Roles of Wnt Target Genes in the Journey of Cancer Stem Cells

 ,
,
Abstract
:
1. Introduction
2. Target Genes of Wnt/β-Catenin Signaling
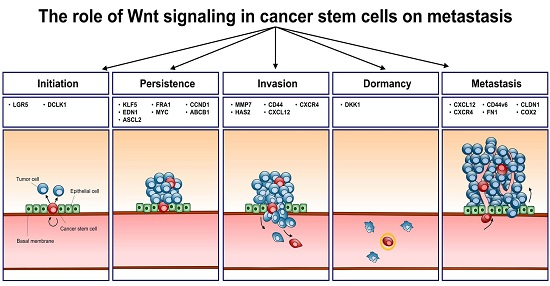
3. Initiation
4. Persistence
5. Invasion and Migration
6. Metastasis
7. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LEF1 | Lymphoid Enhancer-binding factor 1 |
| CSC | Cancer stem cell |
| FZD | Frizzled receptor |
| TCF | T-cell factor |
| APC | Adenomatous polyposis coli |
| LGR5 | Leucine-rich repeat-containing G-protein-coupled receptor 5 |
| DCLK1 | Doublecortin-like kinase 1 |
| EMT | Epithelial-to-mesenchymal transition |
| JNK | c-Jun N-terminal kinases |
| RYK | Receptor tyrosine kinase |
| ROR | Receptor tyrosine kinase-like orphan receptor |
| GSEA | Gene set enrichment analysis |
| ROS | Reactive oxygen species |
| NF-κB | Nuclear transcription factor-κB |
| CD44 | Cluster of differentiation 44 |
| CD133 | Cluster of differentiation 133 |
| ALDH | Aldehyde dehydrogenase |
| KLF5 | Kruppel-like factor |
| HDAC1 | Histone deacetylase 1 |
| EDN1 | Endothelin 1 |
| ASCL2 | Achaete-scute homolog 2 |
| FRA1 | Fos-related antigen-1 |
| CD47 | Cluster of differentiation 47 |
| PD-L1 | Programmed death-ligand 1 |
| CCND1 | Cyclin D1 |
| ABCB1 | ATP-binding cassette subfamily B member 1l |
| MDR | Multiple drug resistance |
| GSK-3β | Glycogen synthase kinase-3β |
| MMP7 | Matrix metalloproteinase-7 |
| ECM | Extracellular matrix |
| MT1-MMP | Membrane type 1-matrix metalloproteinase 1 |
| HA | Hyaluronan |
| HAS-2 | Hyaluronan synthase-2 |
| CXCR4 | C-X-C chemokine receptor 4 |
| CXCL12 | C-X-C motif chemokine ligand 12 |
| Src | Proto-oncogene tyrosine-protein kinase |
| DKK1 | Dickkopf-related protein |
| LRP6 | LDL receptor related protein 6 |
| CLDN1 | Claudin1 |
| TJP | Tight junction protein |
| CD44v | CD44 variant |
| HGF | Hepatocellular growth factor |
| OPN | Ostepontin |
| VEGF | Vascular endothelial growth factor |
| FGF | Fibroblast growth factors |
| FN1 | Fibronectin |
| COX2 | Cyclooxygenase 2 |
| PGE2 | Prostaglandin E2 |
References
- Kahn, M. Can we safely target the WNT pathway? Nature reviews. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Ormanns, S.; Neumann, J.; Horst, D.; Kirchner, T.; Jung, A. WNT signaling and distant metastasis in colon cancer through transcriptional activity of nuclear β-Catenin depend on active PI3K signaling. Oncotarget 2014, 5, 2999–3011. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.; Akiri, G.; Chin, C.; Wisnivesky, J.P.; Beasley, M.B.; Weiser, T.S.; Swanson, S.J.; Aaronson, S.A. Wnt pathway activation predicts increased risk of tumor recurrence in patients with stage I nonsmall cell lung cancer. Ann. Surg. 2013, 257, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Yoshioka, Y.; Isohashi, F.; Seo, Y.; Yoshida, K.; Yamazaki, H. Radiotherapy targeting cancer stem cells: Current views and future perspectives. Anticancer Res. 2013, 33, 747–754. [Google Scholar] [PubMed]
- Yeung, J.; Esposito, M.T.; Gandillet, A.; Zeisig, B.B.; Griessinger, E.; Bonnet, D.; So, C.W. β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010, 18, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Skoda, J.; Hermanova, M.; Loja, T.; Nemec, P.; Neradil, J.; Karasek, P.; Veselska, R. Co-Expression of Cancer Stem Cell Markers Corresponds to a Pro-Tumorigenic Expression Profile in Pancreatic Adenocarcinoma. PLoS ONE 2016, 11, e0159255. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, H.Y.; Park, K.K.; Choi, Y.K.; Nam, J.S.; Hong, I.S. CWP232228 targets liver cancer stem cells through Wnt/β-catenin signaling: A novel therapeutic approach for liver cancer treatment. Oncotarget 2016, 7, 20395–20409. [Google Scholar] [CrossRef] [PubMed]
