Autophagy and Human Neurodegenerative Diseases—A Fly’s Perspective


Abstract
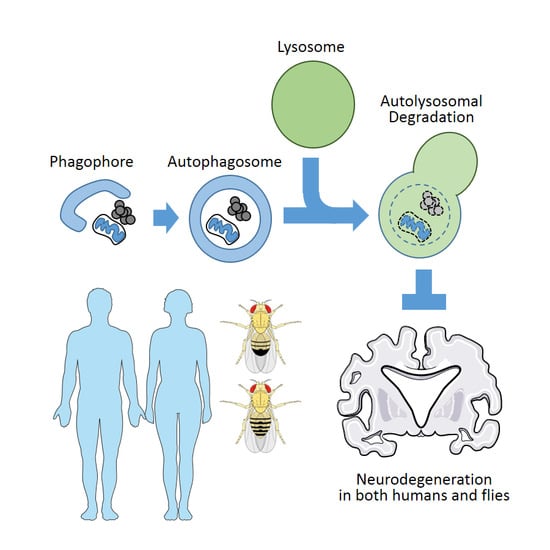
:
1. Autophagy in Human Neurodegenerative Diseases
1.1. Protein Inclusions and Mitochondrial Dysfunction: Hallmarks of Neurodegeneration
1.2. Genetic Mutations that Lead to Protein Inclusions
1.3. Autophagy—One of the Major Homeostatic Processes to Eliminate Protein Aggregates and Damaged Mitochondria
1.4. Role of Autophagy in Neuronal Homeostasis and Preventing Neurodegeneration
1.5. Implication of Autophagy in Alzheimer’s Disease
1.6. Implication of Autophagy in Parkinson’s Disease
2. Drosophila as a Model for Investigating the Autophagy-Neurodegeneration Relationship
2.1. Drosophila as a Genetically Tractable Model Organism
2.2. Drosophila as a Model for Investigating Autophagy
2.3. Drosophila as a Model for Investigating Human Neurodegenerative Diseases
2.4. Drosophila Models of Alzheimer’s Disease: β-amyloid and Tau Flies and Their Autophagic Phenotypes
2.5. Drosophila Models of Parkinson’s Disease: α-Synuclein, PARK Genes and How Autophagic Processes are Compromised in These Models
2.6. Drosophila Models of Polyglutamine Toxicity: Polyglutamine Flies and Their Genetic Interaction with the Autophagic Pathway
2.7. Using Drosophila for Investigating the Pathogenetic Role of Human Autophagy Mutations
3. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| α-syn | α-synuclein |
| Aβ | β-amyloid |
| APP | amyloid precursor protein |
| ATG | autophagy-related |
| FIP200 | FAK family kinase-interacting protein of 200 kDa |
| MAPK | mitogen-activated protein kinases |
| PARK | Parkinson Disease |
| PI3K | phosphatidylinositol 3-kinase |
| TOR | target of rapamycin |
| ULK1 | Unc51-like protein kinase 1 |
References
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A. Polyglutamine pathogenesis: Emergence of unifying mechanisms for huntington’s disease and related disorders. Neuron 2002, 35, 819–822. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Klionsky, D.J. Autophagic processes in yeast: Mechanism, machinery and regulation. Genetics 2013, 194, 341–361. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. The incredible ulks. Cell Commun. Signal. 2012, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Vps15 is required for stress induced and developmentally triggered autophagy and salivary gland protein secretion in Drosophila. Cell Death Differ. 2015, 22, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Proikas-Cezanne, T.; Takacs, Z.; Donnes, P.; Kohlbacher, O. WIPI proteins: Essential Ptdins3P effectors at the nascent autophagosome. J. Cell Sci. 2015, 128, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arencibia, M.; Hochfeld, W.E.; Toh, P.P.; Rubinsztein, D.C. Autophagy, a guardian against neurodegeneration. Semin.Cell Dev. Biol. 2010, 21, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.J.; Garcia-Arencibia, M.; Hochfeld, W.E.; Rubinsztein, D.C. Autophagy and misfolded proteins in neurodegeneration. Exp. Neurol. 