | 3/22 (14%) |
| Left ventricular assist device, n (%) | 1/17 (6%) | 4/22 (18%) |
| Treatment of blocker, n (%) | 6/17 (35%) | 11/22 (50%) |
| Elevated CK, n (%) | 16/17 (94%) | 3/12 (25%) |
| Serum CK (IU/L), mean ± SD | 1086 ± 715 | 148 ± 164 |
| Abnormal ECG, n (%) | 17/17 (100%) | 18/18 (100%) |
| WPW syndrome, n (%) | 9/17 (54%) | 4/18 (22%) |
| Abnormal echocardiogram, n (%) | 17/17 (100%) | 17/18 (94%) |
| Myogenic EMG, n (%) | 11/11 (100%) | 0/6 (0%) |
| Abnormal nerve conduction study, n (%) | 1/12 (8%) | 0/1 (0%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugie, K.; Komaki, H.; Eura, N.; Shiota, T.; Onoue, K.; Tsukaguchi, H.; Minami, N.; Ogawa, M.; Kiriyama, T.; Kataoka, H.; et al. A Nationwide Survey on Danon Disease in Japan. Int. J. Mol. Sci. 2018, 19, 3507. https://doi.org/10.3390/ijms19113507
Sugie K, Komaki H, Eura N, Shiota T, Onoue K, Tsukaguchi H, Minami N, Ogawa M, Kiriyama T, Kataoka H, et al. A Nationwide Survey on Danon Disease in Japan. International Journal of Molecular Sciences. 2018; 19(11):3507. https://doi.org/10.3390/ijms19113507
Chicago/Turabian StyleSugie, Kazuma, Hirofumi Komaki, Nobuyuki Eura, Tomo Shiota, Kenji Onoue, Hiroyasu Tsukaguchi, Narihiro Minami, Megumu Ogawa, Takao Kiriyama, Hiroshi Kataoka, and et al. 2018. "A Nationwide Survey on Danon Disease in Japan" International Journal of Molecular Sciences 19, no. 11: 3507. https://doi.org/10.3390/ijms19113507