The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer

Abstract
:1. Introduction
2. Interfollicular Epidermal Stem Cells
Two Distinct Models for Interfollicular Epidermis Self-Renewal Proposed in Mice
3. The p16INK4a Pathway and Its Regulation
3.1. The p16INK4a/pRb Pathway
3.2. The p16INK4a Expression Regulation
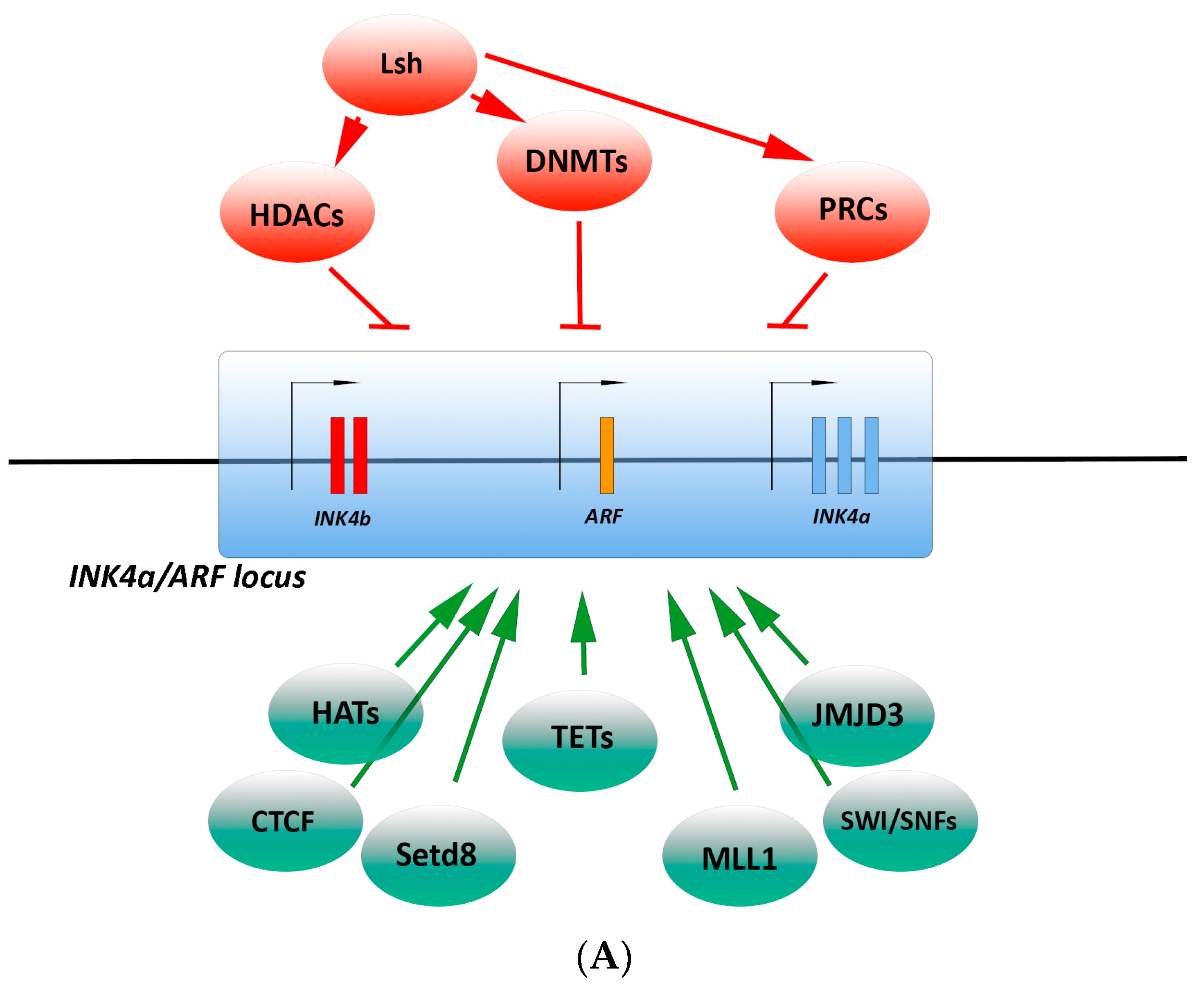
3.2.1. Epigenetic Regulation
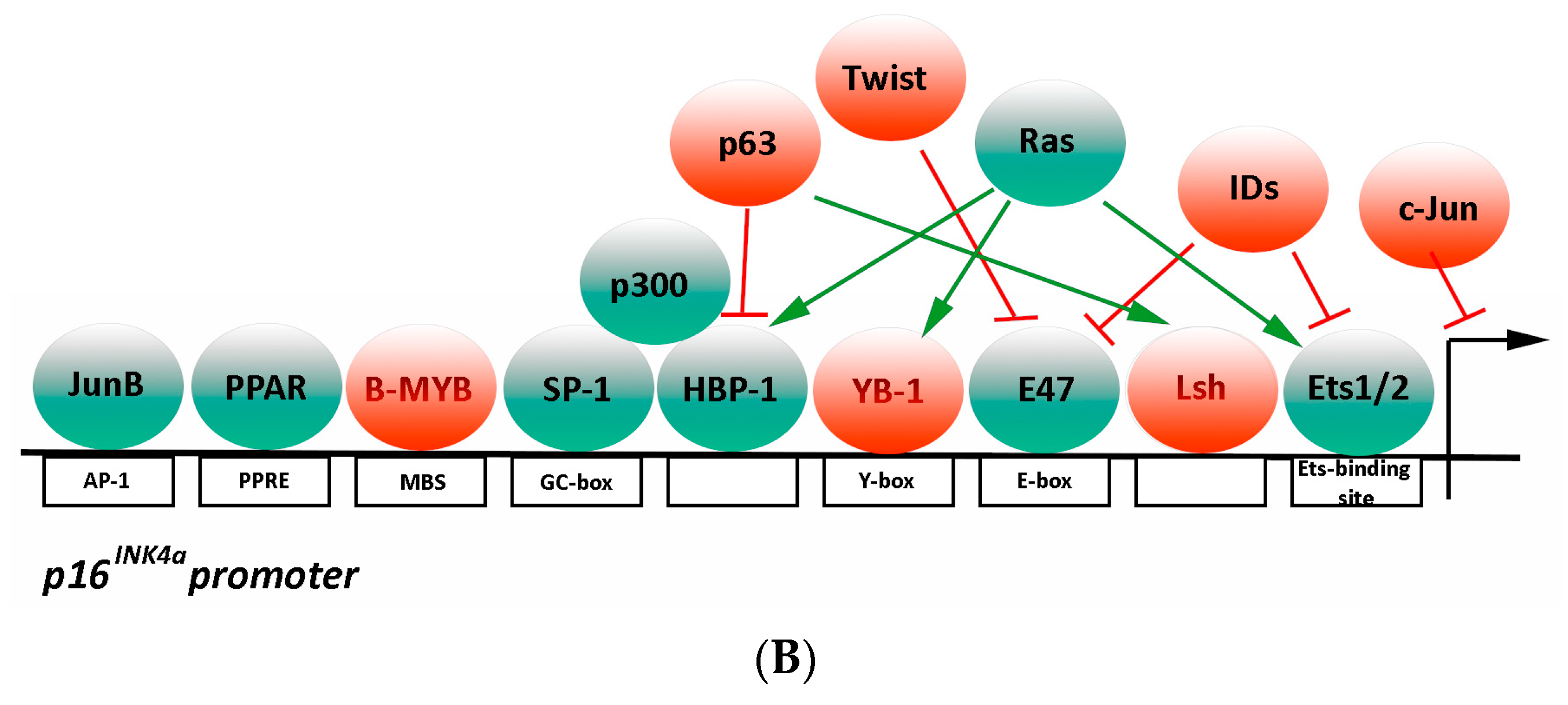
3.2.2. Transcriptional Regulation
4. p16INK4a and Epidermal Homeostasis
4.1. The p16INK4a/pRb Pathway in Epidermal Homeostasis
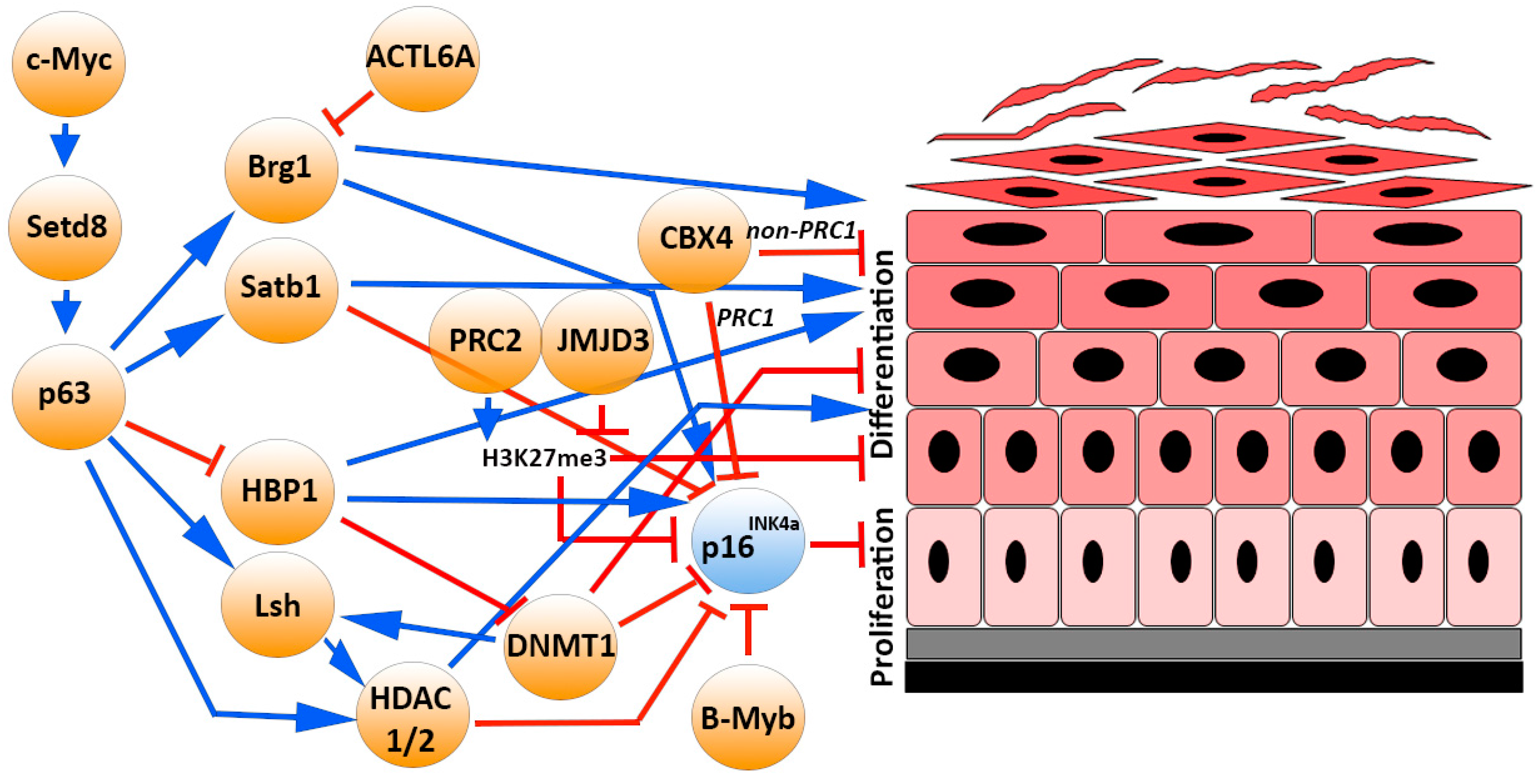
4.2. The p16INK4 Epigenetic Modulators and Transcription Factors in Epidermal Homeostasis
5. p16INK4a and Epidermal Aging
5.1. The p16INK4a/pRb Pathway in Epidermal Aging
5.2. The p16INK4 Epigenetic Modulators and Transcription Factors in Epidermal Aging
6. p16INK4a and Non-Melanoma Skin Cancers
6.1. Oncomine Analysis
6.1.1. p16INK4 Downregulation in Epithelial Tumors
6.1.2. p16INK4 Upregulation in Epithelial Tumors
The p16INK4a/pRb Pathway in Epithelial Tumors
The p16INK4 Epigenetic Modulators and Transcription Factors in Epithelial Tumors
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fuchs, E. Chapter nineteen—Epithelial skin biology: Three decades of developmental biology, a hundred questions answered and a thousand new ones to address. Curr. Top. Dev. Biol. 2016, 116, 357–374. [Google Scholar] [PubMed]
- Mascré, G.; Dekoninck, S.; Drogat, B.; Youssef, K.K.; Brohée, S.; Sotiropoulou, P.A.; Simons, B.D.; Blanpain, C. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 2012, 489, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Potten, C.S. The epidermal proliferative unit: The possible role of the central basal cell. Cell Tissue Kinet. 1974, 7, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, S.; Taichman, L.B. Multiple classes of stem cells in cutaneous epithelium: A lineage analysis of adult mouse skin. EMBO J. 2001, 20, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Clayton, E.; Doupé, D.P.; Klein, A.M.; Winton, D.J.; Simons, B.D.; Jones, P.H. A single type of progenitor cell maintains normal epidermis. Nature 2007, 446, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Doupé, D.P.; Klein, A.M.; Simons, B.D.; Jones, P.H. The ordered architecture of murine ear epidermis is maintained by progenitor cells with random fate. Dev. Cell 2010, 18, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Haase, I.; Nestle, F.O. Mechanisms regulating skin immunity and inflammation. Nat. Rev. Immunol. 2014, 14, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Kolodka, T.M.; Garlick, J.A.; Taichman, L.B. Evidence for keratinocyte stem cells in vitro: Long term engraftment and persistence of transgene expression from retrovirus-transduced keratinocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 4356–4461. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, S.; Taichman, L.B. Organization of stem cells and their progeny in human epidermis. J. Investig. Dermatol. 2005, 124, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Rheinwald, J.G.; Green, H. Serial cultivation of strains of human epidermal keratinocytes: The formation of keratinizing colonies from single cells. Cell 1975, 6, 331–343. [Google Scholar] [CrossRef]
- Barrandon, Y.; Grasset, N.; Zaffalon, A.; Gorostidi, F.; Claudinot, S.; Droz-Georget, S.L.; Nanba, D.; Rochat, A. Capturing epidermal stemness for regenerative medicine. Semin. Cell Dev. Biol. 2012, 23, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.; Ranno, R.; Stracuzzi, G.; Bondanza, S.; Guerra, L.; Zambruno, G.; Micali, G.; de Luca, M. The control of epidermal stem cells (holoclones) in the treatment of massive full-thickness burns with autologous keratinocytes cultured on fibrin. Transplantation 1999, 68, 868–879. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.; Golisano, O.; Paterna, P.; Lambiase, A.; Bonini, S.; Rama, P.; de Luca, M. Location and clonal analysis of stem cells and their differentiated progeny in the human ocular surface. J. Cell Biol. 1999, 145, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Rama, P.; Matuska, S.; Paganoni, G.; Spinelli, A.; de Luca, M.; Pellegrini, G. Limbal stem-cell therapy and long-term corneal regeneration. N. Engl. J. Med. 2010, 363, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Rama, P.; Bonini, S.; Lambiase, A.; Golisano, O.; Paterna, P.; de Luca, M.; Pellegrini, G. Autologous fibrin-cultured limbal stem cells permanently restore the corneal surface of patients with total limbal stem cell deficiency. Transplantation 2001, 72, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, L.; de Luca, M. Cell biology: Dormant and restless skin stem cells. Nature 2012, 489, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Dellambra, E.; Golisano, O.; Bondanza, S.; Siviero, E.; Lacal, P.; Molinari, M.; D’Atri, S.; de Luca, M. Downregulation of 14-3-3σ prevents clonal evolution and leads to immortalization of primary human keratinocytes. J. Cell Biol. 2000, 149, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- Maurelli, R.; Zambruno, G.; Guerra, L.; Abbruzzese, C.; Dimri, G.; Gellini, M.; Bondanza, S.; Dellambra, E. Inactivation of p16INK4a (inhibitor of cyclin-dependent kinase 4A) immortalizes primary human keratinocytes by maintaining cells in the stem cell compartment. FASEB J. 2006, 20, 1516–1518. [Google Scholar] [CrossRef] [PubMed]
- Cordisco, S.; Maurelli, R.; Bondanza, S.; Stefanini, M.; Zambruno, G.; Guerra, L.; Dellambra, E. Bmi-1 reduction plays a key role in physiological and premature aging of primary human keratinocytes. J. Investig. Dermatol. 2010, 130, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat. Res. 2005, 576, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Narita, M. Cellular senescence and chromatin organisation. Br. J. Cancer 2007, 96, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Poi, M.J.; Tsai, M. Regulatory mechanisms of tumor suppressor P16INK4A and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef] [PubMed]
