Anti-NKG2D mAb: A New Treatment for Crohn’s Disease?


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
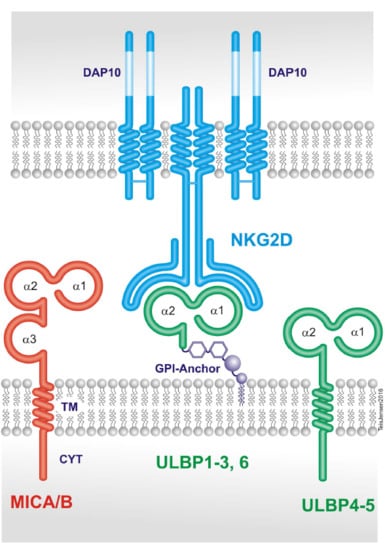
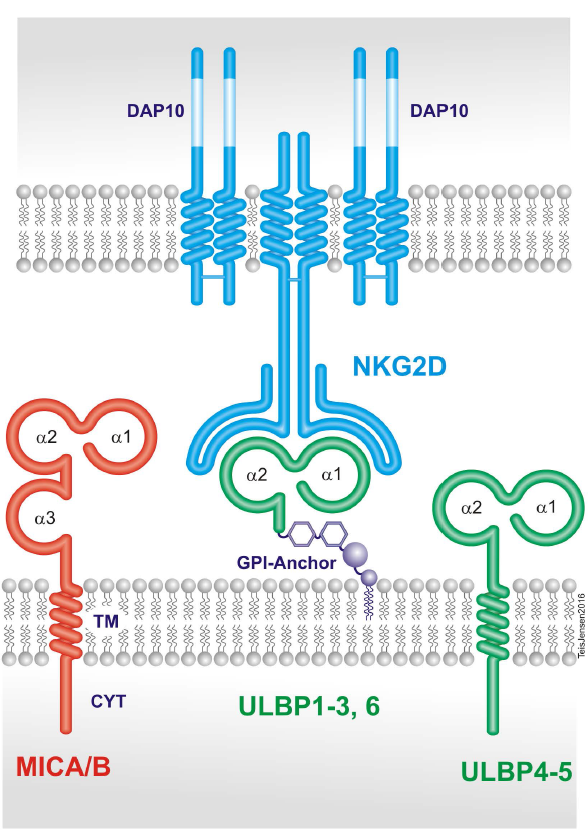
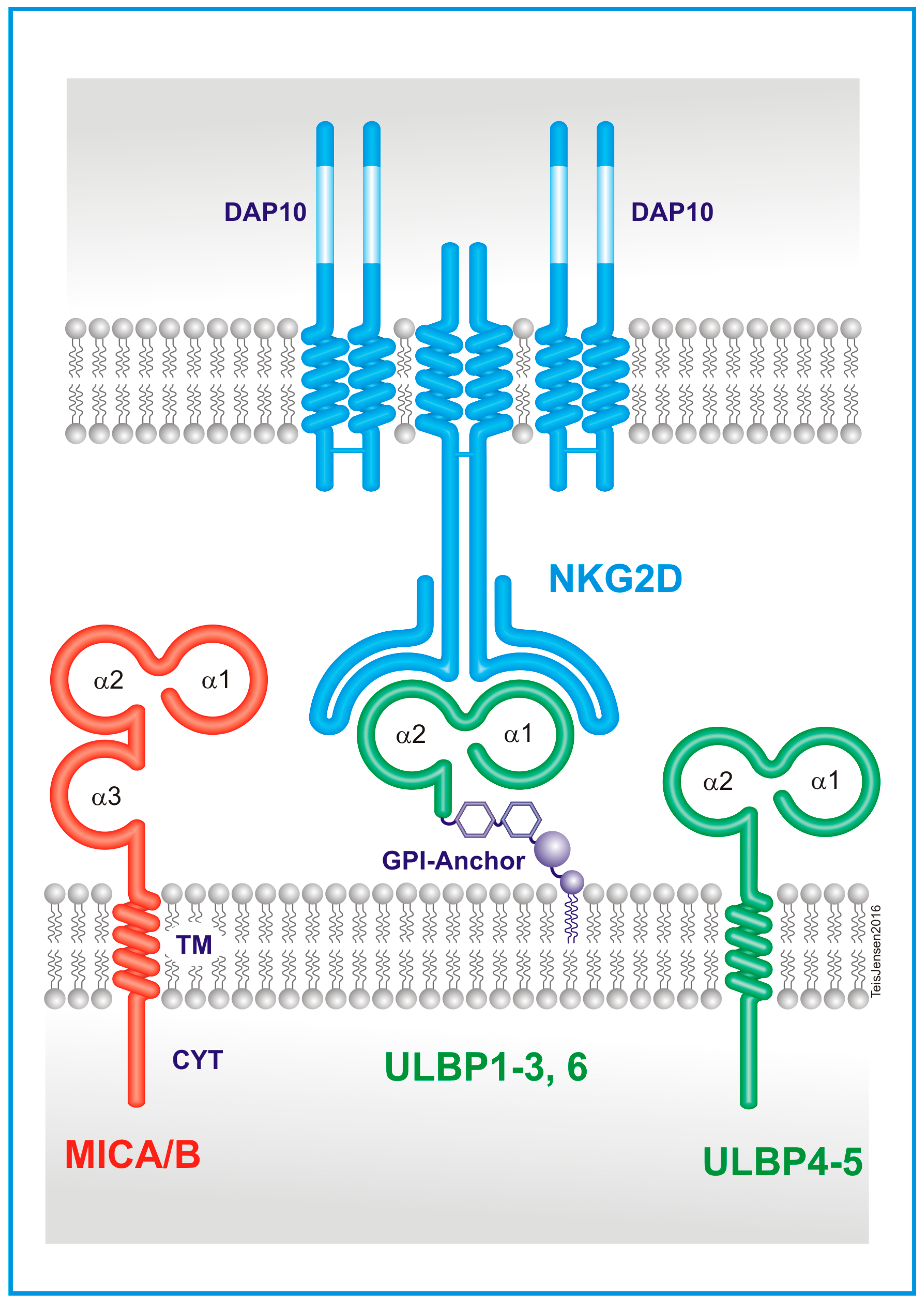
2. The Natural Killer Group 2D (NKG2D) Receptor
3. The NKG2D Ligands
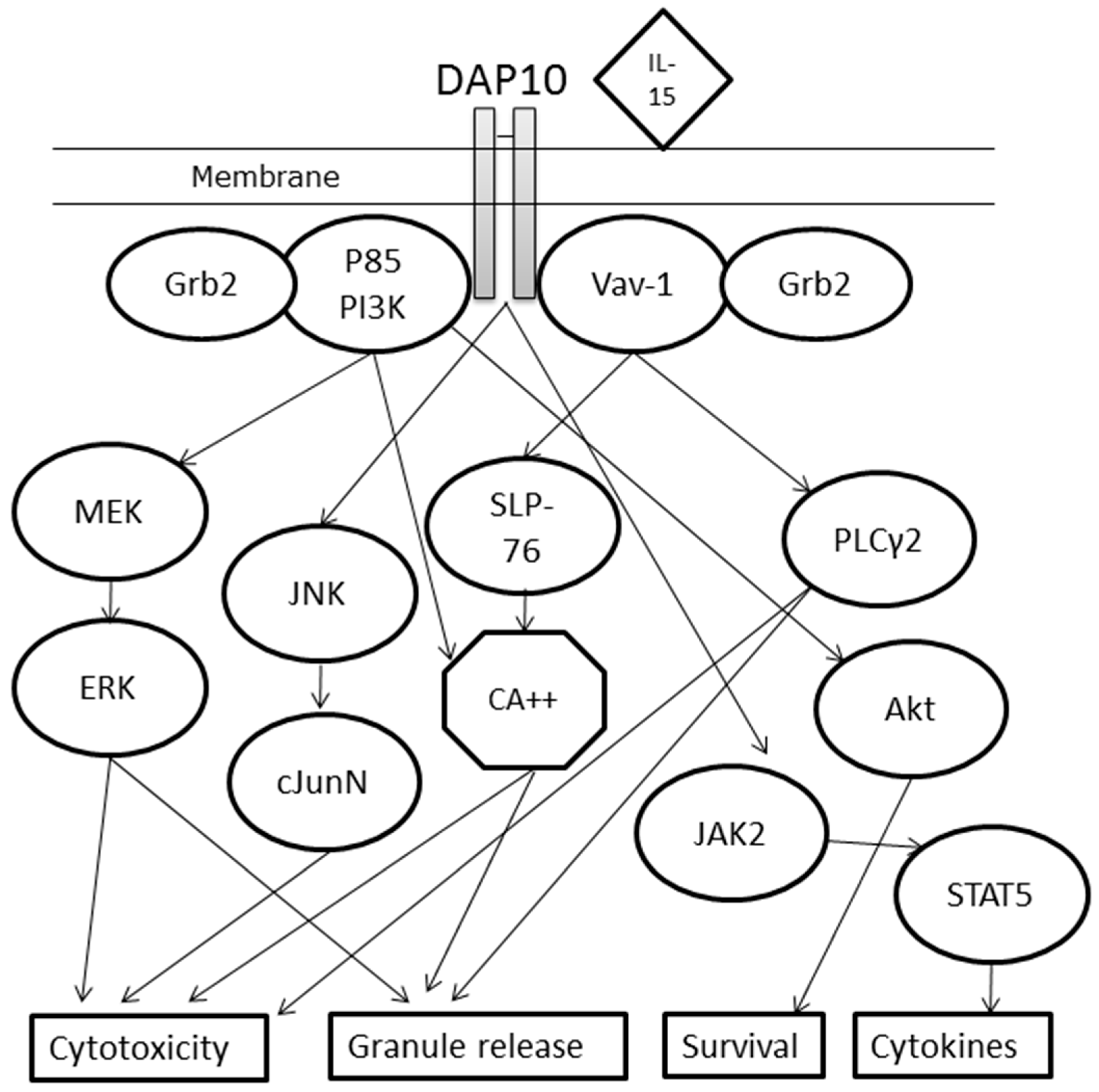
4. Immunological Pathway of NKG2D
5. Cancer Evasion and Infection Control
6. NKG2D in Crohn’s Disease
6.1. Upregulated Ligands in Crohn’s Disease
6.2. NKG2D Pathway Contribution to CD Pathology
7. Clinical Development
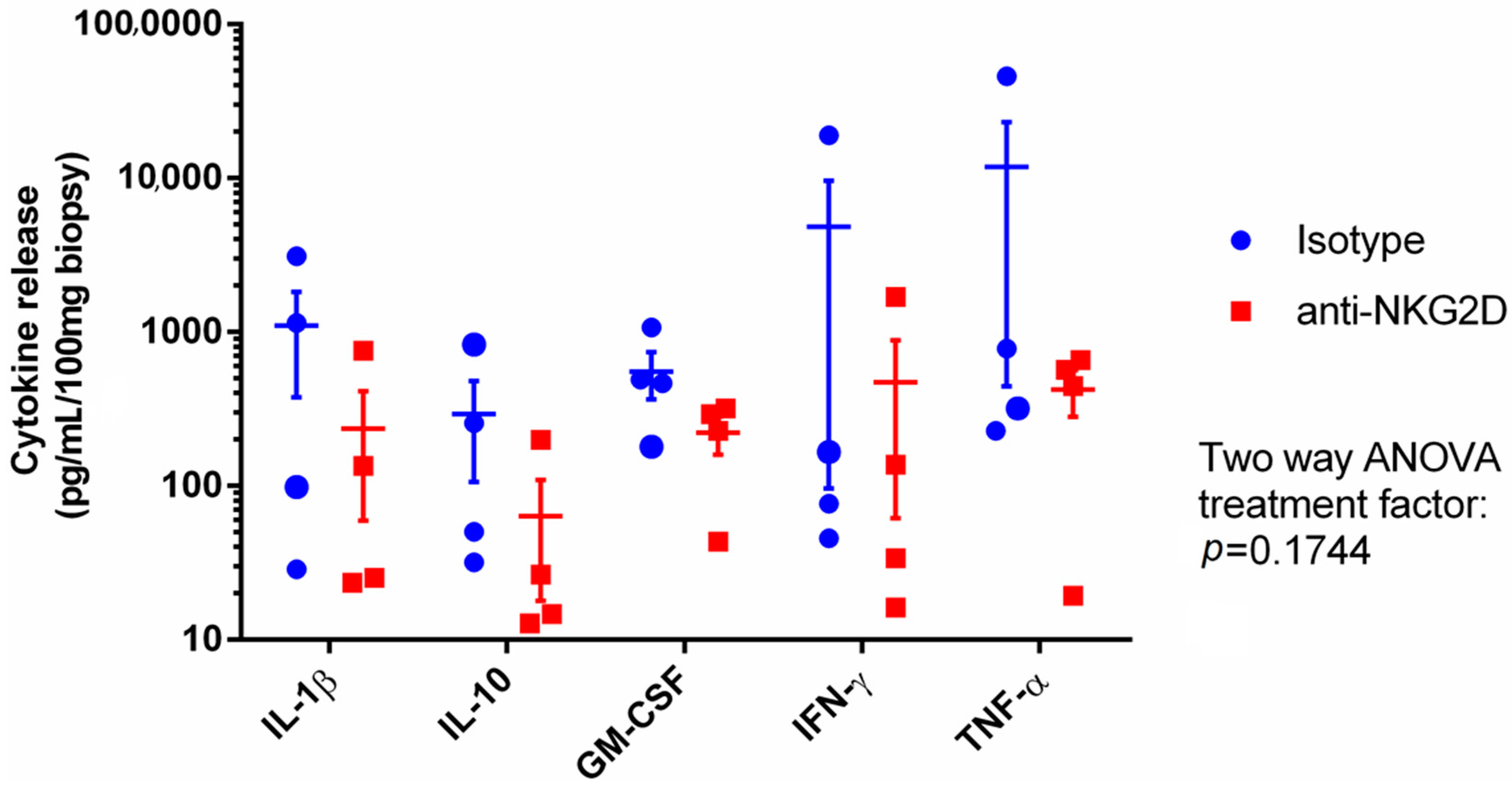
7.1. Phase IIa Results
7.2. Phase IIb Initiation
8. Future Perspectives
9. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| ANOVA | Analysis of variance |
| CD | Crohn’s disease |
| cJunN | c-Jun N-terminal kinase-1 |
| CRP | C-reactive protein |
| DAP10 | DNAX-activating protein of 10 kDa |
| ERK | Extracellular signal-Regulated Kinase |
| Grb2 | Growth factor receptor-bound protein 2 |
| GM-CSF | Granulocyte macrophage colony-stimulating factor |
| GPI | Glycosylphosphatidylinositol |
| IBD | Inflammatory bowel disease |
| IEL | Intraepithelial lymphocyte |
| IFN | Interferon |
| IL | Interleukin |
| ILC | Innate like lymphocyte |
| JAK2 | Janus Kinase 2 |
| JNK | c-Jun N-terminal protein Kinases |
| mAB | Monoclonal antibody |
| MHC | Major histocompatibility complex |
| MIC | MHC class I polypeptide-related sequence |
| MEK | Mitogen-activated protein kinase kinase |
| NK | Natural killer |
| NKG2D | Natural killer group 2D |
| PLCγ2 | Phospholipase C γ-2 |
| P85 PI3K | P85 subunit of phosphoInositide 3-kinase |
| RNA | Ribonucleic acid |
| SEM | Standard error of the mean |
| SLP-76 | SH2 domain-containing leukocyte phosphoprotein of 76 kDa |
| STAT5 | Signal transducer and activator of transcription 5 |
| TCR | T cell receptor |
| Th | T helper cell |
| TM | Transmembrane |
| TNF | Tumor necrosis factor |
| ULBP | UL16 binding protein |
| Vav-1 | Vav guanine nucleotide exchange factor 1 |
References
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Laass, M.W.; Roggenbuck, D.; Conrad, K. Diagnosis and classification of Crohn’s disease. Autoimmun. Rev. 2014, 13, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Busch, K.; Sonnenberg, A.; Bansback, N. Impact of inflammatory bowel disease on disability. Curr. Gastroenterol. Rep. 2014, 16, 414. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.B.; Nanau, R.M.; Delzor, F.; Neuman, M.G. Biologic therapies in inflammatory bowel disease. Transl. Res. J. Lab. Clin. Med. 2014, 163, 533–556. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Cheon, J.H. Pathogenesis of inflammatory bowel disease and recent advances in biologic therapies. Immune Netw. 2017, 17, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Katsanos, K.H.; Papadakis, K.A. Inflammatory bowel disease: Updates on molecular targets for biologics. Gut Liver 2017, 11, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Allez, M.; Tieng, V.; Nakazawa, A.; Treton, X.; Pacault, V.; Dulphy, N.; Caillat-Zucman, S.; Paul, P.; Gornet, J.M.; Douay, C.; et al. Cd4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through mica interactions. Gastroenterology 2007, 132, 2346–2358. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. NKG2D receptor and its ligands in host defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kanai, T.; Totsuka, T.; Okamoto, R.; Tsuchiya, K.; Nemoto, Y.; Yoshioka, A.; Tomita, T.; Nagaishi, T.; Sakamoto, N.; et al. Blockade of NKG2D signaling prevents the development of murine CD4+ T cell-mediated colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G199–G207. [Google Scholar] [CrossRef] [PubMed]