- Malanchi, I.; Peinado, H.; Kassen, D.; Hussenet, T.; Metzger, D.; Chambon, P.; Huber, M.; Hohl, D.; Cano, A.; Birchmeier, W.; et al. Cutaneous cancer stem cell maintenance is dependent on β-catenin signalling. Nature 2008, 452, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Zhang, B.; Ho, Y.; Cook, A.; Li, L.; Wang, Y.; McLaughlin, M.; Bhatia, R. Inhibition of CML Stem Cell Growth By Targeting WNT Signaling Using a Porcupine Inhibitor. Blood 2014, 124, 3130. [Google Scholar]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.B.; Hong, I.S.; Kim, R.J.; Lee, S.Y.; Park, S.J.; Lee, E.S.; Park, J.H.; Yun, C.H.; Chung, J.U.; Lee, K.J.; et al. Wnt/β-Catenin Small-Molecule Inhibitor CWP232228 Preferentially Inhibits the Growth of Breast Cancer Stem-like Cells. Cancer Res. 2015, 75, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, S.-G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Felipe De Sousa, E.M.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Zhu, Q.; Neuenschwander, M.; Specker, E.; Wulf-Goldenberg, A.; Weis, W.I.; Von Kries, J.P.; Birchmeier, W. A small-molecule antagonist of the β-catenin/TCF4 interaction blocks the self-renewal of cancer stem cells and suppresses tumorigenesis. Cancer Res. 2016, 76, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Prasetyanti, P.R.; Zimberlin, C.D.; Bots, M.; Vermeulen, L.; Felipe De Sousa, E.M.; Medema, J.P. Regulation of stem cell self-renewal and differentiation by Wnt and Notch are conserved throughout the adenoma-carcinoma sequence in the colon. Mol. Cancer 2013, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Vadakkan, T.J.; Landua, J.D.; Bu, W.; Wei, W.; Li, F.; Wong, S.T.; Dickinson, M.E.; Rosen, J.M.; Lewis, M.T.; Zhang, M. Wnt-responsive cancer stem cells are located close to distorted blood vessels and not in hypoxic regions in a p53-null mouse model of human breast cancer. Stem Cells Transl. Med. 2014, 3, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Jurinovic, V.; Krebs, S.; Thieme, S.E.; Blum, H.; Goke, B.; Kolligs, F.T. Comprehensive analysis of β-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/β-catenin signalling. BMC Genom. 2014, 15, 74. [Google Scholar] [CrossRef] [PubMed]
- Bottomly, D.; Kyler, S.L.; McWeeney, S.K.; Yochum, G.S. Identification of β-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res. 2010, 38, 5735–5745. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.R.; Sarkar, S.; Luthra, G.K.; Okugawa, Y.; Toiyama, Y.; Gajjar, A.H.; Qiu, S.; Goel, A.; Singh, P. Epigenetic changes and alternate promoter usage by human colon cancers for expressing DCLK1-isoforms: Clinical Implications. Sci. Rep. 2015, 5, 14983. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Xiong, H.; Zhang, Z.; Ren, B. β-Catenin activates the growth factor endothelin-1 in colon cancer cells. Oncogene 2005, 24, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Schuijers, J.; Junker, J.P.; Mokry, M.; Hatzis, P.; Koo, B.K.; Sasselli, V.; van der Flier, L.G.; Cuppen, E.; van Oudenaarden, A.; Clevers, H. Ascl2 acts as an R-spondin/Wnt-responsive switch to control stemness in intestinal crypts. Cell Stem Cell 2015, 16, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Takaoka, A.S.; Naishiro, Y.; Hayashi, R.; Maruyama, K.; Maesawa, C.; Ochiai, A.; Hirohashi, S. Transactivation of the multidrug resistance 1 gene by T-cell factor 4/β-catenin complex in early colorectal carcinogenesis. Cancer Res. 2000, 60, 4761–4766. [Google Scholar] [PubMed]
- Gustavson, M.D.; Crawford, H.C.; Fingleton, B.; Matrisian, L.M. Tcf binding sequence and position determines β-catenin and Lef-1 responsiveness of MMP-7 promoters. Mol. Carcinog. 2004, 41, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, I.; Freudenberger, T.; Twarock, S.; Yamaguchi, Y.; Grandoch, M.; Fischer, J.W. Esophageal squamous cell carcinoma cells modulate chemokine expression and hyaluronan synthesis in fibroblasts. J. Biol. Chem. 2016, 291, 4091–4106. [Google Scholar] [CrossRef] [PubMed]
- Wielenga, V.J.; Smits, R.; Korinek, V.; Smit, L.; Kielman, M.; Fodde, R.; Clevers, H.; Pals, S.T. Expression of CD44 in Apc and Tcfmutant mice implies regulation by the WNT pathway. Am. J. Pathol. 1999, 154, 515–523. [Google Scholar] [CrossRef]
- Holland, J.D.; Györffy, B.; Vogel, R.; Eckert, K.; Valenti, G.; Fang, L.; Lohneis, P.; Elezkurtaj, S.; Ziebold, U.; Birchmeier, W. Combined Wnt/β-catenin, Met, and CXCL12/CXCR4 signals characterize basal breast cancer and predict disease outcome. Cell Rep. 2013, 5, 1214–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choe, Y.; Pleasure, S.J. Wnt signaling regulates intermediate precursor production in the postnatal dentate gyrus by regulating cxcr4 expression. Dev. Neurosci. 2012, 34, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Niida, A.; Hiroko, T.; Kasai, M.; Furukawa, Y.; Nakamura, Y.; Suzuki, Y.; Sugano, S.; Akiyama, T. DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene 2004, 23, 8520–8526. [Google Scholar] [CrossRef] [PubMed]