- LaPak, K.M.; Burd, C.E. The molecular balancing act of p16INK4a in cancer and aging. Mol. Cancer Res. 2014, 12, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.C.; Fuchs, E. The Yin and Yang of chromatin dynamics in stem cell fate selection. Trends Genet. 2016, 32, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Perdigoto, C.N.; Valdes, V.J.; Bardot, E.S.; Ezhkova, E. Epigenetic regulation of epidermal differentiation. Cold Spring Harb. Perspect. Med. 2014, 4, a015263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bardot, E.; Ezhkova, E. Epigenetic regulation of skin: Focus on the polycomb complex. Cell. Mol. Life Sci. 2012, 69, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; O’Loghlen, A. PRC1 complex diversity: Where is it taking us? Trends Cell Biol. 2014, 24, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15INK4B tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Takebayashi, S.I.; Sakamoto, A.; Igata, T.; Nakatsu, Y.; Saitoh, N.; Hino, S.; Nakao, M. The SETD8/PR-Set7 methyltransferase functions as a barrier to prevent senescence-associated metabolic remodeling. Cell Rep. 2017, 18, 2148–2161. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Zeng, Y.; Xiong, Y. DDB1-CUL4 and MLL1 mediate oncogene-induced p16INK4a activation. Cancer Res. 2009, 69, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Pan, K.; Chen, Y.; Huang, C.; Zhang, X. The acetylation of transcription factor HBP1 by p300/CBP enhances p16INK4A expression. Nucleic Acids Res. 2012, 40, 981–995. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A.; Gdula, M.R.; Mardaryev, A.N.; Sharov, A.A.; Fessing, M.Y. Epigenetic regulation of gene expression in keratinocytes. J. Investig. Dermatol. 2012, 132, 2505–2521. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, Y.; Xu, L.; Chen, Y.; Zhang, Y.; Su, D.; Ren, G.; Lu, J.; Huang, B. YY1 restrained cell senescence through repressing the transcription of p16. Biochim. Biophys. Acta 2008, 1783, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Kia, S.K.; Gorski, M.M.; Giannakopoulos, S.; Verrijzer, C.P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 2008, 28, 3457–3464. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Zhuang, X.; Yao, Y.G.; Zhang, R. BRG1 is required for formation of senescence-associated heterochromatin foci induced by oncogenic RAS or BRCA1 loss. Mol. Cell. Biol. 2013, 33, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Muegge, K. Lsh, a guardian of heterochromatin at repeat elements. Biochem. Cell Biol. 2005, 83, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Zhu, H.; Xu, H.; Schmidtmann, A.; Geiman, T.M.; Muegge, K. Lsh controls Hox gene silencing during development. Proc. Natl. Acad. Sci. USA 2007, 104, 14366–14371. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Han, L.; Li, G.; Tong, T. Senescence delay and repression of p16INK4a by Lsh via recruitment of histone deacetylases in human diploid fibroblasts. Nucleic Acids Res. 2009, 37, 5183–5196. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Russo, J.; Kohwi, Y.; Kohwi-Shigematsu, T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008, 452, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Agrelo, R.; Kishimoto, H.; Novatchkova, M.; Peraza, V.; Paolino, M.; Souabni, A.; Wutz, A. SATB1 collaborates with loss of p16 in cellular transformation. Oncogene 2013, 32, 5492–5500. [Google Scholar] [CrossRef] [PubMed]
- Witcher, M.; Emerson, B.M. Epigenetic silencing of the p16INK4a tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol. Cell 2009, 34, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Raisner, R.M.; Madhani, H.D. Patterning chromatin: Form and function for H2A.Z variant nucleosomes. Curr. Opin. Genet. Dev. 2006, 16, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Filippova, G.N. Genetics and epigenetics of the multifunctional protein CTCF. Curr. Top. Dev. Biol. 2008, 80, 337–360. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.A.; Felsenfeld, G. We gather together: Insulators and genome organization. Curr. Opin. Genet. Dev. 2007, 17, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Zebedee, Z.; Huot, T. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 2001, 409, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008, 14, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Peters, G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Ozawa, Y.; Harada, M.; Kitagawa, K.; Niida, H.; Morita, Y.; Tanaka, K.; Suda, T.; Kitagawa, M. YB1 binds to and represses the p16 tumor suppressor gene. Genes Cells 2013, 18, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Cakouros, D.; Isenmann, S.; Cooper, L.; Zannettino, A.; Anderson, P.; Glackin, C.; Gronthos, S. Twist-1 induces Ezh2 recruitment regulating histone methylation along the Ink4A/Arf locus in mesenchymal stem cells. Mol. Cell. Biol. 2012, 32, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Chen, Y.; Roth, M.; Wang, W.; Wang, S.; Yee, A.S.; Zhang, X. HBP1-mediated transcriptional regulation of DNA methyltransferase 1 and its impact on cell senescence. Mol. Cell. Biol. 2013, 33, 887–903. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, J.; Li, R.; Wang, P.; Han, L.; Zhang, Z.; Tong, T. B-MYB delays cell aging by repressing p16INK4α transcription. Cell. Mol. Life Sci. 2011, 68, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Mechta-Grigoriou, F.; Gerald, D.; Yaniv, M. The mammalian Jun proteins: Redundancy and specificity. Oncogene 2001, 20, 2378–2389. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Huang, J.; Zhou, R.; Niu, J.; Zhu, X.; Wang, J.; Zhang, Z.; Tong, T. PPARγ accelerates cellular senescence by inducing p16INK4α expression in human diploid fibroblasts. J. Cell Sci. 2008, 121, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Woods, M.; Pant, R.; Mallya, S.M. Cyclin D1 and cyclin D-dependent kinases enhance oral keratinocyte proliferation but do not block keratinocyte differentiation. Int. J. Oncol. 2010, 37, 1471–1475. [Google Scholar] [PubMed]
- Ivanova, I.A.; D’Souza, S.J.; Dagnino, L. Signalling in the epidermis: The E2F cell cycle regulatory pathway in epidermal morphogenesis, regeneration and transformation. Int. J. Biol. Sci. 2005, 1, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Dellambra, E.; Ciarapica, R.; Capogrossi, M.C. Oxidative stress, microRNAs and cytosolic calcium homeostasis. Cell Calcium 2016, 60, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.; DePinho, R. Telomeres, stem cells, senescence, and cancer. J. Clin. Investig. 2004, 113, 160. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.L.; Reuter, J.A.; Webster, D.E.; Zhu, L.; Khavari, P.A. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 2010, 463, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.L.; Webster, D.E.; Barragan, D.I.; Chang, H.Y.; Khavari, P.A. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev. 2008, 22, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Ezhkova, E.; Lien, W.H.; Stokes, N.; Pasolli, H.A.; Silva, J.M.; Fuchs, E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 2011, 25, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009, 136, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Luis, N.M.; Morey, L.; Mejetta, S.; Pascual, G.; Janich, P.; Kuebler, B.; Cozutto, L.; Roma, G.; Nascimento, E.; Frye, M.; et al. Regulation of human epidermal stem cell proliferation and senescence requires polycomb-dependent and -independent functions of Cbx4. Cell Stem Cell 2011, 9, 233–246. [Google Scholar] [CrossRef] [PubMed]
- LeBoeuf, M.; Terrell, A.; Trivedi, S.; Sinha, S.; Epstein, J.A.; Olson, E.N.; Morrisey, E.E.; Millar, S.E. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev. Cell 2010, 19, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bartolome, R.; Callejas-Valera, J.L.; Gutkind, J.S. Control of the epithelial stem cell epigenome: The shaping of epithelial stem cell identity. Curr. Opin. Cell Biol. 2013, 25, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development 2004, 131, 2737–2748. [Google Scholar] [CrossRef] [PubMed]