- Kjellev, S.; Haase, C.; Lundsgaard, D.; Urso, B.; Tornehave, D.; Markholst, H. Inhibition of NKG2D receptor function by antibody therapy attenuates transfer-induced colitis in scid mice. Eur. J. Immunol. 2007, 37, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Guerra, N.; Pestal, K.; Juarez, T.; Beck, J.; Tkach, K.; Wang, L.; Raulet, D.H. A selective role of NKG2D in inflammatory and autoimmune diseases. Clin. Immunol. 2013, 149, 432–439. [Google Scholar] [CrossRef] [PubMed]
- La Scaleia, R.; Stoppacciaro, A.; Oliva, S.; Morrone, S.; Di Nardo, G.; Santoni, A.; Cucchiara, S.; Palmieri, G. NKG2D/ligand dysregulation and functional alteration of innate immunity cell populations in pediatric IBD. Inflamm. Bowel Dis. 2012, 18, 1910–1922. [Google Scholar] [CrossRef] [PubMed]
- Allez, M.; Skolnick, B.E.; Wisniewska-Jarosinska, M.; Petryka, R.; Overgaard, R.V. Anti-NKG2D monoclonal antibody (NNC0142-0002) in active crohn’s disease: A randomised controlled trial. Gut 2016. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Carapito, R.; Bahram, S. Genetics, genomics, and evolutionary biology of NKG2D ligands. Immunol. Rev. 2015, 267, 88–116. [Google Scholar] [CrossRef] [PubMed]
- Champsaur, M.; Lanier, L.L. Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 2010, 235, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Upshaw, J.L.; Leibson, P.J. NKG2D-mediated activation of cytotoxic lymphocytes: Unique signaling pathways and distinct functional outcomes. Semin. Immunol. 2006, 18, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Glienke, J.; Sobanov, Y.; Brostjan, C.; Steffens, C.; Nguyen, C.; Lehrach, H.; Hofer, E.; Francis, F. The genomic organization of NKG2C, E, F, and D receptor genes in the human natural killer gene complex. Immunogenetics 1998, 48, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Wada, H.; Matsumoto, N.; Maenaka, K.; Suzuki, K.; Yamamoto, K. The inhibitory nk cell receptor CD94/NKG2A and the activating receptor CD94/NKG2C bind the top of HLA-E through mostly shared but partly distinct sets of HLA-E residues. Eur. J. Immunol. 2004, 34, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Maruoka, T.; Otsuka, N.; Kasamatsu, J.; Fugo, K.; Hanzawa, N.; Kasahara, M. Comparative genomic analysis of mammalian NKG2D ligand family genes provides insights into their origin and evolution. Immunogenetics 2010, 62, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Song, Y.; Bakker, A.B.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999, 285, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Zafirova, B.; Wensveen, F.M.; Gulin, M.; Polic, B. Regulation of immune cell function and differentiation by the NKG2D receptor. Cell. Mol. Life Sci. 2011, 68, 3519–3529. [Google Scholar] [CrossRef] [PubMed]
- Verneris, M.R.; Karimi, M.; Baker, J.; Jayaswal, A.; Negrin, R.S. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood 2004, 103, 3065–3072. [Google Scholar] [CrossRef] [PubMed]
- Garrity, D.; Call, M.E.; Feng, J.; Wucherpfennig, K.W. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc. Natl. Acad. Sci. USA 2005, 102, 7641–7646. [Google Scholar] [CrossRef] [PubMed]
- Cerwenka, A.; Lanier, L.L. Ligands for natural killer cell receptors: Redundancy or specificity. Immunol. Rev. 2001, 181, 158–169. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, C.A.; Cerwenka, A.; Willcox, B.E.; Lanier, L.L.; Bjorkman, P.J. Molecular competition for NKG2D: H60 and rae1 compete unequally for NKG2D with dominance of h60. Immunity 2001, 15, 201–211. [Google Scholar] [CrossRef]