- Miwa, N.; Furuse, M.; Tsukita, S.; Niikawa, N.; Nakamura, Y.; Furukawa, Y. Involvement of claudin-1 in the β-catenin/Tcf signaling pathway and its frequent upregulation in human colorectal cancers. Oncol. Res. 2001, 12, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradl, D.; Kühl, M.; Wedlich, D. The Wnt/Wg signal transducer β-catenin controls fibronectin expression. Mol. Cell. Biol. 1999, 19, 5576–5587. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, F.; Bravo, S.; Cruzat, F.; Montecino, M.; De Ferrari, G.V. Wnt/β-catenin signaling enhances cyclooxygenase-2 (COX2) transcriptional activity in gastric cancer cells. PLoS ONE 2011, 6, e18562. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Chen, L.; Li, C.; Zhu, Y. Heterogeneity in cancer stem cells. Cancer Lett. 2015, 357, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.L.; Athineos, D.; et al. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 2013, 12, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage Tracing Reveals Lgr5+ Stem Cell Activity in Mouse Intestinal Adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.B.; Houchen, C.W.; Ali, N. APSA Awardee Submission: Tumor/cancer stem cell marker doublecortin-like kinase 1 in liver diseases. Exp. Biol. Med. 2017, 242, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Wang, M.; Xu, L.; Wen, T.; Liu, J.; An, G. DCLK1 is up-regulated and associated with metastasis and prognosis in colorectal cancer. J. Cancer Res. Clin. Oncol. 2016, 142, 2131–2140. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Tanaka, S.; Akiyama, Y.; Shimada, S.; Adikrisna, R.; Matsumura, S.; Aihara, A.; Mitsunori, Y.; Ban, D.; Ochiai, T.; et al. Dominant Expression of DCLK1 in Human Pancreatic Cancer Stem Cells Accelerates Tumor Invasion and Metastasis. PLoS ONE 2016, 11, e0146564. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Yao, J.; Qu, D.; May, R.; Weygant, N.; Ge, Y.; Ali, N.; Sureban, S.M.; Gude, M.; Vega, K.; et al. Dclk1, a tumor stem cell marker, regulates pro-survival signaling and self-renewal of intestinal tumor cells. Mol. Cancer 2017, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, C.B.; Asfaha, S.; Hayakawa, Y.; Takemoto, Y.; Lukin, D.J.; Nuber, A.H.; Brandtner, A.; Setlik, W.; Remotti, H.; Muley, A.; et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Invest. 2014, 124, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Panneerselvam, J.; Qu, D.; Weygant, N.; May, R.; Bronze, M.S.; Houchen, C.W. Regulatory Roles of Dclk1 in Epithelial Mesenchymal Transition and Cancer Stem Cells. J. Carcinog. Mutagen. 2016, 7, 257. [Google Scholar] [PubMed]
- Qu, D.; May, R.; Sureban, S.M.; Weygant, N.; Chandrakesan, P.; Ali, N.; Li, L.; Barrett, T.; Houchen, C.W. Inhibition of Notch signaling reduces the number of surviving Dclk1+ reserve crypt epithelial stem cells following radiation injury. American journal of physiology. Gastrointest. Liver Physiol. 2014, 306, G404–G411. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, P.; Subramaniam, D.; Paul, S.; Kwatra, D.; Palaniyandi, K.; Islam, S.; Harihar, S.; Ramalingam, S.; Gutheil, W.; Putty, S.; et al. Crocetinic acid inhibits hedgehog signaling to inhibit pancreatic cancer stem cells. Oncotarget 2015, 6, 27661–27673. [Google Scholar] [CrossRef] [PubMed]
- May, R.; Riehl, T.E.; Hunt, C.; Sureban, S.M.; Anant, S.; Houchen, C.W. Identification of a novel putative gastrointestinal stem cell and adenoma stem cell marker, doublecortin and CaM kinase-like-1, following radiation injury and in adenomatous polyposis coli/multiple intestinal neoplasia mice. Stem Cells 2008, 26, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Jakkula, L.U.M.R.; Ahmed, I.; Roy, B.; Anant, S.; Umar, S. Differential Effects of β-catenin and NF-κB Interplay in the Regulation of Cell Proliferation, Inflammation and Tumorigenesis in Response to Bacterial Infection. PLoS ONE 2013, 8, e79432. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Weygant, N.; May, R.; Qu, D.; Chinthalapally, H.R.; Sureban, S.M.; Ali, N.; Lightfoot, S.A.; Umar, S.; Houchen, C.W. DCLK1 facilitates intestinal tumor growth via enhancing pluripotency and epithelial mesenchymal transition. Oncotarget 2014, 5, 9269–9280. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.-B.; Kim, J.-Y.; Cho, S.-D.; Park, K.-S.; Jung, J.-Y.; Lee, H.-Y.; Hong, I.-S.; Nam, J.-S. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Lei, X.; GuAN, Y.; MOu, L.S.; Yuan, Y.-F.; Yue, H.; Lu, Y.; Xu, G.-T.; Qian, J. Maintenance of retinal cancer stem cell-like properties through long-term serum-free culture from human retinoblastoma. Oncol. Rep. 2011, 26, 135. [Google Scholar] [PubMed]