- Paramio, J.M.; Segrelles, C.; Casanova, M.L.; Jorcano, J.L. Opposite functions for E2F1 and E2F4 in human epidermal keratinocyte differentiation. J. Biol. Chem. 2000, 275, 41219–41226. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Santos, M.; Martinez-Fernandez, M.; Lorz, C.; Lazaro, S.; Paramio, J.M. Deregulation of the pRb-E2F4 axis alters epidermal homeostasis and favors tumor development. Oncotarget 2016, 7, 75712–75728. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Paramio, J.M.; Santos, M. Skin tumors Rb(eing) uncovered. Front. Oncol. 2013, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Adhikary, G.; Rorke, E.A.; Chew, Y.C.; Balasubramanian, S. Polycomb group proteins are key regulators of keratinocyte function. J. Investig. Dermatol. 2011, 131, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Benitah, S.A. Chromatin regulators in mammalian epidermis. Semin. Cell Dev. Biol. 2012, 23, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Avgustinova, A.; Benitah, S.A. Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 2016, 17, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, T.X.; Hughes, M.W.; Wu, P.; Yu, J.; Widelitz, R.B.; Fan, G.; Chuong, C.M. Progressive alopecia reveals decreasing stem cell activation probability during aging of mice with epidermal deletion of DNA methyltransferase 1. J. Investig. Dermatol. 2012, 132, 2681–2690. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Beerman, I.; Lien, W.H.; Smith, Z.D.; Gu, H.; Boyle, P.; Gnirke, A.; Fuchs, E.; Rossi, D.J.; Meissner, A. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 2012, 47, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Mulder, K.W.; Wang, X.; Escriu, C.; Ito, Y.; Schwarz, R.F.; Gillis, J.; Sirokmány, G.; Donati, G.; Uribe-Lewis, S.; Pavlidis, P.; et al. Diverse epigenetic strategies interact to control epidermal differentiation. Nat. Cell Biol. 2012, 14, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Bardot, E.S.; Valdes, V.J.; Zhang, J.; Perdigoto, C.N.; Nicolis, S.; Hearn, S.A.; Silva, J.M.; Ezhkova, E. Polycomb subunits Ezh1 and Ezh2 regulate the Merkel cell differentiation program in skin stem cells. EMBO J. 2013, 32, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Dauber, K.L.; Perdigoto, C.N.; Valdes, V.J.; Santoriello, F.J.; Cohen, I.; Ezhkova, E. Dissecting the roles of polycomb repressive complex 2 subunits in the control of skin development. J. Investig. Dermatol. 2016, 136, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Mejetta, S.; Morey, L.; Pascual, G.; Kuebler, B.; Mysliwiec, M.R.; Lee, Y.; Shiekhattar, R.; di Croce, L.; Benitah, S.A. Jarid2 regulates mouse epidermal stem cell activation and differentiation. EMBO J. 2011, 30, 3635–3646. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Mustafi, S.B.; Street, M.; Dey, A.; Dwivedi, S.K.D. Bmi-1: At the crossroads of physiological and pathological biology. Genes Dis. 2015, 2, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Ressler, S.; Bartkova, J.; Niederegger, H.; Bartek, J.; Scharffetter-Kochanek, K.; Jansen-Dürr, P.; Wlaschek, M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 2006, 5, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, C.M.; Uthman, A.; Erovic, B.M.; Pammer, J. Expression of BMI-1 in normal skin and inflammatory and neoplastic skin lesions. J. Cutan. Pathol. 2007, 34, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Lee, K.; Adhikary, G.; Gopalakrishnan, R.; Rorke, E.A.; Eckert, R.L. The Bmi-1 polycomb group gene in skin cancer: Regulation of function by (−)-epigallocatechin-3-gallate. Nutr. Rev. 2008, 66 (Suppl. S1), S65–S68. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Adhikary, G.; Balasubramanian, S.; Gopalakrishnan, R.; McCormick, T.; Dimri, G.P.; Eckert, R.L.; Rorke, E. A expression of Bmi-1 in epidermis enhances cell survival by altering cell cycle regulatory protein expression and inhibiting apoptosis. J. Investig. Dermatol. 2008, 128, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; García, J.M.; Peña, C.; García, V.; Domínguez, G.; Suárez, D.; Camacho, F.I.; Espinosa, R.; Provencio, M.; España, P. Implication of polycomb members Bmi-1, Mel-18, and Hpc-2 in the regulation of p16INK4a, p14ARF, h-TERT, and c-Myc expression in primary breast carcinomas. Clin. Cancer Res. 2006, 12, 6929–6936. [Google Scholar] [CrossRef] [PubMed]
- Roscic, A.; Möller, A.; Calzado, M.A.; Renner, F.; Wimmer, V.C.; Gresko, E.; Lüdi, K.S.; Schmitz, M.L. Phosphorylation-dependent control of Pc2 SUMO E3 ligase activity by its substrate protein HIPK2. Mol. Cell 2006, 24, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Mardaryev, A.N.; Liu, B.; Rapisarda, V.; Poterlowicz, K.; Malashchuk, I.; Rudolf, J.; Sharov, A.A.; Jahoda, C.A.; Fessing, M.Y.; Benitah, S.A.; et al. Cbx4 maintains the epithelial lineage identity and cell proliferation in the developing stratified epithelium. J. Cell Biol. 2016, 212, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Moser, M.A.; Meunier, D.; Fischer, C.; Machat, G.; Mattes, K.; Lichtenberger, B.M.; Brunmeir, R.; Weissmann, S.; Murko, C.; et al. Divergent roles of HDAC1 and HDAC2 in the regulation of epidermal development and tumorigenesis. EMBO J. 2013, 32, 3176–3191. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Fisher, A.G.; Watt, F.M. Epidermal stem cells are defined by global histone modifications that are altered by Myc-induced differentiation. PLoS ONE 2007, 2, e763. [Google Scholar] [CrossRef] [PubMed]
- Markova, N.G.; Karaman-Jurukovska, N.; Pinkas-Sarafova, A.; Marekov, L.N.; Simon, M. Inhibition of histone deacetylation promotes abnormal epidermal differentiation and specifically suppresses the expression of the late differentiation marker profilaggrin. J. Investig. Dermatol. 2007, 127, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef] [PubMed]
- McKeon, F.; Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Driskell, I.; Oda, H.; Blanco, S.; Nascimento, E.; Humphreys, P.; Frye, M. The histone methyltransferase Setd8 acts in concert with c-Myc and is required to maintain skin. EMBO J. 2012, 31, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, S.; Candi, E.; Hu, B.; Dolfini, D.; Ravo, M.; Grober, O.M.V.; Grober, O.M.V.; Weisz, A.; Dotto, G.P.; Melino, G.; et al. The p63 target HBP1 is required for skin differentiation and stratification. Cell Death Differ. 2010, 17, 1896–1907. [Google Scholar] [CrossRef] [PubMed]