- Robinson, J.; Waller, M.J.; Parham, P.; Bodmer, J.G.; Marsh, S.G. IMGT/HLA database—A sequence database for the human major histocompatibility complex. Nucleic Acids Res. 2001, 29, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Radosavljevic, M.; Cuillerier, B.; Wilson, M.J.; Clement, O.; Wicker, S.; Gilfillan, S.; Beck, S.; Trowsdale, J.; Bahram, S. A cluster of ten novel MHC class I related genes on human chromosome 6q24.2-q25.3. Genomics 2002, 79, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Eagle, R.A.; Trowsdale, J. Post-translational modification of the NKG2D ligand raet1g leads to cell surface expression of a glycosylphosphatidylinositol-linked isoform. J. Biol. Chem. 2010, 285, 16408–16415. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Messina, L.; Ashiru, O.; Aguera-Gonzalez, S.; Reyburn, H.T.; Vales-Gomez, M. The human NKG2D ligand ULBP2 can be expressed at the cell surface with or without a GPI anchor and both forms can activate nk cells. J. Cell Sci. 2011, 124, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Amroun, H.; Djoudi, H.; Busson, M.; Allat, R.; El Sherbini, S.M.; Sloma, I.; Ramasawmy, R.; Brun, M.; Dulphy, N.; Krishnamoorthy, R.; et al. Early-onset ankylosing spondylitis is associated with a functional mica polymorphism. Hum. Immunol. 2005, 66, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Steinle, A.; Li, P.; Morris, D.L.; Groh, V.; Lanier, L.L.; Strong, R.K.; Spies, T. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics 2001, 53, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Douik, H.; Ben Chaaben, A.; Attia Romdhane, N.; Romdhane, H.B.; Mamoghli, T.; Fortier, C.; Boukouaci, W.; Harzallah, L.; Ghanem, A.; Gritli, S.; et al. Association of MICA-129 polymorphism with nasopharyngeal cancer risk in a tunisian population. Hum. Immunol. 2009, 70, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mariuzza, R.A. Structural basis for recognition of cellular and viral ligands by NK cell receptors. Front. Immunol. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Schrambach, S.; Ardizzone, M.; Leymarie, V.; Sibilia, J.; Bahram, S. In vivo expression pattern of mica and micb and its relevance to auto-immunity and cancer. PLoS ONE 2007, 2, e518. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.; Lopez-Soto, A.; Suarez-Alvarez, B.; Lopez-Vazquez, A.; Lopez-Larrea, C. NKG2D ligands: Key targets of the immune response. Trends Immunol. 2008, 29, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.; Groh, V.; Spies, T. Immunobiology of human NKG2D and its ligands. Curr. Top. Microbiol. Immunol. 2006, 298, 121–138. [Google Scholar] [PubMed]
- Zou, Z.; Nomura, M.; Takihara, Y.; Yasunaga, T.; Shimada, K. Isolation and characterization of retinoic acid-inducible cDNA clones in F9 cells: A novel cDNA family encodes cell surface proteins sharing partial homology with MHC class I molecules. J. Biochem. 1996, 119, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, K.; Benjamin, J.; Takaki, R.; Phillips, J.H.; Lanier, L.L. Function of NKG2D in natural killer cell-mediated rejection of mouse bone marrow grafts. Nat. Immunol. 2005, 6, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Hsiung, B.; Pestal, K.; Procyk, E.; Raulet, D.H. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J. Exp. Med. 2012, 209, 2409–2422. [Google Scholar] [CrossRef] [PubMed]