- Kondo, T.; Setoguchi, T.; Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl. Acad. Sci. USA 2004, 101, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Cidado, J.; Wong, H.Y.; Rosen, D.M.; Cimino-Mathews, A.; Garay, J.P.; Fessler, A.G.; Rasheed, Z.A.; Hicks, J.; Cochran, R.L.; Croessmann, S. Ki-67 is required for maintenance of cancer stem cells but not cell proliferation. Oncotarget 2016, 7, 6281. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Ogawa, S.; Manabe, I.; Tanaka, M.; Sanada, M.; Sato, T.; Taketo, M.M.; Nakao, K.; Clevers, H.; Fukayama, M.; et al. KLF5 Regulates the Integrity and Oncogenicity of Intestinal Stem Cells. Cancer Res. 2014, 74, 2882–2891. [Google Scholar] [CrossRef] [PubMed]
- Maehara, O.; Sato, F.; Natsuizaka, M.; Asano, A.; Kubota, Y.; Itoh, J.; Tsunematsu, S.; Terashita, K.; Tsukuda, Y.; Nakai, M.; et al. A pivotal role of Kruppel-like factor 5 in regulation of cancer stem-like cells in hepatocellular carcinoma. Cancer Biol. Ther. 2015, 16, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-W.; Liao, C.-Y.; Yang, W.-Y.; Lin, Y.-M.; Jin, S.-L.C.; Wang, H.-D.; Yuh, C.-H. Overexpression of Endothelin 1 Triggers Hepatocarcinogenesis in Zebrafish and Promotes Cell Proliferation and Migration through the AKT Pathway. PLoS ONE 2014, 9, e85318. [Google Scholar] [CrossRef] [PubMed]
- Rosano, L.; Cianfrocca, R.; Spinella, F.; Di Castro, V.; Nicotra, M.R.; Lucidi, A.; Ferrandina, G.; Natali, P.G.; Bagnato, A. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin. Cancer Res. 2011, 17, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, L.G.; van Gijn, M.E.; Hatzis, P.; Kujala, P.; Haegebarth, A.; Stange, D.E.; Begthel, H.; van den Born, M.; Guryev, V.; Oving, I.; et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell 2009, 136, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Jubb, A.M.; Chalasani, S.; Frantz, G.D.; Smits, R.; Grabsch, H.I.; Kavi, V.; Maughan, N.J.; Hillan, K.J.; Quirke, P.; Koeppen, H. Achaete-scute like 2 (ascl2) is a target of Wnt signalling and is upregulated in intestinal neoplasia. Oncogene 2006, 25, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Stange, D.E.; Engel, F.; Longerich, T.; Koo, B.K.; Koch, M.; Delhomme, N.; Aigner, M.; Toedt, G.; Schirmacher, P.; Lichter, P.; et al. Expression of an ASCL2 related stem cell signature and IGF2 in colorectal cancer liver metastases with 11p15.5 gain. Gut 2010, 59, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Yang, Y.; Tian, Y.; Bai, J.; Zhang, X.; Li, X.; Peng, Z.; He, Y.; Chen, L.; Pan, Q.; et al. Ascl2 knockdown results in tumor growth arrest by miRNA-302b-related inhibition of colon cancer progenitor cells. PLoS ONE 2012, 7, e32170. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Pan, Q.; Shang, Y.; Zhu, R.; Ye, J.; Liu, Y.; Zhong, X.; Li, S.; He, Y.; Chen, L.; et al. MicroRNA-200 (miR-200) cluster regulation by achaete scute-like 2 (Ascl2): Impact on the epithelial-mesenchymal transition in colon cancer cells. J. Biol. Chem. 2014, 289, 36101–36115. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.H.; Cui, Y.H.; Guo, Q.N.; Zhou, Y. Elevated ASCL2 expression is associated with metastasis of osteosarcoma and predicts poor prognosis of the patients. Am. J. Cancer Res. 2016, 6, 1431–1440. [Google Scholar] [PubMed]
- Kwon, O.H.; Park, J.L.; Baek, S.J.; Noh, S.M.; Song, K.S.; Kim, S.Y.; Kim, Y.S. Aberrant upregulation of ASCL2 by promoter demethylation promotes the growth and resistance to 5-fluorouracil of gastric cancer cells. Cancer Sci. 2013, 104, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Chen, X.; Fang, X.; Wang, D.; Xie, M.; Chen, Q. Fra-1 is upregulated in lung cancer tissues and inhibits the apoptosis of lung cancer cells by the P53 signaling pathway. Oncol. Rep. 2016, 35, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, H.; Mu, X.; Cui, J.; Peng, Z. Dysregulation of Fra1 expression by Wnt/β-catenin signalling promotes glioma aggressiveness through epithelial-mesenchymal transition. Biosci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Motrich, R.D.; Castro, G.M.; Caputto, B.L. Old players with a newly defined function: Fra-1 and c-Fos support growth of human malignant breast tumors by activating membrane biogenesis at the cytoplasm. PLoS ONE 2013, 8, e53211. [Google Scholar] [CrossRef] [PubMed]