- Keyes, W.M.; Pecoraro, M.; Aranda, V.; Vernersson-Lindahl, E.; Li, W.; Vogel, H.; Guo, X.; Garcia, E.L.; Michurina, T.V.; Enikolopov, G.; et al. A ΔNp63α is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 2011, 8, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Fessing, M.Y.; Mardaryev, A.N.; Gdula, M.R.; Sharov, A.A.; Sharova, T.Y.; Rapisarda, V.; Gordon, K.B.; Smorodchenko, A.D.; Poterlowicz, K.; Ferone, G.; et al. p63 regulates Satb1 to control tissue-specific chromatin remodeling during development of the epidermis. J. Cell Biol. 2011, 194, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Lena, A.M.; Mancini, M.; Rivetti di Val Cervo, P.; Saintigny, G.; Mahé, C.; Melino, G.; Candi, E. Micro RNA-191 triggers keratinocytes senescence by SATB1 and CDK6 downregulation. Biochem. Biophys. Res. Commun. 2012, 423, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Mardaryev, A.N.; Gdula, M.R.; Yarker, J.L.; Emelianov, V.U.; Emelianov, V.N.; Poterlowicz, K.; Sharov, A.A.; Sharova, T.Y.; Scarpa, J.A.; Joffe, B.; et al. p63 and Brg1 control developmentally regulated higher-order chromatin remodelling at the epidermal differentiation complex locus in epidermal progenitor cells. Development 2014, 141, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Indra, A.K.; Dupé, V.; Bornert, J.M.; Messaddeq, N.; Yaniv, M.; Mark, M.; Chambon, P.; Metzger, D. Temporally controlled targeted somatic mutagenesis in embryonic surface ectoderm and fetal epidermal keratinocytes unveils two distinct developmental functions of BRG1 in limb morphogenesis and skin barrier formation. Development 2005, 132, 4533–4544. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Tang, J.; Lopez-Pajares, V.; Tao, S.; Qu, K.; Crabtree, G.R.; Khavari, P.A. ACTL6a enforces the epidermal progenitor state by suppressing SWI/SNF-dependent induction of KLF4. Cell Stem Cell 2013, 12, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Garrido, M.; Ceballos, L.; Alonso-Lecue, P.; Abraira, C.; Delgado, M.D.; Gandarillas, A. A cell cycle role for the epigenetic factor CTCF-L/BORIS. PLoS ONE 2012, 7, e39371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, P.; Chin, S.S.; Wang, D.; Liu, S.; Sinha, S.; Garrett-Sinha, L.A. Ets1 blocks terminal differentiation of keratinocytes and induces expression of matrix metalloproteases and innate immune mediators. J. Cell Sci. 2010, 123, 3566–3575. [Google Scholar] [CrossRef] [PubMed]
- Chin, S.S.; Romano, R.A.; Nagarajan, P.; Sinha, S.; Garrett-Sinha, L.A. Aberrant epidermal differentiation and disrupted ΔNp63/Notch regulatory axis in Ets1 transgenic mice. Biol. Open 2013, 2, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Langlands, K.; Down, G.A.; Kealey, T. Id proteins are dynamically expressed in normal epidermis and dysregulated in squamous cell carcinoma. Cancer Res. 2000, 60, 5929–5933. [Google Scholar] [PubMed]
- Maruyama, H.; Ishitsuka, Y.; Fujisawa, Y.; Furuta, J.; Sekido, M.; Kawachi, Y. B-Myb enhances proliferation and suppresses differentiation of keratinocytes in three-dimensional cell culture. Arch. Dermatol. Res. 2014, 306, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Adhikary, G.; Young, C.A.; Jans, R.; Crish, J.F.; Xu, W.; Rorke, E.A. AP1 transcription factors in epidermal differentiation and skin cancer. J. Skin Cancer 2013, 2013, 537028. [Google Scholar] [CrossRef] [PubMed]
- Wurm, S.; Zhang, J.; Guinea-Viniegra, J.; Garcia, F.; Munoz, J.; Bakiri, L.; Ezhkova, E.; Wagner, E.F. Terminal epidermal differentiation is regulated by the interaction of Fra-2/AP-1 with Ezh2 and ERK1/2. Genes Dev. 2015, 29, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Yamada, A.; Kimura, S.; Peters, J.M.; Gonzalez, F.J. Alterations in skin and stratified epithelia by constitutively activated PPARalpha. J. Investig. Dermatol. 2006, 126, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; dePinho, R.A. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol. 2007, 8, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Giangreco, A.; Qin, M.; Pintar, J.E.; Watt, F.M. Epidermal stem cells are retained in vivo throughout skin aging. Aging Cell 2008, 7, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.C.; Bickenbach, J.R. Aging epidermis is maintained by changes in transit-amplifying cell kinetics, not stem cell kinetics. J. Investig. Dermatol. 2009, 129, 2541–2543. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.M.; Bickenbach, J.R. Epidermal stem cells are resistant to cellular aging. Aging Cell 2007, 6, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Gallico, G.G.; O’Connor, N.E.; Compton, C.C.; Kehinde, O.; Green, H. Permanent coverage of large burn wounds with autologous cultured human epithelium. N. Engl. J. Med. 1984, 311, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Charruyer, A.; Barland, C.O.; Yue, L.; Wessendorf, H.B.; Lu, Y.; Jeffrey Lawrence, H.; Mancianti, M.L.; Ghadially, R. Transit-amplifying cell frequency and cell cycle kinetics are altered in aged epidermis. J. Investig. Dermatol. 2009, 129, 2574–2583. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Ramsey, M.R.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N.E. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.E.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006, 443, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Janzen, V.; Forkert, R.; Fleming, H.E.; Saito, Y.; Waring, M.T.; Dombkowski, D.M.; Cheng, T.; dePinho, R.A.; Sharpless, N.E.; Scadden, D.T. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 2006, 443, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16INK4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of p16INK4a in peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Muss, H.B.J.K.; Alston, S.M.; Lacy, A.C.; Jolly, T.A.; Williams, G.; Carey, L.A.; Dees, E.C.; Anders, C.K.; Irvin, W.J.S.N. Chemotherapy in older women with breast cancer. N. Engl. J. Med. 2009, 361, 1023–1024. [Google Scholar] [CrossRef]
- Boquoi, A.; Arora, S.; Chen, T.; Litwin, S.; Koh, J.; Enders, G.H. Reversible cell cycle inhibition and premature aging features imposed by conditional expression of p16INK4a. Aging Cell 2015, 14, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Waaijer, M.E.C.; Parish, W.E.; Strongitharm, B.H.; van Heemst, D.; Slagboom, P.E.; de Craen, A.J.M.; Sedivy, J.M.; Westendorp, R.G.J.; Gunn, D.A.; Maier, A.B. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, 11, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Adamus, J.; Aho, S.; Meldrum, H.; Bosko, C.; Lee, J.M. p16INK4a influences the aging phenotype in the living skin equivalent. J. Investig. Dermatol. 2014, 134, 1131–1133. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Kajiya, H.; Ozeki, S.; Okabe, K.; Ikebe, T. Reactive oxygen species promotes cellular senescence in normal human epidermal keratinocytes through epigenetic regulation of p16INK4a. Biochem. Biophys. Res. Commun. 