- Caillat-Zucman, S. How NKG2D ligands trigger autoimmunity? Hum. Immunol. 2006, 67, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Bruhl, A.; El-Gabalawy, H.; Nelson, J.L.; Spies, T. Stimulation of t cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2003, 100, 9452–9457. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Lavender, H.; Soilleux, E.J.; O’Callaghan, C.A. NF-kappaB regulates mica gene transcription in endothelial cell through a genetically inhibitable control site. J. Boil. Chem. 2012, 287, 4299–4310. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, Y.L.; Bondar, C.; Guzman, L.; Cueto Rua, E.; Chopita, N.; Fuertes, M.; Zwirner, N.W.; Chirdo, F.G. Broad MICA/B expression in the small bowel mucosa: A link between cellular stress and celiac disease. PLoS ONE 2013, 8, e73658. [Google Scholar] [CrossRef] [PubMed]
- Hue, S.; Mention, J.J.; Monteiro, R.C.; Zhang, S.; Cellier, C.; Schmitz, J.; Verkarre, V.; Fodil, N.; Bahram, S.; Cerf-Bensussan, N.; et al. A direct role for NKG2D/mica interaction in villous atrophy during celiac disease. Immunity 2004, 21, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible mica. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, L.I.; Ogasawara, K.; Hamerman, J.A.; Takaki, R.; Zingoni, A.; Allison, J.P.; Lanier, L.L. Engagement of NKG2D by cognate ligand or antibody alone is insufficient to mediate costimulation of human and mouse CD8+ T cells. J. Immunol. 2005, 174, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Boukouaci, W.; Al-Daccak, R.; Dulphy, N.; Lauden, L.; Amokrane, K.; Fortier, C.; Marzais, F.; Bennabi, M.; Peffault de Latour, R.; Socie, G.; et al. Soluble mica-NKG2D interaction upregulates IFN-γ production by activated Cd3-Cd56+ NK cells: Potential impact on chronic graft versus host disease. Hum. Immunol. 2013, 74, 1536–1541. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Zocchi, M.R. Antigen presenting cells and stromal cells trigger human natural killer lymphocytes to autoreactivity: Evidence for the involvement of natural cytotoxicity receptors (NCR) and NKG2D. Clin. Dev. Immunol. 2006, 13, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Whitman, E.; Barber, A. NKG2D receptor activation of Nf-κB enhances inflammatory cytokine production in murine effector CD8(+) T cells. Mol. Immunol. 2015, 63, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. Dap10- and dap12-associated receptors in innate immunity. Immunol. Rev. 2009, 227, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Upshaw, J.L.; Arneson, L.N.; Schoon, R.A.; Dick, C.J.; Billadeau, D.D.; Leibson, P.J. NKG2D-mediated signaling requires a DAP10-bound GRB2-VAV1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat. Immunol. 2006, 7, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Jelencic, V.; Lenartic, M.; Wensveen, F.M.; Polic, B. NKG2D: A versatile player in the immune system. Immunol. Lett. 2017. [Google Scholar]
- Cosman, D.; Mullberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to cmv glycoprotein ul16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Campbell, J.A.; Trossman, D.S.; Yokoyama, W.M.; Carayannopoulos, L.N. Zoonotic orthopoxviruses encode a high-affinity antagonist of NKG2D. J. Exp. Med. 2007, 204, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Spear, P.; Wu, M.R.; Sentman, M.L.; Sentman, C.L. NKG2D ligands as therapeutic targets. Cancer Immun. 2013, 13, 8. [Google Scholar] [PubMed]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, B.K.; Yim, D.; Chow, I.T.; Gonzalez, S.; Dai, Z.; Mann, H.H.; Strong, R.K.; Groh, V.; Spies, T. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 2007, 447, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Baragano Raneros, A.; Suarez-Alvarez, B.; Lopez-Larrea, C. Secretory pathways generating immunosuppressive NKG2D ligands: New targets for therapeutic intervention. Oncoimmunology 2014, 3, e28497. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, E.; Koch, J.; Cerwenka, A.; Steinle, A. New prospects on the NKG2D/NKG2Dl system for oncology. Oncoimmunology 2013, 2, e26097. [Google Scholar] [CrossRef] [PubMed]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor beta 1 inhibits expression of NKP30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-β downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro-Oncology 2010, 12, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Lee, K.M.; Kim, D.W.; Heo, D.S. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef] [PubMed]
