Cytoprotective Effect of the UCP2-SIRT3 Signaling Pathway by Decreasing Mitochondrial Oxidative Stress on Cerebral Ischemia–Reperfusion Injury

Abstract
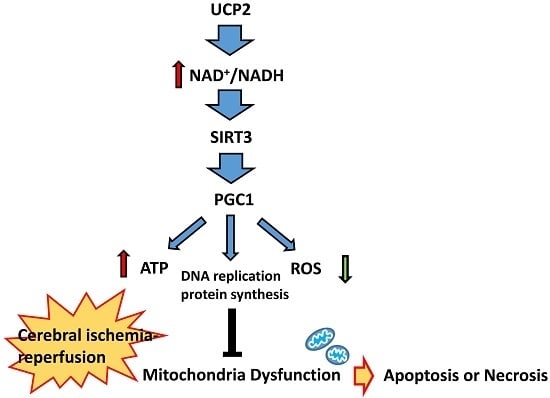
:
{kind=link}
{kind=link}
1. Introduction
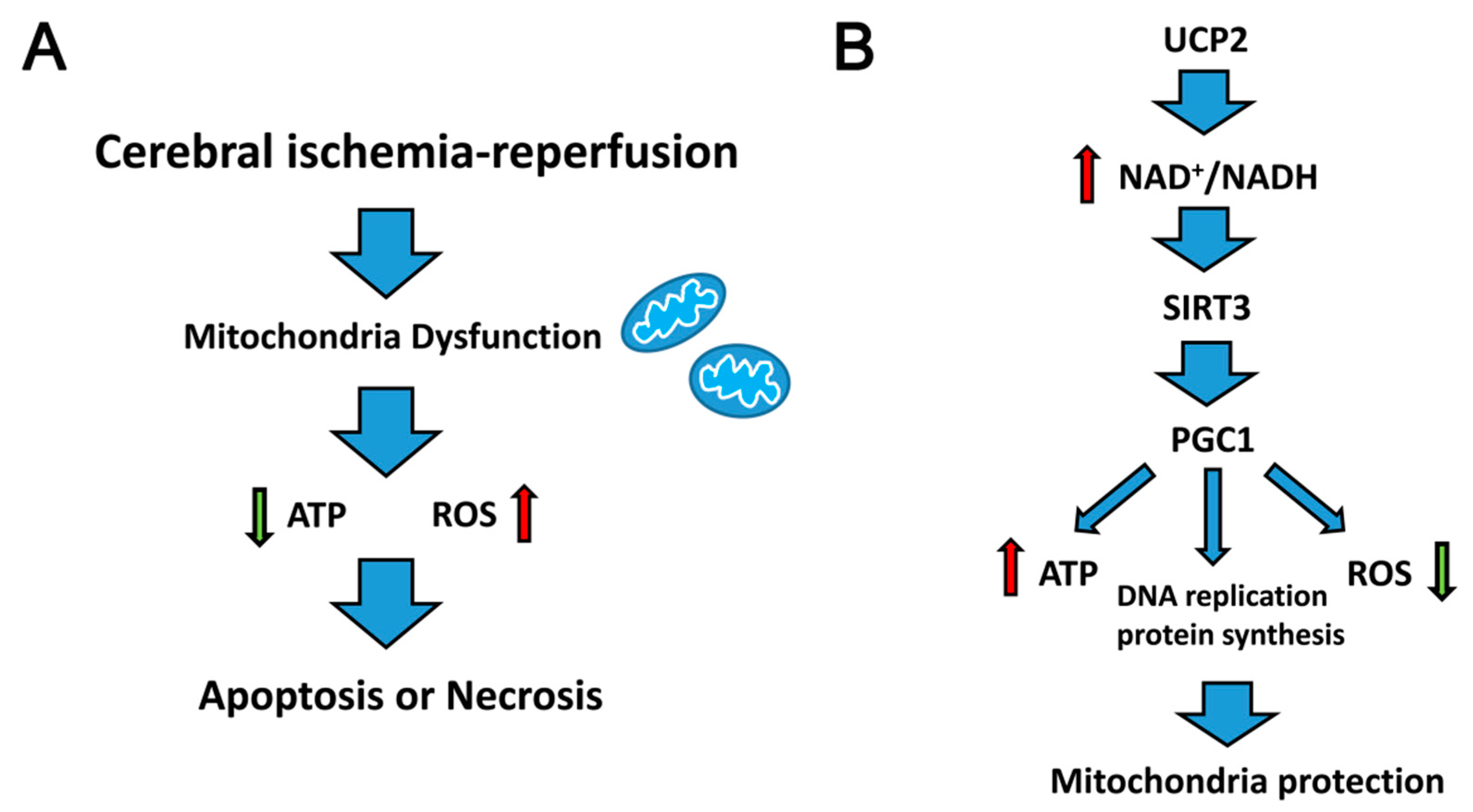
2. Mitochondrial Dysfunction and Reperfusion Injury
3. Sirtuin 3 (SIRT3) and Mitochondrial Protection
- (I)
- SIRT3 can increase ATP production and reduce ROS accumulation by regulating the activity of various important enzymes in the energy metabolism pathways in the mitochondria. Studies have shown that SIRT3 activates long-chain acyl-CoA dehydrogenase (LCAD) in fatty acid β-oxidation, to regulate the lipid metabolism balance [23] However, SIRT3 also promotes energy generation by regulating several key metabolism molecules, such as isocitrate dehydrogenase 2 (IDH2) in the tricarboxylic acid cycle [24], succinate dehydrogenase (SDH) of complex II [25], NADH dehydrogenase and ATP synthase [26]. It also activates acetyl-CoA synthetase 2 (AceCS2) by deacetylation, which promotes the formation of acetyl-coA [23,27]. In addition, SIRT3 also activates LKB1-AMPK pathway to increase ATP production [28].