2014, 452, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Cao, R.; Viatour, P.; Sage, J.; Zhang, Y.; Xiong, Y. pRB family proteins are required for H3K27 trimethylation and polycomb repression complexes binding to and silencing p16INK4a tumor suppressor gene. Genes Dev. 2007, 21, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Rivetti di val Cervo, P.; Lena, A.M.; Nicoloso, M.; Rossi, S.; Mancini, M.; Zhou, H.; Saintigny, G.; Dellambra, E.; Odorisio, T.; Mahé, C.; et al. p63-microRNA feedback in keratinocyte senescence. Proc. Natl. Acad. Sci. USA 2012, 109, 1133–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyes, W.M.; Wu, Y.; Vogel, H.; Guo, X.; Lowe, S.W.; Mills, A.A. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005, 19, 1986–1999. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.Q. Growth retardation and premature aging phenotypes in mice with disruption of the SNF2-like gene, PASG. Genes Dev. 2004, 18, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Chen, T.; Han, L.; Wang, P.; Li, N.; Tong, T. Transcriptional activation of the senescence regulator Lsh by E2F1. Mech. Ageing Dev. 2011, 132, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, W.; Liu, X.; Paulson, K.E.; Yee, A.S.; Zhang, X. Transcriptional factor HBP1 targets p16INK4a, upregulating its expression and consequently is involved in Ras-induced premature senescence. Oncogene 2010, 29, 5083–5094. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, B.J.; Chaturvedi, V.; Bacon, P.; Qin, J.Z.; Denning, M.F.; Diaz, M.O. Id-1 delays senescence but does not immortalize keratinocytes. J. Biol. Chem. 2000, 275, 27501–27504. [Google Scholar] [CrossRef] [PubMed]
- Alani, R.M.; Hasskarl, J.; Grace, M.; Hernandez, M.C.; Israel, M.A.; Münger, K. Immortalization of primary human keratinocytes by the helix-loop-helix protein, Id-1. Proc. Natl. Acad. Sci. USA 1999, 96, 9637–9641. [Google Scholar] [CrossRef] [PubMed]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenführ, M.; Maertens, G.; Banck, M.; Zhou, M.M.; Walsh, M.J.; et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Mowla, S.N.; Lam, E.W.F.; Jat, P.S. Cellular senescence and aging: The role of B-MYB. Aging Cell 2014, 13, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Lear, J.T.; Szeimies, R. Non-melanoma skin cancer. Lancet 2010, 375, 673–685. [Google Scholar] [CrossRef]
- Diffey, B.L.; Langtry, J.A.A. Skin cancer incidence and the ageing population. Br. J. Dermatol. 2005, 153, 679–680. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Robert, L. Cell senescence: role in aging and age-related diseases. Interdiscip. Top. Gerontol. 2014, 39, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Ruini, C.; Witkowski, A.M.; Cesinaro, A.; Teixeira de Carvalho, N.; Pellacani, G. From actinic keratosis to squamous cell carcinoma: Evidence of morphologic and biologic progression. J. Am. Acad. Dermatol. 2015, 72, S8–S10. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chi, S.; Xie, J. Hedgehog signaling in skin cancers. Cell Signal. 2011, 23, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Cretnik, M.; Poje, G.; Musani, V.; Kruslin, B.; Ozretic, P.; Tomas, D.; Situm, M.; Levanat, S. Involvement of p16 and PTCH in pathogenesis of melanoma and basal cell carcinoma. Int. J. Oncol. 2009, 34, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16INK4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; dePinho, R.A. Loss of p16INK4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Mortier, L.; Marchetti, P.; Delaporte, E.; Martin de Lassalle, E.; Thomas, P.; Piette, F.; Formstecher, P.; Polakowska, R.; Danzé, P.M. Progression of actinic keratosis to squamous cell carcinoma of the skin correlates with deletion of the 9p21 region encoding the p16INK4a tumor suppressor. Cancer Lett. 2002, 176, 205–214. [Google Scholar] [CrossRef]
- Chang, T.G.; Wang, J.; Chen, L.W.; Hsu, C.Y.; Chang, H.W.; Chen, J.S.; Cho, C.L. Loss of expression of the p16 gene is frequent in malignant skin tumors. Biochem. Biophys. Res. Commun. 1997, 230, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Kalof, A.N.; Evans, M.F.; Simmons-Arnold, L.; Beatty, B.G.; Cooper, K. p16INK4a immunoexpression and HPV in situ hybridization signal patterns: Potential markers of high-grade cervical intraepithelial neoplasia. Am. J. Surg. Pathol. 2005, 29, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Saridaki, Z.; Liloglou, T.; Zafiropoulos, A.; Koumantaki, E.; Zoras, O.; Spandidos, D.A. Mutational analysis of CDKN2A genes in patients with squamous cell carcinoma of the skin. Br. J. Dermatol. 2003, 148, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Hodges, A.; Smoller, B.R. Immunohistochemical comparison of p16 expression in actinic keratoses and squamous cell carcinomas of the skin. Mod. Pathol. 2002, 15, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Salama, M.E.; Mahmood, M.N.; Qureshi, H.S.; Ma, C.; Zarbo, R.J.; Ormsby, A.H. p16INK4a expression in actinic keratosis and Bowen’s disease. Br. J. Dermatol. 2003, 149, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Blokx, W.A.M.; de Jong, E.M.G.J.; de Wilde, P.C.M.; Bulten, J.; Link, M.M.G.M.; Ruiter, D.J.; van de Kerkhof, P.C.M. p16 and p53 expression in (pre)malignant epidermal tumors of renal transplant recipients and immunocompetent individuals. Mod. Pathol. 2003, 16, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Cabral, L.S.; Festa Neto, C.; Sanches, J.A.; Ruiz, I.R.G. Genomic instability in human actinic keratosis and squamous cell carcinoma. Clinics (Sao Paulo) 2011, 66, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Kanellou, P.; Zaravinos, A.; Zioga, M.; Spandidos, D.A. Deregulation of the tumour suppressor genes p14(ARF), p15(INK4b), p16(INK4a) and p53 in basal cell carcinoma. Br. J. Dermatol. 2009, 160, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Brasanac, D.; Stojkovic-Filipovic, J.; Bosic, M.; Tomanovic, N.; Manojlovic-Gacic, E. Expression of G1/S-cyclins and cyclin-dependent kinase inhibitors in actinic keratosis and squamous cell carcinoma. J. Cutan. Pathol. 2016, 43, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Bagazgoitia, L.; Cuevas, J.; Juarranz, A. Expression of p53 and p16 in actinic keratosis, bowenoid actinic keratosis and Bowen’s disease. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, K.; Svensson, S.; Landberg, G. Retinoblastoma protein function and p16INK4a expression in actinic keratosis, squamous cell carcinoma in situ and invasive squamous cell carcinoma of the skin and links between p16INK4a expression and infiltrative behavior. Mod. Pathol. 