- Orchard, T.R.; Dhar, A.; Simmons, J.D.; Vaughan, R.; Welsh, K.I.; Jewell, D.P. MHC class I chain-like gene a (MICA) and its associations with inflammatory bowel disease and peripheral arthropathy. Clin. Exp. Immunol. 2001, 126, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Chamorro, A.; Moreno, A.; Gomez-Garcia, M.; Cabello, M.J.; Martin, J.; Lopez-Nevot, M.A. Mica*A4 protects against ulcerative colitis, whereas MICA*A5.1 is associated with abscess formation and age of onset. Clin. Exp. Immunol. 2016, 184, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Hernandez, R.; Valdes, M.; Lucas, D.; Campillo, J.A.; Martinez-Garcia, P.; Salama, H.; Lopez, M.; Salgado, G.; Botella, C.; Minguela, A.; et al. Association analysis of MICA gene polymorphism and MICA-129 dimorphism with inflammatory bowel disease susceptibility in a Spanish population. Hum. Immunol. 2010, 71, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Xu, C.T.; Pan, B.R. Epidemiology and gene markers of ulcerative colitis in the Chinese. World J. Gastroenterol. WJG 2009, 15, 788–803. [Google Scholar] [CrossRef] [PubMed]
- Perera, L.; Shao, L.; Patel, A.; Evans, K.; Meresse, B.; Blumberg, R.; Geraghty, D.; Groh, V.; Spies, T.; Jabri, B.; et al. Expression of nonclassical class I molecules by intestinal epithelial cells. Inflamm. Bowel Dis. 2007, 13, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Vadstrup, K.; Galsgaard, E.D.; Jensen, H.; Lanier, L.L.; Ryan, J.C.; Chen, S.Y.; Nolan, G.P.; Vester-Andersen, M.K.; Pedersen, J.S.; Gerwien, J.; et al. NKG2D ligand expression in crohn’s disease and NKG2D-dependent stimulation of CD8+ T cell migration. Exp. Mol. Pathol. 2017, 103, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Maasho, K.; Opoku-Anane, J.; Marusina, A.I.; Coligan, J.E.; Borrego, F. NKG2D is a costimulatory receptor for human naive CD8+ T cells. J. Immunol. 2005, 174, 4480–4484. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.P.; Choi, S.C.; Kiesler, P.; Gil-Krzewska, A.; Borrego, F.; Weck, J.; Krzewski, K.; Coligan, J.E. Complex regulation of human NKG2D-DAP10 cell surface expression: Opposing roles of the gammac cytokines and TGF-β1. Blood 2011, 118, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.I.; Lee, L.; Schwarz, E.; Groh, V.; Spies, T.; Ebert, E.C.; Jabri, B. NKG2D receptors induced by il-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J. Immunol. 2001, 167, 5527–5530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, J.; Niu, J.; Zhou, Z.; Zhang, J.; Tian, Z. Interleukin-12 improves cytotoxicity of natural killer cells via upregulated expression of NKG2D. Hum. Immunol. 2008, 69, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.J.; Marusina, A.I.; Pathmanathan, I.; Borrego, F.; Coligan, J.E. IL-21 down-regulates NKG2D/DAP10 expression on human NK and CD8+ T cells. J. Immunol. 2006, 176, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Magri, G.; Pende, D.; Angulo, A.; Lopez-Botet, M. Inhibition of NKG2D expression in nk cells by cytokines secreted in response to human cytomegalovirus infection. Blood 2010, 115, 5170–5179. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef]