- (II)
- SIRT3 improves ROS removal from the mitochondria by enhancing the activity of various enzymes in the antioxidant system. Previously, studies have suggested that SIRT3 can increase the expression of manganese superoxide dismutase (MnSOD) and CAT by promoting the transcription of forkhead box O3 (FOXO3a) [29]. It can also deacetylate MnSOD and peroxisome proliferator-activated receptor–gamma coactivator 1α (PGC1α) directly, thereby enhancing the activity of the downstream antioxidant proteins and improving the ability to remove mitochondrial ROS. Moreover, SIRT3 activates glutamate dehydrogenase (GDH) and IDH2 in amino acid metabolism through deacetylation, to promote NADPH production, which in turn provides H+ for the glutathione reduction reaction, eventually increasing ROS hydrolysis [30,31].
- (III)
- SIRT3 promotes the steady state of the mitochondrial environment. It mediates the deacetylation of Cyclophilin D (CypD), a modulatory component of the mitochondrial permeability transition pore (mPTP), which inhibits mPTP opening and delays mitochondrial swelling [30]. Furthermore, SIRT3 prevented cell death by promoting the interaction of Ku70 and Bax, which decreased Bax transfer into the mitochondria from the cytoplasm [32]. In addition, SIRT3 activates PGC1α, which is the transcription co-activator and transcription factor of many nuclear receptors. SIRT3 interacts with PGC1, resulting in acetylation, which plays an important role in mitochondrial DNA replication, transcription and protein synthesis [33].
4. Uncoupling Protein 2 (UCP2) and Cerebral Ischemia–Reperfusion Injury
5. UCP2-NAD+/NADH-SIRT3 Signaling Pathway
6. Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha (PGC1α) Mediated Mitochondrial Biosynthesis
7. Conclusions and Further Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anzell, A.R.; Maizy, R.; Przyklenk, K.; Sanderson, T.H. Mitochondrial quality control and disease: Insights into ischemia-reperfusion injury. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Doyle, K.P.; Simon, R.P.; Stenzel-Poore, M.P. Mechanisms of ischemic brain damage. Neuropharmacology 2008, 55, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Huttemann, M. Molecular mechanisms of ischemia-reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Guaragnella, N.; Giannattasio, S.; Moro, L. Mitochondrial dysfunction in cancer chemoresistance. Biochem. Pharmacol. 2014, 92, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Schrepfer, E.; Scorrano, L. Mitofusins, from mitochondria to metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Albaugh, B.N.; Denu, J.M. Where in the cell is SIRT3?—Functional localization of an NAD+-dependent protein deacetylase. Biochem. J. 2008, 411, e11–e13. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Germain, D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Salvatori, I.; Valle, C.; Ferri, A.; Carrì, M.T. SIRT3 and mitochondrial metabolism in neurodegenerative diseases. Neurochem. Int. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Vassilopoulos, A.; Parisiadou, L.; Yan, Y.; Gius, D. Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid. Redox Signal. 2014, 20, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.H.; Zhu, Y.M.; Park, S.H.; Liu, G.X.; O’Brien, J.; Jiang, H.Y.; Gius, D. SIRT3-mediated dimerization of IDH2 directs cancer cell metabolism and tumor growth. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Rahman, M.S.; Saha, S.K.; Saikot, F.K.; Deep, A.; Kim, K.H. Function of theSIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 2017, 16, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M.; Farbood, Y.; Moghaddam, H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017, 38, 1167–1186. [Google Scholar] [CrossRef] [PubMed]