- Sayan, A.E.; Stanford, R.; Vickery, R.; Grigorenko, E.; Diesch, J.; Kulbicki, K.; Edwards, R.; Pal, R.; Greaves, P.; Jariel-Encontre, I.; et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene 2012, 31, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Luo, S.; Lechler, T.; Zhang, J.Y. FRA1 promotes squamous cell carcinoma growth and metastasis through distinct AKT and c-Jun dependent mechanisms. Oncotarget 2016, 7, 34371–34383. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.V. The role of c-myc in cellular growth control. Oncogene 1999, 18, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- de Alboran, I.M.; O’Hagan, R.C.; Gärtner, F.; Malynn, B.; Davidson, L.; Rickert, R.; Rajewsky, K.; DePinho, R.A.; Alt, F.W. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity 2001, 14, 45–55. [Google Scholar] [CrossRef]
- Schuhmacher, M.; Staege, M.S.; Pajic, A.; Polack, A.; Weidle, U.H.; Bornkamm, G.W.; Eick, D.; Kohlhuber, F. Control of cell growth by c-Myc in the absence of cell division. Curr. Biol. 1999, 9, 1255–1258. [Google Scholar] [CrossRef]
- Kim, S.; Li, Q.; Dang, C.V.; Lee, L.A. Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 11198–11202. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2016, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Hasan, M.R. Cancer metabolism and drug resistance. Metabolites 2015, 5, 571–600. [Google Scholar] [CrossRef] [PubMed]
- Sattler, U.G.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. 2010, 94, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T.F.; Kline, J. Myc—A thorn in the side of cancer immunity. Cell Res. 2016, 26, 639–640. [Google Scholar] [CrossRef] [PubMed]
- Stacey, D.W. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr. Opin. Cell Biol. 2003, 15, 158–163. [Google Scholar] [CrossRef]
- Zhang, J.; Gill, A.J.; Issacs, J.D.; Atmore, B.; Johns, A.; Delbridge, L.W.; Lai, R.; McMullen, T.P. The Wnt/β-catenin pathway drives increased cyclin D1 levels in lymph node metastasis in papillary thyroid cancer. Hum. Pathol. 2012, 43, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-L.; Jiang, L.-M.; Han, W.-D. Wnt/β-catenin signaling pathway in lung cancer stem cells is a potential target for the development of novel anticancer drugs. J. BUON 2015, 20, 1094–1100. [Google Scholar] [PubMed]
- Musgrove, E.A.; Hunter, L.J.; Lee, C.S.; Swarbrick, A.; Hui, R.; Sutherland, R.L. Cyclin D1 overexpression induces progestin resistance in T-47D breast cancer cells despite p27Kip1 association with cyclin E-Cdk2. J. Biol. Chem. 2001, 276, 47675–47683. [Google Scholar] [CrossRef] [PubMed]
- Biliran, H., Jr.; Wang, Y.; Banerjee, S.; Xu, H.; Heng, H.; Thakur, A.; Bollig, A.; Sarkar, F.H.; Liao, J.D. Overexpression of cyclin D1 promotes tumor cell growth and confers resistance to cisplatin-mediated apoptosis in an elastase-myc transgene-expressing pancreatic tumor cell line. Clin. Cancer Res. 2005, 11, 6075–6086. [Google Scholar] [CrossRef] [PubMed]
- Flahaut, M.; Meier, R.; Coulon, A.; Nardou, K.A.; Niggli, F.K.; Martinet, D.; Beckmann, J.S.; Joseph, J.M.; Muhlethaler-Mottet, A.; Gross, N. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wnt/β-catenin pathway. Oncogene 2009, 28, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Xia, W.; Wong, G. Hyaluronan-mediated CD44 interaction with p300 and SIRT1 regulates β-catenin signaling and NFκB-specific transcription activity leading to MDR1 and Bcl-xL gene expression and chemoresistance in breast tumor cells. J. Biol. Chem. 2009, 284, 2657–2671. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.; Binato, R.; Du Rocher, B.; Castelo-Branco, M.T.; Pizzatti, L.; Abdelhay, E. Wnt/β-catenin pathway regulates ABCB1 transcription in chronic myeloid leukemia. BMC Cancer 2012, 12, 303. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Weinberg, R.A. In Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Du, L.; Chen, D.; Ye, Z.; Duan, H.; Tu, T.; Feng, J.; Yang, Y.; Chen, Q.; Yan, X. Reduced CD146 expression promotes tumorigenesis and cancer stemness in colorectal cancer through activating Wnt/β-catenin signaling. Oncotarget 2016, 7, 40704. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Iljin, K.; Sara, H.; Mpindi, J.P.; Mirtti, T.; Vainio, P.; Rantala, J.; Alanen, K.; Nees, M.; Kallioniemi, O. FZD4 as a mediator of ERG oncogene-induced WNT signaling and epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2010, 70, 6735–6745. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-W.; Su, Y.-J.; Hsiao, M.; Wei, K.-C.; Lin, W.-H.; Liang, C.-J.; Chen, S.-C.; Lee, J.-L. Diverse targets of β-catenin during the epithelial–mesenchymal transition define cancer stem cells and predict disease relapse. Cancer Res. 2015, 75, 3398–3410. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Thiagarajan, P.S.; Rai, V.; Reizes, O.; Lathia, J.; Egelhoff, T. Increased cancer stem cell invasion is mediated by myosin IIB and nuclear translocation. Oncotarget 2016, 7, 47586. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Hur, W.; Hong, S.W.; Kim, J.-H.; Kim, S.M.; Lee, E.B.; Yoon, S.K. ELK3 promotes the migration and invasion of liver cancer stem cells by targeting HIF-1α. Oncol. Rep. 2017, 37, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Yonekawa, A.; Harada, C.; Hamada, M.; Katagiri, W.; Nakazawa, M.; Yura, Y. Involvement of the Wnt-β-catenin pathway in invasion and migration of oral squamous carcinoma cells. Int. J. Oncol. 2010, 37, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Said, A.H.; Raufman, J.-P.; Xie, G. The role of matrix metalloproteinases in colorectal cancer. Cancers 2014, 6, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Reunanen, N.; Kähäri, V. Matrix metalloproteinases in cancer cell invasion. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2002; pp. 1–19. [Google Scholar]
- Yamamoto, H.; Itoh, F.; Iku, S.; Adachi, Y.; Fukushima, H.; Sasaki, S.; Mukaiya, M.; Hirata, K.; Imai, K. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human pancreatic adenocarcinomas: Clinicopathologic and prognostic significance of matrilysin expression. J. Clin. Oncol. 2001, 19, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Yamamoto, H.; Itoh, F.; Hinoda, Y.; Okada, Y.; Imai, K. Contribution of matrilysin (MMP-7) to the metastatic pathway of human colorectal cancers. Gut 1999, 45, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Aihara, R.; Mochiki, E.; Nakabayashi, T.; Akazawa, K.; Asao, T.; Kuwano, H. Clinical significance of mucin phenotype, β-catenin and matrix metalloproteinase 7 in early undifferentiated gastric carcinoma. Br. J. Surg. 2005, 92, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Hlubek, F.; Spaderna, S.; Jung, A.; Kirchner, T.; Brabletz, T. β-Catenin activates a coordinated expression of the proinvasive factors laminin-5 γ2 chain and MT1-MMP in colorectal carcinomas. Int. J. Cancer 2004, 108, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rossé, C.; Chavrier, P. Cellular and molecular mechanisms of MT1-MMP-dependent cancer cell invasion. Annu. Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Li, Q.; Li, S.; He, J.; Cao, W.; Lan, J.; Sun, B.; Zou, H.; Wang, C.; Liu, R.; et al. Membrane type 1-matrix metalloproteinase induces epithelial-to-mesenchymal transition in esophageal squamous cell carcinoma: Observations from clinical and in vitro analyses. Sci. Rep. 2016, 6, 22179. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Zhang, X.; Mizumoto, Y.; Maida, Y.; Bono, Y.; Takakura, M.; Kyo, S. Molecular characterization of CD133+ cancer stem-like cells in endometrial cancer. Int. J. Oncol. 2014, 44, 669–677. [Google Scholar] [PubMed]
- Dunn, K.M.B.; Lee, P.K.; Wilson, C.M.; Iida, J.; Wasiluk, K.R.; Hugger, M.; McCarthy, J.B. Inhibition of hyaluronan synthases decreases matrix metalloproteinase-7 (MMP-7) expression and activity. Surgery 2009, 145, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Bernert, B.; Porsch, H.; Heldin, P. Hyaluronan synthase 2 (HAS2) promotes breast cancer cell invasion by suppression of tissue metalloproteinase inhibitor 1 (TIMP-1). J. Biol. Chem. 2011, 286, 42349–42359. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Kobayashi, A.; Xia, B.; Watabe, M.; Pai, S.K.; Hirota, S.; Xing, F.; Liu, W.; Pandey, P.R.; Fukuda, K. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res. 2012, 72, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Boil. 2003, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, U.P.; Grizzle, W.E.; Lillard, J.W. CXCL12–CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab. Investig. 2004, 84, 1666–1676. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V. CD44 Acts as a Signaling Platform Controlling Tumor Progression and Metastasis. Front. Immunol. 2015, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Ping, Y.F.; Yao, X.H.; Jiang, J.Y.; Zhao, L.T.; Yu, S.C.; Jiang, T.; Lin, M.; Chen, J.H.; Wang, B.; Zhang, R. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Liekens, S.; Schols, D.; Hatse, S. CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr. Pharm. Des. 2010, 16, 3903–3920. [Google Scholar] [CrossRef] [PubMed]
- Würth, R.; Bajetto, A.; Harrison, J.K.; Barbieri, F.; Florio, T. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Front. Cell. Neurosci. 2014, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulos, P.A.; Polioudaki, H.; Agelaki, S.; Kallergi, G.; Saridaki, Z.; Mavroudis, D.; Georgoulias, V. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett. 2010, 288, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, H.; Watanabe, T.; Mimori, K.; Adachi, M.; Hayashi, N.; Tamura, J.; Matsuda, K.; Fukushima, R.; Okinaga, K.; Sasako, M. Clinical significance of circulating tumor cells, including cancer stem-like cells, in peripheral blood for recurrence and prognosis in patients with Dukes’ stage B and C colorectal cancer. J. Clin. Oncol. 2011, 29, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.-F.; Wang, Q.; Gerald, W.; Hudis, C.A.; Norton, L.; Smid, M.; Foekens, J.A.; Massagué, J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 2009, 16, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, W.; King, T.D.; Liu, C.C.; Bijur, G.N.; Bu, G. Dkk1 stabilizes Wnt co-receptor LRP6: Implication for Wnt ligand-induced LRP6 down-regulation. PLoS ONE 2010, 5, e11014. [Google Scholar] [CrossRef] [PubMed]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massague, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Chiang, A.C.; Zhang, X.H.-F.; Kim, J.Y.; Kris, M.G.; Ladanyi, M.; Gerald, W.L.; Massagué, J. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 2009, 138, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G. Mechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Wolfer, A.; Wittner, B.S.; Irimia, D.; Flavin, R.J.; Lupien, M.; Gunawardane, R.N.; Meyer, C.A.; Lightcap, E.S.; Tamayo, P.; Mesirov, J.P. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc. Natl. Acad. Sci. USA 2010, 107, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Markov, A.G.; Aschenbach, J.R.; Amasheh, S. Claudin clusters as determinants of epithelial barrier function. IUBMB Life 2015, 67, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Salvador, E.; Burek, M.; Förster, C.Y. Tight Junctions and the Tumor Microenvironment. Curr. Pathobiol. Rep. 2016, 4, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Tabariès, S.; Siegel, P. The role of claudins in cancer metastasis. Oncogene 2016, 36, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.-C.; Pan, S.-H.; Yang, S.-C.; Yu, S.-L.; Che, T.-F.; Lin, C.-W.; Tsai, M.-S.; Chang, G.-C.; Wu, C.-H.; Wu, Y.-Y. Claudin-1 is a metastasis suppressor and correlates with clinical outcome in lung adenocarcinoma. Am. J. Respir. Crit. Care Med. 2009, 179, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, J.; Filipović, A.; Giamas, G. Claudin-1 as a promoter of EMT in hepatocellular carcinoma. Oncogene 2013, 32, 4871–4872. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; He, C.; Qu, Y.; Li, J.; Zhang, J.; Du T, C.X.; Yu, Y.; Liu, B.; Zhu, Z. Claudin-1 enhances tumor proliferation and metastasis by regulating cell anoikis in gastric cancer. Oncotarget 2015, 6, 1652. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Singh, A.B.; Deane, N.G.; No, Y.; Shiou, S.-R.; Schmidt, C.; Neff, J.; Washington, M.K.; Beauchamp, R.D. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J. Clin. Investig. 2005, 115, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, T.; Akagi, Y.; Ochi, T.; Tanaka, N.; Kawahara, A.; Ishibashi, Y.; Gotanda, Y.; Yamaguchi, K.; Shiratuchi, I.; Oka, Y. Increased claudin-1 protein expression in hepatic metastatic lesions of colorectal cancer. Anticancer Res. 2012, 32, 2309–2314. [Google Scholar] [PubMed]