- Liu, W.; Huber, S.A. Cross-talk between CD1D-restricted NKT cells and γΔ cells in T regulatory cell response. Virol. J. 2011, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Kagnoff, M.F. Current concepts in mucosal immunity. III. Ontogeny and function of gamma delta t cells in the intestine. Am. J. Physiol. 1998, 274, G455–G458. [Google Scholar] [PubMed]
- Lundqvist, C.; Hammarstrom, M.L.; Athlin, L.; Hammarstrom, S. Isolation of functionally active intraepithelial lymphocytes and enterocytes from human small and large intestine. J. Immunol. Methods 1992, 152, 253–263. [Google Scholar] [CrossRef]
- Kuhl, A.A.; Loddenkemper, C.; Westermann, J.; Hoffmann, J.C. Role of gamma delta T cells in inflammatory bowel disease. Pathobiology 2002, 70, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Cheroutre, H.; Lambolez, F.; Mucida, D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol. 2011, 11, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Moretta, L.; Montaldo, E.; Vacca, P.; del Zotto, G.; Moretta, F.; Merli, P.; Locatelli, F.; Mingari, M.C. Human natural killer cells: Origin, receptors, function, and clinical applications. Int. Arch. Allergy Immunol. 2014, 164, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Perez, M.I.; Jave-Suarez, L.F.; Ortiz-Lazareno, P.C.; Bravo-Cuellar, A.; Gonzalez-Ramella, O.; Aguilar-Lemarroy, A.; Hernandez-Flores, G.; Pereira-Suarez, A.L.; Daneri-Navarro, A.; del Toro-Arreola, S. Cervical cancer cell lines expressing NKG2D-ligands are able to down-modulate the NKG2D receptor on NKL cells with functional implications. BMC Immunol. 2012, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Lundholm, M.; Schroder, M.; Nagaeva, O.; Baranov, V.; Widmark, A.; Mincheva-Nilsson, L.; Wikstrom, P. Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PLoS ONE 2014, 9, e108925. [Google Scholar] [CrossRef] [PubMed]
- Mincheva-Nilsson, L.; Baranov, V. Cancer exosomes and NKG2D receptor-ligand interactions: Impairing NKG2D-mediated cytotoxicity and anti-tumour immune surveillance. Semin. Cancer Biol. 2014, 28, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Eagle, R.A.; Jafferji, I.; Barrow, A.D. Beyond stressed self: Evidence for NKG2D ligand expression on healthy cells. Curr. Immunol. Rev. 2009, 5, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Bahram, S.; Bauer, S.; Herman, A.; Beauchamp, M.; Spies, T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc. Natl. Acad. Sci. USA 1996, 93, 12445–12450. [Google Scholar] [CrossRef] [PubMed]
- Bilbao, J.R.; Martin-Pagola, A.; Perez De Nanclares, G.; Calvo, B.; Vitoria, J.C.; Vazquez, F.; Castano, L. HLA-DRB1 and mica in autoimmunity: Common associated alleles in autoimmune disorders. Ann. N. Y. Acad. Sci. 2003, 1005, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.K.; Jung, M.H.; Park, S.H.; Baek, I.C.; Choi, H.B.; Kim, T.G.; Suh, B.K. Association of mica alleles with autoimmune thyroid disease in korean children. Int. J. Endocrinol. 2012, 2012, 235680. [Google Scholar] [CrossRef] [PubMed]
- Glas, J.; Martin, K.; Brunnler, G.; Kopp, R.; Folwaczny, C.; Weiss, E.H.; Albert, E.D. MICA, MICB and C1_4_1 polymorphism in Crohn’s disease and ulcerative colitis. Tissue Antigens 2001, 58, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Cox, S.T.; Madrigal, J.A.; Saudemont, A. Diversity and characterization of polymorphic 5′ promoter haplotypes of mica and MICB genes. Tissue Antigens 2014, 84, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.Q.; Jiang, T.; Zhao, J.; Chen, Z.T.; Zhou, F.; Xia, B. Upregulated mRNA expression of major histocompatibility complex class I chain-related gene A in colon and activated natural killer cells of Chinese patients with ulcerative colitis. J. Dig. Dis. 2011, 12, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xia, B.; Li, J.; Ye, M.; Zhang, X.; Tan, Q. MICB microsatellite polymorphism is associated with ulcerative colitis in Chinese population. Clin. Immunol. 