2012, 238, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.L.; Klionsky, D.J. How to live long and prosper: Autophagy, mitochondria, and aging. Physiology 2008, 23, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, G.; Erdi, B.; Sass, M.; Neufeld, T.P. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007, 21, 3061–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Sandford, E.; Gatica, D.; Qiu, Y.; Liu, X.; Zheng, Y.; Schulman, B.A.; Xu, J.; Semple, I.; Ro, S.H.; et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife 2016, 5, e12245. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, H.L.; Park, H.W.; Ro, S.H.; Nam, S.G.; Reed, J.M.; Guan, J.L.; Lee, J.H. Drosophila Fip200 is an essential regulator of autophagy that attenuates both growth and aging. Autophagy 2013, 9, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Wang, C.; Peng, X.; Gan, B.; Guan, J.L. Neural-specific deletion of Fip200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J. Biol. Chem. 2010, 285, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Esselens, C.; Oorschot, V.; Baert, V.; Raemaekers, T.; Spittaels, K.; Serneels, L.; Zheng, H.; Saftig, P.; De Strooper, B.; Klumperman, J.; et al. Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J. Cell Biol. 2004, 166, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.A.; Murphy, D.D.; Giasson, B.I.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Degradative organelles containing mislocalized α-and β-synuclein proliferate in presenilin-1 null neurons. J. Cell Biol. 2004, 165, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.; Bandyopadhyay, U.; Jiang, Y.; et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011, 134, 258–277. [Google Scholar] [CrossRef] [PubMed]
- Ling, D.; Song, H.J.; Garza, D.; Neufeld, T.P.; Salvaterra, P.M. Aβ42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS ONE 2009, 4, e4201. [Google Scholar] [CrossRef] [PubMed]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The role of autophagy in parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Martinez-Vicente, M.; Caldwell, G.A.; Caldwell, K.A.; Yue, Z.; Cookson, M.R.; Klein, C.; Vila, M.; Bezard, E. Lysosomal impairment in parkinson’s disease. Mov. Disord. 2013, 28, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Kabuta, T.; Wada, K. Insights into links between familial and sporadic parkinson’s disease: Physical relationship between UCH-L1 variants and chaperone-mediated autophagy. Autophagy 2008, 4, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F. Parkin and mitochondrial quality control: Toward assembling the puzzle. Trends Cell Biol. 2014, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Bier, E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 2005, 6, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. Dis. Models Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, G.; Csikos, G.; Sinka, R.; Erdelyi, M.; Sass, M. The Drosophila homolog of Aut1 is essential for autophagy and development. FEBS Lett. 2003, 543, 154–158. [Google Scholar] [CrossRef]