2004, 17, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Grønhøj Larsen, C.; Gyldenløve, M.; Jensen, D.H.; Therkildsen, M.H.; Kiss, K.; Norrild, B.; Konge, L.; von Buchwald, C. Correlation between human papillomavirus and p16 overexpression in oropharyngeal tumours: A systematic review. Br. J. Cancer 2014, 110, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Beadle, B.M.; William, W.N.; McLemore, M.S.; Sturgis, E.M.; Williams, M.D. p16 Expression in cutaneous squamous carcinomas with neck metastases: A potential pitfall in identifying unknown primaries of the head and neck. Head Neck 2013, 35, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.S.; Patrick, D.R.; Edwards, G.; Goodhart, P.J.; Huber, H.E.; Miles, L.; Garsky, V.M.; Oliff, A.; Heimbrook, D.C. Protein domains governing interactions between E2F, the retinoblastoma gene product, and human papillomavirus type 16 E7 protein. Mol. Cell. Biol. 1993, 13, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Romagosa, C.; Simonetti, S.; López-Vicente, L.; Mazo, A.; Lleonart, M.E.; Castellvi, J.; Ramon y Cajal, S. p16(Ink4a) overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 2011, 30, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Küsters-Vandevelde, H.V.N.; de Koning, M.N.C.; Melchers, W.J.G.; Quint, W.G.V.; de Wilde, P.C.M.; de Jong, E.M.G.J.; van de Kerkhof, P.C.M.; Blokx, W.A.M. Expression of p14ARF, p16INK4a and p53 in relation to HPV in (pre-)malignant squamous skin tumours. J. Cell. Mol. Med. 2009, 13, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Mao, X.; Talbot, I.C. Aberrant cytological localization of p16 and CDK4 in colorectal epithelia in the normal adenoma carcinoma sequence. World J. Gastroenterol. 2006, 12, 6391–6396. [Google Scholar] [CrossRef] [PubMed]
- Conscience, I.; Jovenin, N.; Coissard, C.; Lorenzato, M.; Durlach, A.; Grange, F.; Birembaut, P.; Clavel, C.; Bernard, P. p16 is overexpressed in cutaneous carcinomas located on sun-exposed areas. Eur. J. Dermatology 2006, 16, 518–522. [Google Scholar] [CrossRef]
- Eshkoor, S.A.; Ismail, P.; Rahman, S.A.; Oshkour, S.A. p16 gene expression in basal cell carcinoma. Arch. Med. Res. 2008, 39, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Svensson, S.; Nilsson, K.; Ringberg, A.; Landberg, G. Invade or proliferate? Two contrasting events in malignant behavior governed by p16(INK4a) and an intact Rb pathway illustrated by a model system of basal cell carcinoma. Cancer Res. 2003, 63, 1737–1742. [Google Scholar] [PubMed]
- Nandakumar, V.; Vaid, M.; Tollefsbol, T.O.; Katiyar, S.K. Aberrant DNA hypermethylation patterns lead to transcriptional silencing of tumor suppressor genes in UVB-exposed skin and UVB-induced skin tumors of mice. Carcinogenesis 2011, 32, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Biasiotta, A.; D’Arcangelo, D.; Passarelli, F.; Nicodemi, E.M.; Facchiano, A. Ion channels expression and function are strongly modified in solid tumors and vascular malformations. J. Transl. Med. 2016, 14, 285. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, D.; Facchiano, F.; Nassa, G.; Stancato, A.; Antonini, A.; Rossi, S.; Senatore, C.; Cordella, M.; Tabolacci, C.; Salvati, A.; et al. PDGFR-alpha inhibits melanoma growth via CXCL10/IP-10: A multi-omics approach. Oncotarget 2016, 7, 77257–77275. [Google Scholar] [CrossRef] [PubMed]
- Nindl, I.; Dang, C.; Forschner, T.; Kuban, R.J.; Meyer, T.; Sterry, W.; Stockfleth, E. Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol. Cancer 2006, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Riker, A.I.; Enkemann, S.A.; Fodstad, O.; Liu, S.; Ren, S.; Morris, C.; Xi, Y.; Howell, P.; Metge, B.; Samant, R.S.; et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genomics 2008, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Ginos, M.A.; Page, G.P.; Michalowicz, B.S.; Patel, K.J.; Volker, S.E.; Pambuccian, S.E.; Ondrey, F.G.; Adams, G.L.; Gaffney, P.M. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004, 64, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Cromer, A.; Carles, A.; Millon, R.; Ganguli, G.; Chalmel, F.; Lemaire, F.; Young, J.; Dembélé, D.; Thibault, C.; Muller, D.; et al. Identification of genes associated with tumorigenesis and metastatic potential of hypopharyngeal cancer by microarray analysis. Oncogene 2004, 23, 2484–2498. [Google Scholar] [CrossRef] [PubMed]
- Toruner, G.A.; Ulger, C.; Alkan, M.; Galante, A.T.; Rinaggio, J.; Wilk, R.; Tian, B.; Soteropoulos, P.; Hameed, M.R.; Schwalb, M.N.; et al. Association between gene expression profile and tumor invasion in oral squamous cell carcinoma. Cancer Genet. Cytogenet. 2004, 154, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.H.; Liao, C.T.; Peng, S.C.; Chen, Y.J.; Cheng, A.J.; Juang, J.L.; Tsai, C.Y.; Chen, T.C.; Chuang, Y.J.; Tang, C.Y.; et al. A novel molecular signature identified by systems genetics approach predicts prognosis in oral squamous cell carcinoma. PLoS ONE 2011, 6, e23452. [Google Scholar] [CrossRef] [PubMed]
- Lazarov, M.; Kubo, Y.; Cai, T.; Dajee, M.; Tarutani, M.; Lin, Q.; Fang, M.; Tao, S.; Green, C.L.; Khavari, P. A CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat. Med. 2002, 8, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Aldabagh, B.; Yu, J.; Arron, S.T. Role of human papillomavirus in cutaneous squamous cell carcinoma: A meta-analysis. J. Am. Acad. Dermatol. 2014, 70, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, J.H.; Hauck, F.; Barros, M.H.M.; Niedobitek, G. pRb and CyclinD1 Complement p16 as Immunohistochemical Surrogate Markers of HPV Infection in Head and Neck Cancer. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.J.; Dicker, A.J.; Dahler, A.L.; Saunders, N.A. E2F as a Regulator of Keratinocyte Proliferation: Implications for Skin Tumor Development. J. Investig. Dermatol. 1997, 109, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Sohng, S.H.; Shin, D.H.; Choi, J.S.; Bae, Y.K. Immunohistochemical expression of cytokeratin 15, cytokeratin 19, follistatin, and Bmi-1 in basal cell carcinoma. Int. J. Dermatol. 2016, 55, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.L.; You, P.; Su, J.; Zhu, M.H.; Xie, D.F.; Zhu, H.Y.; He, Z.Y.; Li, J.X.; Ding, X.Y.; et al. Oncoprotein BMI-1 induces the malignant transformation of HaCaT cells. J. Cell. Biochem. 2009, 106, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, G.; Grun, D.; Kerr, C.; Balasubramanian, S.; Rorke, E.A.; Vemuri, M.; Boucher, S.; Bickenbach, J.R.; Hornyak, T.; Xu, W.; et al. Identification of a population of epidermal squamous cell carcinoma cells with enhanced potential for tumor formation. PLoS ONE 2013, 8, e84324. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, G.; Grun, D.; Balasubramanian, S.; Kerr, C.; Huang, J.M.; Eckert, R.L. Survival of skin cancer stem cells requires the Ezh2 polycomb group protein. Carcinogenesis 2015, 36, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Abba, M.C.; Molinolo, A.A.; Vitale-Cross, L.; Wang, Z.; Zaida, M.; Delic, N.C.; Samuels, Y.; Lyons, G.J.; Gutkind, J.S. The head and neck cancer cell oncogenome: A platform for the development of precision molecular therapies. Oncotarget 2014, 5, 8906–8923. [Google Scholar] [CrossRef] [PubMed]