- Liu, T.; Cheng, W.; Lai, D.; Huang, Y.; Guo, L. Characterization of primary ovarian cancer cells in different culture systems. Oncol. Rep. 2010, 23, 1277. [Google Scholar] [PubMed]
- Bhat, A.A.; Sharma, A.; Pope, J.; Krishnan, M.; Washington, M.K.; Singh, A.B.; Dhawan, P. Caudal homeobox protein Cdx-2 cooperates with Wnt pathway to regulate claudin-1 expression in colon cancer cells. PLoS ONE 2012, 7, e37174. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.-T.; Ye, Y.-P.; Deng, Y.-J.; Bian, X.-W.; Ding, Y.-Q. Metastatic cancer stem cells: From the concept to therapeutics. Am. J. Stem Cells 2014, 3, 46–62. [Google Scholar] [PubMed]
- Diehn, M.; Majeti, R. Metastatic cancer stem cells: An opportunity for improving cancer treatment? Cell Stem Cell 2010, 6, 502–503. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Motiani, K.; Giridhar, P.V.; Kasper, S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp. Biol. Med. 2013, 238, 324–338. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; von Maltzahn, J.; Soleimani, V.D.; Yin, H.; Rudnicki, M.A. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell 2013, 12, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.-A.; Delaloye, J.-F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012, 481, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived IL-1β stimulates Wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin D3. Oncogene 2009, 28, 3892–3902. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.K.; Zou, Y.; Ghosh, A. Wnts acting through canonical and noncanonical signaling pathways exert opposite effects on hippocampal synapse formation. Neural Dev. 2008, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shu, W.; Lu, M.M.; Morrisey, E.E. Wnt7b activates canonical signaling in epithelial and vascular smooth muscle cells through interactions with Fzd1, Fzd10, and LRP5. Mol. Cell. Biol. 2005, 25, 5022–5030. [Google Scholar] [CrossRef] [PubMed]
- Avgustinova, A.; Iravani, M.; Robertson, D.; Fearns, A.; Gao, Q.; Klingbeil, P.; Hanby, A.M.; Speirs, V.; Sahai, E.; Calvo, F. Tumour cell-derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat. Commun. 2016, 7, 10305. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Karavitis, J.; Zhang, M. COX2 regulation of breast cancer bone metastasis. Oncoimmunology 2013, 2, e23129. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Biological Function | Gene | Full Name | Direct/Indirect Target | Reference |
|---|---|---|---|---|
| Initiation | LGR5 | Leucine-rich repeat-containing G-protein-coupled receptor 5 | Direct | [21] |
| DCLK1 | Doublecortin-like kinase | Direct | [22] | |
| Persistence | KLF5 | Krueppel-like factor 5 | Direct | [21] |
| EDN1 | Endothelin-1 | Direct | [23] | |
| ASCL2 | Achaete-scute homolog 2 | Direct | [24] | |
| FRA1 | Fos-related antigen 1 | Direct | [21] | |
| MYC | Myc proto-oncogene protein | Direct | [21] | |
| CCND1 | CyclinD1 | Direct | [21] | |
| ABCB1 | ABC multidrug transporter | Direct | [25] | |
| Invasion | MMP7 | Matrix Metallopeptidase 7 | Direct | [26] |
| HAS2 | Hyaluronan synthase-2 | Direct | [27] | |
| CD44 | Cluster of differentiation 44 | Indirect | [28] | |
| CXCL12 | C-X-C motif chemokine ligand 12 | Direct | [29] | |
| CXCR4 | Chemokine receptor type 4 | Direct | [30] | |
| Metastasis | CXCL12 | C-X-C motif chemokine ligand 12 | Direct | [29] |
| CXCR4 | Chemokine receptor type 4 | Direct | [30] | |
| DKK1 | Dickkopf-related protein 1 | Direct | [31] | |
| CLDN1 | Claudin-1 | Direct | [32] | |
| CD44v6 | Cluster of differentiation 44 variant exon 6 | Indirect | [33] | |
| FN1 | Fibronectin | Direct | [34] | |
| COX2 | Cyclooxygenase-2 | Direct | [35] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Park, S.-Y.; Jun, Y.; Kim, J.-Y.; Nam, J.-S. Roles of Wnt Target Genes in the Journey of Cancer Stem Cells. Int. J. Mol. Sci. 2017, 18, 1604. https://doi.org/10.3390/ijms18081604
Kim J-H, Park S-Y, Jun Y, Kim J-Y, Nam J-S. Roles of Wnt Target Genes in the Journey of Cancer Stem Cells. International Journal of Molecular Sciences. 2017; 18(8):1604. https://doi.org/10.3390/ijms18081604
Chicago/Turabian StyleKim, Jee-Heun, So-Yeon Park, Youngsoo Jun, Ji-Young Kim, and Jeong-Seok Nam. 2017. "Roles of Wnt Target Genes in the Journey of Cancer Stem Cells" International Journal of Molecular Sciences 18, no. 8: 1604. https://doi.org/10.3390/ijms18081604