- Scott, R.C.; Schuldiner, O.; Neufeld, T.P. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell 2004, 7, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.L.; Baehrecke, E.H. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007, 131, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Lindmo, K.; Juhasz, G.; Sass, M.; Seglen, P.O.; Brech, A.; Stenmark, H. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev. Cell 2004, 7, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Ro, S.H.; Semple, I.A.; Park, H.; Park, H.; Park, H.W.; Kim, M.; Kim, J.S.; Lee, J.H. Sestrin2 promotes Unc-51-like kinase 1 (ULK1)-mediated phosphorylation of p62/sequestosome-1. FEBS J. 2014, 281, 3816–3827. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Malik, S.A.; Morselli, E.; Kepp, O.; Criollo, A.; Mouchel, P.L.; Carnuccio, R.; Kroemer, G. Stimulation of autophagy by the p53 target gene sestrin2. Cell Cycle 2009, 8, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Park, E.J.; Birse, R.; Kim, T.E.; Perkins, G.A.; Ocorr, K.; Ellisman, M.H.; Bodmer, R.; Bier, E.; et al. Sestrin as a feedback inhibitor of tor that prevents age-related pathologies. Science 2010, 327, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lao, U.; Edgar, B.A. Tor-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J. Cell Biol. 2009, 186, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Hegedus, K.; Pircs, K.; Varga, A.; Juhasz, G. Different effects of Atg2 and Atg18 mutations on Atg8a and Atg9 trafficking during starvation in Drosophila. FEBS Lett. 2014, 588, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Varga, K.; Nagy, P.; Arsikin Csordas, K.; Kovacs, A.L.; Hegedus, K.; Juhasz, G. Loss of Atg16 delays the alcohol-induced sedation response via regulation of corazonin neuropeptide production in Drosophila. Sci. Rep. 2016, 6, 34641. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Karpati, M.; Varga, A.; Pircs, K.; Venkei, Z.; Takats, S.; Varga, K.; Erdi, B.; Hegedus, K.; Juhasz, G. Atg17/FIP200 localizes to perilysosomal Ref(2)P aggregates and promotes autophagy by activation of Atg1 in Drosophila. Autophagy 2014, 10, 453–467. [Google Scholar] [CrossRef] [PubMed]
- McGurk, L.; Berson, A.; Bonini, N.M. Drosophila as an in vivo model for human neurodegenerative disease. Genetics 2015, 201, 377–402. [Google Scholar] [CrossRef] [PubMed]
- Krench, M.; Littleton, J.T. Neurotoxicity pathways in drosophila models of the polyglutamine disorders. Curr. Top. Dev. Biol. 2017, 121, 201–223. [Google Scholar] [PubMed]