- Missero, C.; Antonini, D. Crosstalk among p53 family members in cutaneous carcinoma. Exp. Dermatol. 2014, 23, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Bosic, M.M.; Brasanac, D.C.; Stojkovic-Filipovic, J.M.; Zaletel, I.V.; Gardner, J.M.; Cirovic, S.L. Expression of p300 and p300/CBP associated factor (PCAF) in actinic keratosis and squamous cell carcinoma of the skin. Exp. Mol. Pathol. 2016, 100, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Cai, M.Y.; Luo, R.Z.; Tian, X.; Liao, Q.M.; Zhang, X.Y.; Han, J.D. Overexpression of p300 correlates with poor prognosis in patients with cutaneous squamous cell carcinoma. Br. J. Dermatol. 2015, 172, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, M.R.; He, L.; Forster, N.; Ory, B.; Ellisen, L.W. Physical association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Res. 2011, 71, 4373–4379. [Google Scholar] [CrossRef] [PubMed]
- Torkamandi, S.; Moghbeli, M.; Farshchian, M.; Rad, A.; Abbaszadegan, M.R. Role of Brg1 in progression of esophageal squamous cell carcinoma. Iran. J. Basic Med. Sci. 2014, 17, 912–917. [Google Scholar] [PubMed]
- Li, Y.C.; Bu, L.L.; Mao, L.; Ma, S.R.; Liu, J.F.; Yu, G.T.; Deng, W.W.; Zhang, W.F.; Sun, Z.J. SATB1 promotes tumor metastasis and invasiveness in oral squamous cell carcinoma. Oral Dis. 2017, 23, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Cong, Q.X.; Zhang, H.; Sun, S.X.; Li, H.F.; Wang, Y.; Jian, S. Pilot study special AT-rich sequence-binding protein 1 investigating as a potential biomarker for esophageal squamous cell carcinoma. Dis. Esophagus 2016, 29, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.D.; Ji, W.; Zhang, W.; He, L.X.; Yang, J.; Liang, H.J.; Wang, L.L. Overexpression of SATB1 in laryngeal squamous cell carcinoma. ORL J. Otorhinolaryngol. Relat. Spec. 2010, 72, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Schramek, D.; Adam, R.C.; Keyes, B.E.; Wang, P.; Zheng, D.; Fuchs, E. ETS family transcriptional regulators drive chromatin dynamics and malignancy in squamous cell carcinomas. Elife 2015, 4, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, V.; Cesnjaj, M.; Bacon, P.; Panella, J.; Choubey, D.; Diaz, M.O.; Nickoloff, B.J. Role of INK4a/Arf locus-encoded senescent checkpoints activated in normal and psoriatic keratinocytes. Am. J. Pathol. 2003, 162, 161–170. [Google Scholar] [CrossRef]
- Lin, J.; Guan, Z.; Wang, C.; Feng, L.; Zheng, Y.; Caicedo, E.; Bearth, E.; Peng, J.R.; Gaffney, P.; Ondrey, F.G. Inhibitor of differentiation 1 contributes to head and neck squamous cell carcinoma survival via the NF-kappaB/survivin and phosphoinositide 3-kinase/Akt signaling pathways. Clin. Cancer Res. 2010, 16, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Pan, H.; Li, J.; Zhong, Q.; Chen, X.; Dry, S.M.; Wang, C.Y. Epigenetic Activation of AP1 Promotes Squamous Cell Carcinoma Metastasis. Sci. Signal. 2013, 6, ra28. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.Y.; Ke, H.; Hall, R.P.; Zhang, J. Y. c-Jun Promotes whereas JunB Inhibits Epidermal Neoplasia. J. Investig. Dermatol. 2011, 131, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Hyakusoku, H.; Sano, D.; Takahashi, H.; Hatano, T.; Isono, Y.; Shimada, S.; Ito, Y.; Myers, J.N.; Oridate, N. JunB promotes cell invasion, migration and distant metastasis of head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 6. [Google Scholar] [CrossRef] [PubMed]
- Mangone, F.R.R.; Brentani, M.M.; Nonogaki, S.; Begnami, M.D.F.S.; Campos, A.H.J.F.M.; Walder, F.; Carvalho, M.B.; Soares, F.A.; Torloni, H.; Kowalski, L.P.; et al. Overexpression of Fos-related antigen-1 in head and neck squamous cell carcinoma. Int. J. Exp. Pathol. 2005, 86, 205–212. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, S.O.M.; Mesquita, R.A.; Pinto, D.S.; Gutkind, S. Immunolocalization of c-Fos and c-Jun in human oral mucosa and in oral squamous cell carcinoma. J. Oral Pathol. Med. 2002, 31, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Ye, D.X.; Zhang, W.B.; Pan, H.Y.; Zhang, Z.Y.; Zhang, L. Overexpression of c-fos promotes cell invasion and migration via CD44 pathway in oral squamous cell carcinoma. J. Oral Pathol. Med. 2015, 44, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Bhattacharya, S.; Steele, R.; Phillips, N.; Ray, R.B. Involvement of c-Fos in the Promotion of Cancer Stem-like Cell Properties in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2017, 23, 3120–3128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Luo, S.; Lechler, T.; Zhang, J.Y. FRA1 promotes squamous cell carcinoma growth and metastasis through distinct AKT and c-Jun dependent mechanisms. Oncotarget 2016, 7, 34371–34383. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.T.; Zhu, P.Q.; Luo, H.L.; Zhang, Y.; Hao, T.F.; Xia, G.F.; Zhu, Z.M.; Qiu, C. Long noncoding RNA ANRIL: A potential novel prognostic marker in cancer: A meta-analysis. Minerva Med. 2016, 107, 77–83. [Google Scholar] [PubMed]
- Li, Z.; Yu, X.; Shen, J. ANRIL: A pivotal tumor suppressor long non-coding RNA in human cancers. Tumour Biol. 2016, 37, 5657–5661. [Google Scholar] [CrossRef] [PubMed]
- Sand, M.; Bechara, F.G.; Sand, D.; Gambichler, T.; Hahn, S.A.; Bromba, M.; Stockfleth, E.; Hessam, S. Long-noncoding RNAs in basal cell carcinoma. Tumor Biol. 2016, 37, 10595–10608. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Nindl Dataset [178] | Riker Dataset [179] | Ginos Dataset [180] | Cromer Dataset [181] | Peng Dataset [183] | Toruner Dataset [182] | TOT |
|---|---|---|---|---|---|---|---|
| Normal Tissue | 6 | 4 | 13 | 4 | 22 | 4 | 53 |
| Actinic (Solar) Keratosis | 4 | 0 | 0 | 0 | 0 | 0 | 4 |
| Skin Squamous Cell Carcinoma | 5 | 11 | 0 | 0 | 0 | 0 | 16 |
| Skin Basal Cell Carcinoma | 0 | 15 | 0 | 0 | 0 | 0 | 15 |
| Head and Neck Squamous Cell Carcinoma | 0 | 0 | 41 | 34 | 0 | 0 | 75 |
| Oral Cavity Squamous Cell Carcinoma | 0 | 0 | 0 | 0 | 57 | 16 | 73 |
| Actinic (solar) Keratosis | Skin Squamous Cell Carcinoma | Skin Basal Cell Carcinoma | Skin Squamous Cell Carcinoma | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nindl Dataset | Riker Dataset | |||||||||
| Cancer vs. Normal | Cancer vs. Normal | Cancer vs. Normal | Cancer vs. Normal | |||||||
| n | Protein | Gene | Fold Change | p Value | Fold Change | p Value | Fold Change | p Value | Fold Change | p Value |
| 1 | p16INK4a | CDKN2A | 2.218 | 0.004 | 3.553 | 0.003 | 2.297 | 0.009 | 4.404 | 4.09 × 10−4 |
| p16INK4a Downstream Genes | ||||||||||
| 2 | CDK4 | CDK4 | −1.033 | 0.602 | 1.125 | 0.163 | 1.976 | 5.34 × 10−5 | 1.713 | 8.07 × 10−4 |
| 3 | Cyclin D1 | CCND1 | 2.393 | 0.053 | 1.513 | 0.226 | 1.006 | 0.492 | 1.002 | 0.496 |
| 4 | pRB | RB1 | 1.974 | 0.05 | 1.349 | 0.17 | 1.251 | 0.017 | 1.579 | 5.48 × 10−4 |
| 5 | p107 | RBL1 | n.a. | n.a. | n.a. | n.a. | 1.307 | 0.284 | 1.9 | 0.11 |
| 6 | p130 | RBL2 | 1.632 | 0.152 | 1.676 | 0.163 | 1.367 | 0.141 | 1.41 | 0.046 |
| 7 | E2F1 | E2F1 | 1.885 | 0.0037 | 2.071 | 0.028 | 1.604 | 0.011 | 1.749 | 0.004 |
| 8 | E2F2 | E2F2 | 1.918 | 0.065 | 2.655 | 0.052 | 1.277 | 0.16 | 1.769 | 0.028 |
| 9 | E2F3 | E2F3 | 1.759 | 0.006 | 1.78 | 0.006 | 3.28 | 2.41 × 10−5 | 2.617 | 3.08 × 10−4 |
| 10 | E2F4 | E2F4 | −1.072 | 0.638 | 1.114 | 0.089 | −1.139 | 0.702 | 1.03 | 0.452 |
| Epigenetic Regulator Genes | ||||||||||