2006, 120, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, B.; Lu, M.; Ge, L.; Zhang, X. Micb0106 gene polymorphism is associated with ulcerative colitis in central china. Int. J. Colorectal Dis. 2010, 25, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Bisping, G.; Lugering, N.; Lutke-Brintrup, S.; Pauels, H.G.; Schurmann, G.; Domschke, W.; Kucharzik, T. Patients with inflammatory bowel disease (IBD) reveal increased induction capacity of intracellular interferon-gamma (IFN-gamma) in peripheral CD8+ lymphocytes co-cultured with intestinal epithelial cells. Clin. Exp. Immunol. 2001, 123, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Lory, J.; Corazza, N.; Griffiths, G.M.; Z’Graggen, K.; Mazzucchelli, L.; Kappeler, A.; Mueller, C. Activated CD4+ and CD8+ cytotoxic cells are present in increased numbers in the intestinal mucosa from patients with active inflammatory bowel disease. Am. J. Pathol. 1998, 152, 261–268. [Google Scholar] [PubMed]
- Camus, M.; Esses, S.; Pariente, B.; Le Bourhis, L.; Douay, C.; Chardiny, V.; Mocan, I.; Benlagha, K.; Clave, E.; Toubert, A.; et al. Oligoclonal expansions of mucosal t cells in crohn’s disease predominate in NKG2D-expressing CD4 T cells. Mucosal Immunol. 2014, 7, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Pariente, B.; Mocan, I.; Camus, M.; Dutertre, C.A.; Ettersperger, J.; Cattan, P.; Gornet, J.M.; Dulphy, N.; Charron, D.; Lemann, M.; et al. Activation of the receptor NKG2D leads to production of Th17 cytokines in CD4+ T cells of patients with crohn’s disease. Gastroenterology 2011, 141, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Sandborn, W.J.; D’Haens, G.; Panes, J.; Kaser, A.; Ferrante, M.; Louis, E.; Franchimont, D.; Dewit, O.; Seidler, U.; et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe crohn’s disease: A randomised, double-blind, placebo-controlled phase 2 study. Lancet 2017, 389, 1699–1709. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as induction and maintenance therapy for crohn’s disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef] [PubMed]
- Meresse, B.; Chen, Z.; Ciszewski, C.; Tretiakova, M.; Bhagat, G.; Krausz, T.N.; Raulet, D.H.; Lanier, L.L.; Groh, V.; Spies, T.; et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 2004, 21, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Allez, M.; Petryka, R.; Skolnick, B.E.; Wisniewska-Jarosinska, M.A. Mo1213 efficacy and safety of NNC0142-0002, a novel human monoclonal antibody targeting NKG2D: A randomized, double-blind, single-dose phase 2 trial in patients with crohn’s disease. Gastroenterology 2014, 146, S-587. [Google Scholar] [CrossRef]
- Vadstrup, K.; Galsgaard, E.D.; Gerwien, J.; Vester-Andersen, M.K.; Pedersen, J.S.; Rasmussen, J.; Neermark, S.; Kiszka-Kanowitz, M.; Jensen, T.; Bendtsen, F. Validation and optimization of an ex vivo assay of intestinal mucosal biopsies in Crohn’s disease: Reflects inflammation and drug effects. PLoS ONE 2016, 11, e0155335. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Safety and efficacy study of jnj-64304500 in participants with moderately to severely active crohn's disease (trident). Available online: https://clinicaltrials.gov/ct2/show/study/NCT02877134?term=JNJ-64304500&rank=2&show_locs=Y#locn (06–06) (accessed on 14 September 2017).