- Hewitt, V.L.; Whitworth, A.J. Mechanisms of parkinson’s disease: Lessons from Drosophila. Curr. Top. Dev. Biol. 2017, 121, 173–200. [Google Scholar] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Crowther, D.C.; Kinghorn, K.J.; Miranda, E.; Page, R.; Curry, J.A.; Duthie, F.A.; Gubb, D.C.; Lomas, D.A. Intraneuronal Aβ, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 2005, 132, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human Wild-type Tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Shulman, J.M.; Feany, M.B. Genetic modifiers of tauopathy in Drosophila. Genetics 2003, 165, 1233–1242. [Google Scholar] [PubMed]
- Ambegaokar, S.S.; Jackson, G.R. Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from Tau phosphorylation. Hum. Mol. Genet. 2011, 20, 4947–4977. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Folwell, J.; Cowan, C.M.; Ubhi, K.K.; Shiabh, H.; Newman, T.A.; Shepherd, D.; Mudher, A. Aβ exacerbates the neuronal dysfunction caused by human tau expression in a Drosophila model of alzheimer’s disease. Exp. Neurol. 2010, 223, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Fulga, T.A.; Elson-Schwab, I.; Khurana, V.; Steinhilb, M.L.; Spires, T.L.; Hyman, B.T.; Feany, M.B. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 2007, 9, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Feany, M.B.; Bender, W.W. A Drosophila model of parkinson’s disease. Nature 2000, 404, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.C.; Whitworth, A.J.; Andrews, L.A.; Parker, T.J.; Pallanck, L.J. Genetic and genomic studies of drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum. Mol. Genet. 2005, 14, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [PubMed]
- Cha, G.H.; Kim, S.; Park, J.; Lee, E.; Kim, M.; Lee, S.B.; Kim, J.M.; Chung, J.; Cho, K.S. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc. Natl. Acad. Sci. USA 2005, 102, 10345–10350. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila pink1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Meulener, M.; Whitworth, A.J.; Armstrong-Gold, C.E.; Rizzu, P.; Heutink, P.; Wes, P.D.; Pallanck, L.J.; Bonini, N.M. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with parkinson’s disease. Curr. Biol. 2005, 15, 1572–1577. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, S.Y.; Cha, G.H.; Lee, S.B.; Kim, S.; Chung, J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene 2005, 361, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Yenisetti, S.C.; Min, K.T. Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr. Biol. 2005, 15, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Kim, W.; Lee, S.; Chung, J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in drosophila. Biochem. Biophys. Res. Commun. 2007, 358, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, X.; Yu, Y.; Li, X.; Wang, T.; Jiang, H.; Ren, Q.; Jiao, Y.; Sawa, A.; Moran, T.; et al. A drosophila model for LRRK2-linked parkinsonism. Proc. Natl. Acad. Sci. USA 2008, 105, 2693–2698. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Gehrke, S.; Wang, H.Q.; Takahashi, R.; Hasegawa, K.; Oota, E.; Lu, B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008, 27, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Semple, I.; Kim, B.; Kiers, A.; Nam, S.; Park, H.W.; Park, H.; Ro, S.H.; Kim, J.S.; Juhasz, G.; et al. Drosophila Gyf/Grb10 interacting GYF protein is an autophagy regulator that controls neuron and muscle homeostasis. Autophagy 2015, 11, 1358–1372. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, J.; Kim, S.; Song, S.; Kwon, S.K.; Lee, S.H.; Kitada, T.; Kim, J.M.; Chung, J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem. Biophys. Res. Commun. 2008, 377, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ding, W.X. Targeting PINK1-Parkin-mediated mitophagy for treating liver injury. Pharmacol. Res. 2015, 102, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Finkel, T. Autophagy as a regulator of cardiovascular redox homeostasis. Free Radic. Biol. Med. 2016, 109, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type atpase ATP13a2/PARK9 function induces general lysosomal deficiency and leads to parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef] [PubMed]
- Usenovic, M.; Krainc, D. Lysosomal dysfunction in neurodegeneration: The role of ATP13A2/PARK9. Autophagy 2012, 8, 987–988. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, Y.; Fujikake, N.; Okamoto, Y.; Popiel, H.A.; Hatanaka, Y.; Ueyama, M.; Suzuki, M.; Gaumer, S.; Murata, M.; Wada, K.; et al. P62 plays a protective role in the autophagic degradation of polyglutamine protein oligomers in polyglutamine disease model flies. J. Biol. Chem. 2015, 290, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for huntington’s disease in an mtor-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Overvatn, A.; Stenmark, H.; Bjorkoy, G.; Simonsen, A.; et al. P62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Birkeland, H.C.; Gillooly, D.J.; Mizushima, N.; Kuma, A.; Yoshimori, T.; Slagsvold, T.; Brech, A.; Stenmark, H. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 2004, 117, 4239–4251. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Cumming, R.C.; Lindmo, K.; Galaviz, V.; Cheng, S.; Rusten, T.E.; Finley, K.D. Genetic modifiers of the drosophila blue cheese gene link defects in lysosomal transport with decreased life span and altered ubiquitinated-protein profiles. Genetics 2007, 176, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Finley, K.D.; Edeen, P.T.; Cumming, R.C.; Mardahl-Dumesnil, M.D.; Taylor, B.J.; Rodriguez, M.H.; Hwang, C.E.; Benedetti, M.; McKeown, M. Blue cheese mutations define a novel, conserved gene involved in progressive neural degeneration. J. Neurosci. 2003, 23, 1254–1264. [Google Scholar] [PubMed]