- Rekuviene, E.; Ivanoviene, L.; Borutaite, V.; Morkuniene, R. Rotenone decreases ischemia-induced injury by inhibiting mitochondrial permeability transition in mature brains. Neurosci. Lett. 2017, 653, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Leak, R.K.; Gao, Y.; Chen, J. The dynamics of the mitochondrial organelle as a potential therapeutic target. J. Cereb. Blood Flow Metab. 2013, 33, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Yue, R.; Xia, X.; Jiang, J.; Yang, D.; Han, Y.; Chen, X.; Cai, Y.; Li, L.; Wang, W.E.; Zeng, C. Mitochondrial DNA oxidative damage contributes to cardiomyocyte ischemia/reperfusion-injury in rats: Cardioprotective role of lycopene. J. Cell. Physiol. 2015, 230, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- Narne, P.; Pandey, V.; Phanithi, P.B. Interplay between mitochondrial metabolism and oxidative stress in ischemic stroke: An epigenetic connection. Mol. Cell. Neurosci. 2017, 82, 176–194. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Liu, Y.; Xie, Y.; Wang, Y.; Qi, S. Remote ischemic postconditioning confers neuroprotective effects via inhibition of the BID-mediated mitochondrial apoptotic pathway. Mol. Med. Rep. 2017, 16, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, O.; de Cristóbal, J.; Sánchez, V.; Lizasoain, I.; Cárdenas, A.; Pereira, M.P.; Colado, M.I.; Leza, J.C.; Lorenzo, P.; Moro, M.A. Inhibition of glutamate release by delaying ATP fall accounts for neuroprotective effects of antioxidants in experimental stroke. FASEB J. 2003, 17, 2082–2084. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. Mech. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wei, W.T.; Jiang, Y.B.; Peng, H.; Cai, J.H.; Mao, C.; Dai, H.; Choy, W.; Bemis, J.E.; Jirousek, M.R.; et al. Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J. Biol. Chem. 2009, 284, 24394–24405. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.Y.; Kim, H.S.; Flynn, C.R.; Hill, S.; McDonald, W.H.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Dittenhaferreed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.S.; Haas, W.; Desquiretdumas, V.; Wallace, D.C.; Procaccio, V.; Gygi, S.P.; Haigis, M.C. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE 2011, 6, e23295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilopoulos, A.; Pennington, J.D.; Andresson, T.; Rees, D.M.; Bosley, A.D.; Fearnley, I.M.; Ham, A.; Flynn, C.R.; Hill, S.; Rose, K.L.; et al. SIRT3 deacetylates ATP synthase F1 complex proteins in response to nutrient- and exercise-induced stress. Antioxid. Redox Signal. 2014, 21, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Lee, S.; Denu, J.M. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. USA 2006, 103, 10230–10235. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, B.; Bossywetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Hirschey, M.D.; Finley, L.W.; Haigis, M.C. Sirtuin regulation of mitochondria: Energy production, apoptosis, and signaling. Trends Biochem. Sci. 2010, 35, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Samant, A.S.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [PubMed]
- Berardi, M.J.; Chou, J.J. Fatty acid flippase activity of UCP2 is essential for its proton transport in mitochondria. Cell Metab. 2014, 20, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Azzu, V.; Jastroch, M.; Divakaruni, A.S.; Brand, M.D. The regulation and turnover of mitochondrial uncoupling proteins. Biochim. Biophys. Acta 2010, 1797, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Bouillaud, F.; Alves-Guerra, M.C.; Ricquier, D. UCPs, at the interface between bioenergetics and metabolism. Biochim. Biophys. Acta 2016, 1863, 2443–2456. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Fu, A.; Robson-Doucette, C.; Allister, E.M.; Wheeler, M.B.; Screaton, R.; Harper, M.E. Glutathionylation state of uncoupling protein-2 and the control of glucose-stimulated insulin secretion. J. Biol. Chem. 2012, 287, 39673–39685. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, A.P.; Cheng, G.; Sutter, A.G.; Evans, Z.P.; Polito, C.C.; Jin, L.; Liu, J.; Schmidt, M.G.; Chavin, K.D. Mitochondrial uncoupling protein 2 induces cell cycle arrest and necrotic cell death. Metab. Syndr. Relat. D 2014, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Giardina, T.M.; Steer, J.H.; Lo, S.Z.; Joyce, D.A. Uncoupling protein-2 accumulates rapidly in the inner mitochondrial membrane during mitochondrial reactive oxygen stress in macrophages. Biochim. Biophys. Acta 2008, 1777, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, I.; Diano, S.; Warden, C.H.; Bartfai, T.; Nitsch, R.; Horvath, T.L. Brain mitochondrial uncoupling protein 2 (UCP2): A protective stress signal in neuronal injury. Biochem. Pharmacol. 2002, 64, 363–367. [Google Scholar] [CrossRef]