- ClinicalTrials.gov. A study to investigate the safety, tolerability, pharmacokinetics and pharmacodynamics following subcutaneous injection of jnj-64304500 in healthy japanese and caucasian male participants. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03002025?term=NKG2D&rank=12&view=record (06–06) (accessed on 14 September 2017).
- Van Dullemen, H.M.; van Deventer, S.J.; Hommes, D.W.; Bijl, H.A.; Jansen, J.; Tytgat, G.N.; Woody, J. Treatment of crohn’s disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2). Gastroenterology 1995, 109, 129–135. [Google Scholar] [CrossRef]
- Ben-Horin, S.; Chowers, Y. Review article: Loss of response to anti-TNF treatments in Crohn’s disease. Aliment. Pharmacol. Ther. 2011, 33, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, M.A.; Wise, E.L.; Buchwald, Z.S.; Pinto, A.K.; Zafirova, B.; Polic, B.; Shaw, A.S. Rae1epsilon ligand expressed on pancreatic islets recruits NKG2D receptor-expressing cytotoxic T cells independent of T cell receptor recognition. Immunity 2012, 36, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Hart, A.L.; Ng, S.C.; Mann, E.; Al-Hassi, H.O.; Bernardo, D.; Knight, S.C. Homing of immune cells: Role in homeostasis and intestinal inflammation. Inflamm. Bowel Dis. 2010, 16, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Hokari, R.; Miura, S.; Fujimori, H.; Koseki, S.; Tsuzuki, Y.; Kimura, H.; Higuchi, H.; Serizawa, H.; Granger, D.N.; Ishii, H. Altered migration of gut-derived T lymphocytes after activation with concanavalin a. Am. J. Physiol. 1999, 277, G763–G772. [Google Scholar] [PubMed]
- Ruck, T.; Bittner, S.; Gross, C.C.; Breuer, J.; Albrecht, S.; Korr, S.; Gobel, K.; Pankratz, S.; Henschel, C.M.; Schwab, N.; et al. CD4+NKG2D+ T cells exhibit enhanced migratory and encephalitogenic properties in neuroinflammation. PLoS ONE 2013, 8, e81455. [Google Scholar] [CrossRef]
- Mayer, L.; Sandborn, W.J.; Stepanov, Y.; Geboes, K.; Hardi, R.; Yellin, M.; Tao, X.; Xu, L.A.; Salter-Cid, L.; Gujrathi, S.; et al. Anti-IP-10 antibody (bms-936557) for ulcerative colitis: A phase II randomised study. Gut 2014, 63, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Villablanca, E.J.; Cassani, B.; von Andrian, U.H.; Mora, J.R. Blocking lymphocyte localization to the gastrointestinal mucosa as a therapeutic strategy for inflammatory bowel diseases. Gastroenterology 2011, 140, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Isernhagen, A.; Schilling, D.; Monecke, S.; Shah, P.; Elsner, L.; Walter, L.; Multhoff, G.; Dressel, R. The mica-129Met/Val dimorphism affects plasma membrane expression and shedding of the NKG2D ligand mica. Immunogenetics 2016, 68, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Wittenbrink, M.; Spreu, J.; Steinle, A. Differential NKG2D binding to highly related human NKG2D ligands ULBP2 and RAET1G is determined by a single amino acid in the ALPHA2 domain. Eur. J. Immunol. 2009, 39, 1642–1651. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.L.; Nguyen, V.H.; Ichimura, H.; Pham, T.T.; Nguyen, C.H.; Pham, T.V.; Elbadry, M.I.; Yoshioka, K.; Tanaka, J.; Trung, L.Q.; et al. A functional polymorphism in the NKG2D gene modulates NK-cell cytotoxicity and is associated with susceptibility to human papilloma virus-related cancers. Sci. Rep. 2016, 6, 39231. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vadstrup, K.; Bendtsen, F. Anti-NKG2D mAb: A New Treatment for Crohn’s Disease? Int. J. Mol. Sci. 2017, 18, 1997. https://doi.org/10.3390/ijms18091997
Vadstrup K, Bendtsen F. Anti-NKG2D mAb: A New Treatment for Crohn’s Disease? International Journal of Molecular Sciences. 2017; 18(9):1997. https://doi.org/10.3390/ijms18091997
Chicago/Turabian StyleVadstrup, Kasper, and Flemming Bendtsen. 2017. "Anti-NKG2D mAb: A New Treatment for Crohn’s Disease?" International Journal of Molecular Sciences 18, no. 9: 1997. https://doi.org/10.3390/ijms18091997