- Lim, A.; Kraut, R. The Drosophila BEACH family protein, blue cheese, links lysosomal axon transport with motor neuron degeneration. J. Neurosci. 2009, 29, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013, 45, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Ratliff, E.P.; Mauntz, R.E.; Kotzebue, R.W.; Gonzalez, A.; Achal, M.; Barekat, A.; Finley, K.A.; Sparhawk, J.M.; Robinson, J.E.; Herr, D.R.; et al. Aging and autophagic function influences the progressive decline of adult drosophila behaviors. PLoS ONE 2015, 10, e0132768. [Google Scholar] [CrossRef] [PubMed]
- Barekat, A.; Gonzalez, A.; Mauntz, R.E.; Kotzebue, R.W.; Molina, B.; El-Mecharrafie, N.; Conner, C.J.; Garza, S.; Melkani, G.C.; Joiner, W.J.; et al. Using Drosophila as an integrated model to study mild repetitive traumatic brain injury. Sci. Rep. 2016, 6, 25252. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mtor induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Huebener, J.; Renna, M.; Bonin, M.; Riess, O.; Rubinsztein, D.C. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 2010, 133, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Park, H.; Semple, I.A.; Jang, I.; Ro, S.H.; Kim, M.; Cazares, V.A.; Stuenkel, E.L.; Kim, J.J.; Kim, J.S.; et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun. 2014, 5, 4834. [Google Scholar] [CrossRef] [PubMed]


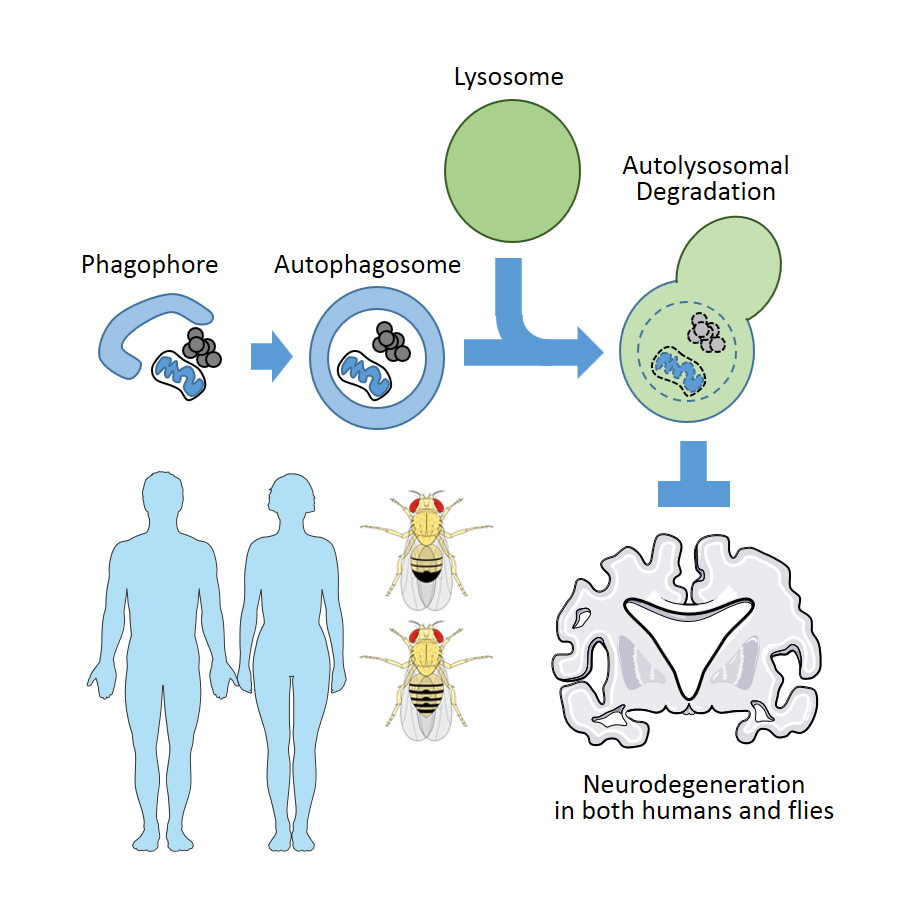{kind=link}
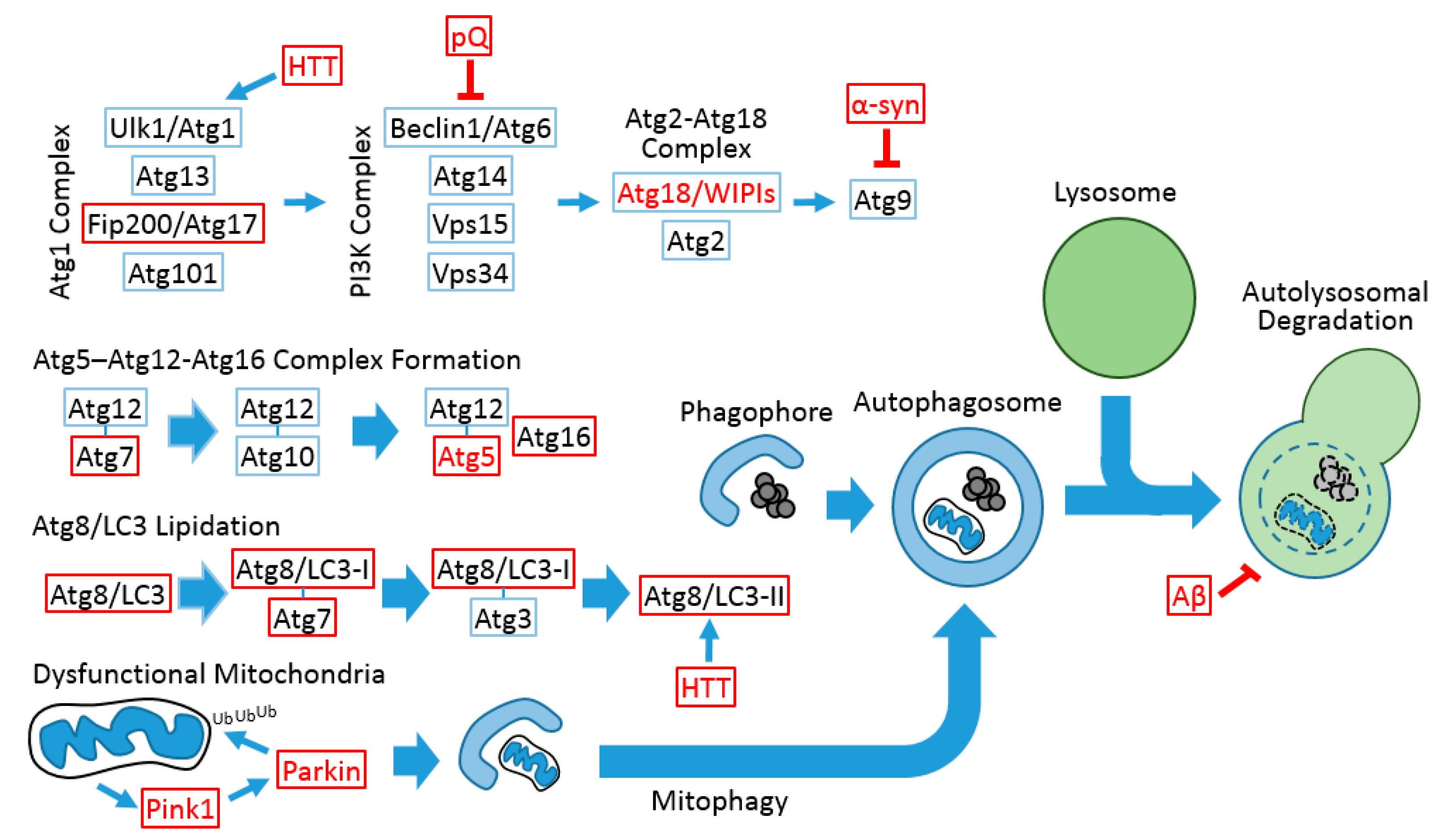{kind=link}
| Gene | Neurodegenerative Phenotype(s) Exhibited by Null Mutants or Hypomorphs |
|---|---|
| Atg5 | Defective locomoter activity, p62/Ref(2)P accumulation in brain, neuronal cell death [22] |
| Atg7 | Defective locomotor activity, ubiquitin accumulation in brain, neuronal inclusion body formation, neuronal cell death, brain vacuolization [21] |
| Retinal degeneration [47] | |
| Atg8a | Ubiquitin accumulation in brain [48] |
| p62/Ref(2)P inclusion formation in neurons [49] | |
| Atg16 | Neuronal inclusion body formation, p62/Ref(2)P accumulation in brain [50] |
| Atg17/Fip200 | Defective locomotor activity, defective wing posture, ubiquitin accumulation in brain, neuronal inclusion body formation, mitochondrial dysfunction, brain vacuolization [23,51] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Ho, A.; Lee, J.H. Autophagy and Human Neurodegenerative Diseases—A Fly’s Perspective. Int. J. Mol. Sci. 2017, 18, 1596. https://doi.org/10.3390/ijms18071596
Kim M, Ho A, Lee JH. Autophagy and Human Neurodegenerative Diseases—A Fly’s Perspective. International Journal of Molecular Sciences. 2017; 18(7):1596. https://doi.org/10.3390/ijms18071596
Chicago/Turabian StyleKim, Myungjin, Allison Ho, and Jun Hee Lee. 2017. "Autophagy and Human Neurodegenerative Diseases—A Fly’s Perspective" International Journal of Molecular Sciences 18, no. 7: 1596. https://doi.org/10.3390/ijms18071596