- Haines, B.; Li, P.A. Overexpression of Mitochondrial Uncoupling Protein 2 Inhibits Inflammatory Cytokines and Activates Cell Survival Factors after Cerebral Ischemia. PLoS ONE 2012, 7, e31739. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Menzies, K.; Auwerx, J. NAD+ metabolism and the control of energy homeostasis—A balancing act between mitochondria and the nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, L.; Fu, B.; Bai, X.; Zhang, B.; Wu, L.; Cui, J.; Cui, S.; Wei, R.; Chen, X.; Cai, G. NAD Blocks High Glucose Induced Mesangial Hypertrophy via Activation of the Sirtuins-AMPK-mTOR Pathway. Cell. Physiol. Biochem. 2011, 27, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Karamanlidis, G.; Lee, C.F.; Garciamenendez, L.; Kolwicz, S.C.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Rong, T. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart Failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Rowe, G.C.; Jiang, A.; Arany, Z. PGC-1 coactivators in cardiac development and disease. Circ. Res. 2010, 107, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The role of PGC1α in cancer metabolism and its therapeutic implications. Mol. Cancer Ther. 2016, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lebleu, V.S.; O’Connell, J.T.; Herrera, K.N.G.; Wikman, H.; Pantel, K.; Haigis, M.C.; Carvalho, F.M.D.; Damascena, A.; Chinen, L.T.D.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1015. [Google Scholar] [CrossRef] [PubMed]
- Do, M.T.; Kim, H.G.; Choi, J.H.; Jeong, H.G. Metformin induces microRNA-34a to downregulate the Sirt1/Pgc-1α/Nrf2 pathway, leading to increased susceptibility of wild-type p53 cancer cells to oxidative stress and therapeutic agents. Free Radic. Biol. Med. 2014, 74, 21–34. [Google Scholar] [PubMed]
- Fisher, K.W.; Das, B.; Kortum, R.L.; Chaika, O.V.; Lewis, R.E. Kinase suppressor of Ras 1 (KSR1) regulates PGC1α and estrogen-related receptor α to promote oncogenic Ras-dependent anchorage-independent growth. Mol. Cell. Biol. 2011, 31, 2453–2461. [Google Scholar] [CrossRef] [PubMed]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Herzog, B.; Cardenas, J.; Hall, R.K.; Villena, J.A.; Budge, P.J.; Giguère, V.; Granner, D.K.; Kralli, A. Estrogen-related receptor alpha is a repressor of phosphoenolpyruvate carboxykinase gene transcription. J. Biol. Chem. 2006, 281, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.X.; Wang, S.S.; Ge, M.; Wang, D.J. The suppression of PGC-1α is associated with hypoxia-induced endothelial dysfunction and provides a new therapeutic target in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L1233–L1242. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Yang, D.; Lin, T.K.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ischemia. Int. J. Mol. Sci. 2011, 12, 7199–7215. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, J.; Liu, J.; Yan, X.-Y.; Zhang, Y.; Zhang, J.-J.; Zhang, L.-C.; Sun, L.-K. Cytoprotective Effect of the UCP2-SIRT3 Signaling Pathway by Decreasing Mitochondrial Oxidative Stress on Cerebral Ischemia–Reperfusion Injury. Int. J. Mol. Sci. 2017, 18, 1599. https://doi.org/10.3390/ijms18071599
Su J, Liu J, Yan X-Y, Zhang Y, Zhang J-J, Zhang L-C, Sun L-K. Cytoprotective Effect of the UCP2-SIRT3 Signaling Pathway by Decreasing Mitochondrial Oxidative Stress on Cerebral Ischemia–Reperfusion Injury. International Journal of Molecular Sciences. 2017; 18(7):1599. https://doi.org/10.3390/ijms18071599
Chicago/Turabian StyleSu, Jing, Jie Liu, Xiao-Yu Yan, Yong Zhang, Juan-Juan Zhang, Li-Chao Zhang, and Lian-Kun Sun. 2017. "Cytoprotective Effect of the UCP2-SIRT3 Signaling Pathway by Decreasing Mitochondrial Oxidative Stress on Cerebral Ischemia–Reperfusion Injury" International Journal of Molecular Sciences 18, no. 7: 1599. https://doi.org/10.3390/ijms18071599