| 11 | DNMT1 | DNMT1 | 1.473 | 0.068 | 1.81 | 0.05 | 1.945 | 0.001 | 2.221 | 1.27 × 10−4 |
| 12 | DNMT3A | DNMT3A | −2.54 | 0.894 | −3.426 | 0.969 | 3.818 | 1.55 × 10−8 | 1.703 | 3.19 × 10−4 |
| 13 | DNMT3B | DNMT3B | 1.256 | 0.008 | 2.032 | 0.008 | 1.079 | 0.258 | 1.48 | 0.011 |
| 14 | Bmi-1 | BMI1 | −1.347 | 0.874 | −1.536 | 0.939 | 1.713 | 0.001 | 1.324 | 0.24 |
| 15 | Ezh1 | EZH1 | 1.397 | 0.259 | 1.619 | 0.272 | 1.611 | 0.251 | 1.379 | 0.331 |
| 16 | Ezh2 | EZH2 | 1.671 | 0.059 | 2.31 | 0.017 | 2.016 | 2.24 × 10−4 | 2.112 | 8.79 × 10−5 |
| 17 | EED | EED | −1.293 | 0.659 | −1.043 | 0.567 | 1.231 | 0.074 | 1.411 | 0.016 |
| 18 | CBX4 | CBX4 | −1.269 | 0.64 | 1.072 | 0.456 | −1.082 | 0.762 | 1.006 | 0.486 |
| 19 | CBX7 | CBX7 | −1.315 | 0.855 | −1.993 | 0.997 | −2.078 | 0.992 | −2.923 | 0.999 |
| 20 | Jarid2 | JARID2 | 1.167 | 0.223 | 1.409 | 0.021 | 1.091 | 0.195 | 1.382 | 0.006 |
| 21 | JMJD3 | JMJD3 | 1.568 | 0.018 | 1.562 | 0.017 | 2.822 | 0.075 | 1.853 | 0.171 |
| 22 | JDP2 | JDP2 | n.a. | n.a. | n.a. | n.a. | 1.264 | 0.052 | −1.147 | 0.821 |
| 23 | SETD8 | KMT5A | −1.282 | 0.629 | 1.411 | 0.102 | 1.06 | 0.346 | 1.505 | 0.004 |
| 24 | KMT2A | MLL | 1.657 | 0.004 | 1.479 | 0.043 | 1.413 | 0.01 | 1.184 | 0.076 |
| 25 | HAT p300 | EP300 | 1.692 | 0.041 | 1.855 | 0.025 | 1.112 | 0.039 | 1.143 | 0.03 |
| 26 | HDAC1 | HDAC1 | 1.066 | 0.414 | 1.632 | 0.025 | 1.636 | 1.39 × 10−5 | 2.309 | 2.89 × 10−7 |
| 27 | HDAC2 | HDAC2 | −1.461 | 0.909 | 1.007 | 0.486 | 2.223 | 1.19 × 10−4 | 1.682 | 0.001 |
| 28 | p63 | TP63 | 1.452 | 0.234 | 2.905 | 0.008 | 1.273 | 0.022 | 1.512 | 0.002 |
| 29 | BRG1 | SMARCA4 | 2.03 | 0.003 | 1.826 | 2.27 × 10−4 | 1.666 | 2.08 × 10−6 | 1.767 | 0.002 |
| 30 | LSH | HELLS | 4.504 | 0.039 | 8.371 | 0.016 | 5.292 | 3.02 × 10−4 | 1.948 | 0.003 |
| 31 | Satb1 | SATB1 | −1.125 | 0.853 | −1.17 | 0.835 | −1.342 | 0.865 | 1.25 | 0.813 |
| 32 | CTCF | CTCF | −1.083 | 0.756 | 1.063 | 0.35 | 1.54 | 1.72 × 10−4 | 1.277 | 0.006 |
| Transcriptional Regulator Genes | ||||||||||
| 33 | Ets1 | ETS1 | 1.31 | 0.759 | 1.446 | 0.13 | 1.139 | 0.329 | 2.421 | 0.011 |
| 34 | Ets2 | ETS2 | 1.247 | 0.102 | 1.696 | 0.007 | −1.463 | 0.823 | 1.106 | 0.393 |
| 35 | ID1 | ID1 | 1.23 | 0.243 | 1.48 | 0.103 | −1.824 | 0.995 | 1.434 | 0.041 |
| 36 | ID2 | ID2 | 1.508 | 0.042 | −1.117 | 0.698 | 1.281 | 0.163 | 1.165 | 0.262 |
| 37 | ID3 | ID3 | 1.224 | 0.184 | −1.512 | 0.92 | −2.316 | 0.999 | −1.137 | 0.687 |
| 38 | YB1 | YXB1 | 1.126 | 0.164 | 1.117 | 0.271 | 1.314 | 0.005 | 1.48 | 0.001 |
| 39 | SP1 | SP1 | -1.699 | 0.769 | 1.847 | 0.082 | 1.048 | 0.223 | 1.112 | 0.317 |
| 40 | HBP1 | PBRM1 | 1.167 | 0.156 | 1.146 | 0.192 | 1.513 | 5.96 × 10−6 | 1.984 | 0.031 |
| 41 | b-myb | MYB | −1.66 | 0.898 | 1.348 | 0.925 | −1.14 | 0.593 | 1.592 | 0.204 |
| 42 | JUNB | JUNB | 1.107 | 0.292 | 1.375 | 0.078 | −1.989 | 0.927 | −1.09 | 0.586 |
| 43 | c-Jun | C-JUN | 1.635 | 0.016 | 1.517 | 0.014 | 1.055 | 0.455 | 1.04 | 0.468 |
| 44 | JunD | JUND | 1.16 | 0.121 | 1.058 | 0.401 | −1.428 | 0.742 | −1.558 | 0.8 |
| 45 | c-Fos | C-FOS | −2.149 | 0.834 | −3.068 | 0.937 | −6.298 | 0.972 | −1.95 | 0.814 |
| 46 | FosB | FOSB | 1.265 | 0.401 | −6.929 | 0.96 | −11.992 | 0.923 | −8.365 | 0.896 |
| 47 | FRA1 | FOSL1 | 2.66 | 0.048 | 2.937 | 0.021 | 1.12 | 0.366 | 1.867 | 0.058 |
| 48 | FRA2 | FOSL2 | 1.883 | 0.015 | 2.38 | 0.005 | 1.339 | 0.065 | 1.658 | 0.013 |
| 49 | PPARalpha | PPARA | −1.556 | 0.877 | 1.132 | 0.352 | 1.229 | 0.378 | 1.153 | 0.198 |
| 50 | ANRIL | CDKN2BAS | n.a. | n.a. | n.a. | n.a. | −1.109 | 0.697 | −1.152 | 0.744 |
| Head and Neck Squamous Cell Carcinoma | Oral Cavity SquamousCell Carcinoma | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ginos Dataset | Cromer Dataset | Peng Dataset | Toruner dataset | |||||||
| Cancer vs. Normal | Cancer vs. Normal | Cancer vs. Normal | Cancer vs. Normal | |||||||
| n | Protein | Gene | Fold Change | p Value | Fold Change | p Value | Fold Change | p Value | Fold Change | p Value |
| 1 | p16INK4a | CDKN2A | 2.297 | 6.27 × 10−5 | −1.415 | 0.712 | 1.684 | 1.21 × 10−7 | 1.372 | 0.047 |
| p16INK4a Downstream Genes | ||||||||||
| 2 | CDK4 | CDK4 | 1.842 | 8.84 × 10−12 | 2.63 | 8.00 × 10−6 | 1.498 | 3.76E−08 | 2.13 | 6.31 × 10−4 |
| 3 | Cyclin D1 | CCND1 | 1.465 | 0.023 | 2.031 | 5.97 × 10−5 | −1.112 | 0.907 | 1.201 | 0.164 |
| 4 | pRB | RB1 | 1.372 | 8.92 × 10−4 | 1.165 | 0.429 | 1.2 | 0.018 | 1.579 | 0.005 |
| 5 | p107 | RBL1 | n.a. | n.a. | −2.032 | 0.97 | 1.334 | 4.47 × 10−4 | n.a. | n.a. |
| 6 | p130 | RBL2 | 1.081 | 0.172 | −1.449 | 0.774 | −1.228 | 0.997 | 1.141 | 0.046 |
| 7 | E2F1 | E2F1 | 1.347 | 0.046 | 1.754 | 1.00 × 10−6 | 1.032 | 0.313 | 1.083 | 0.05 |
| 8 | E2F2 | E2F2 | −1.329 | 0.856 | −1.147 | 0.813 | −1.424 | 1 | 1.044 | 0.172 |
| 9 | E2F3 | E2F3 | 2.945 | 7.20 × 10−8 | 1.948 | 0.159 | 1.099 | 4.19 × 10−15 | 1.231 | 0.284 |
| 10 | E2F4 | E2F4 | 1.131 | 0.025 | 1.462 | 0.014 | 1.415 | 1.28 × 10−11 | −1.178 | 1.00 |
| Epigenetic Regulator Genes | ||||||||||
| 11 | DNMT1 | DNMT1 | 1.706 | 2.00 × 10−7 | 1.581 | 0.124 | 1.499 | 1.87 × 10−15 | 1.457 | 2.00 × 10−3 |
| 12 | DNMT3A | DNMT3A | −1.046 | 0.584 | n.a. | n.a. | 1.01 | 0.37 | −1.038 | 0.644 |
| 13 | DNMT3B | DNMT3B | 1.37 | 5.19 × 10−4 | n.a. | n.a. | 1.512 | 3.82 × 10−8 | 1.296 | 0.009 |
| 14 | Bmi-1 | BMI1 | 1.818 | 3.23 × 10−7 | 1.627 | 5.16 × 10−5 | −1.046 | 0.689 | 1.708 | 2.78 × 10−7 |
| 15 | Ezh1 | EZH1 | 1.238 | 0.011 | 1.383 | 0.083 | 1.131 | 6.00 × 10−2 | 1.259 | 0.03 |
| 16 | Ezh2 | EZH2 | −1.325 | 0.88 | −1.151 | 0.833 | −1.401 | 1.000 | 1.021 | 0.4 |
| 17 | EED | EED | 1.495 | 3.89 × 10−4 | 1.118 | 0.286 | 1.349 | 3.12 × 10−7 | 1.112 | 0.013 |
| 18 | CBX4 | CBX4 | −1.133 | 0.750 | −1.132 | 0.613 | −1.183 | 0.999 | −1.04 | 0.817 |
| 19 | CBX7 | CBX7 | −1.429 | 0.992 | n.a. | n.a. | −1.311 | 1 | −1.24 | 1 |
| 20 | Jarid2 | JARID2 | 1.123 | 0.045 | 1.17 | 0.039 | 1.2 | 0.002 | 1.365 | 0. 948 |
| 21 | JMJD3 | JMJD3 | 1.039 | 0.292 | 1.127 | 0.116 | 1.088 | 0.174 | −1.229 | 0.924 |
| 22 | JDP2 | JDP2 | n.a. | n.a. | n.a. | n.a. | −1.214 | 1 | n.a. | n.a. |
| 23 | SETD8 | KMT5A | −1.375 | 0.999 | n.a. | n.a. | 1.058 | 0.214 | 1.097 | 2.19 × 10−4 |
| 24 | KMT2A | MLL | −1.185 | 0.974 | −1.252 | 0.901 | 1.179 | 0.005 | 1.11 | 0.215 |
| 25 | HAT p300 | EP300 | 1.054 | 0.327 | −1.196 | 0.737 | 1.141 | 0.006 | −1.146 | 0.904 |
| 26 | HDAC | HDAC1 | −1.075 | 0.83 | 1.361 | 0.054 | 1.174 | 0.016 | 1.883 | 1.42 × 10−4 |
| 27 | HDAC | HDAC2 | 1.613 | 1.41 × 10−4 | 1.181 | 0.241 | 1.066 | 0.151 | 1.265 | 0.029 |
| 28 | p63 | TP63 | 1.491 | 0.001 | 1.717 | 0.019 | 1.238 | 2.68 × 10−4 | 1.12 | 0.021 |
| 29 | BRG1 | SMARCA4 | 1.047 | 0.309 | −1.543 | 0.960 | 1.001 | 0.416 | 1.175 | 9.28 × 10−4 |
| 30 | LSH | HELLS | 2.463 | 0.013 | n.a. | n.a. | 1.617 | 6.24 × 10−6 | 1.445 | 0.004 |
| 31 | Satb1 | SATB1 | 1.025 | 0.396 | −2.039 | 0.92 | −1.496 | 0.997 | −1.685 | 1 |
| 32 | CTCF | CTCF | 1.003 | 0.478 | 1.292 | 0.018 | −1.114 | 0.998 | 1.29 | 0.097 |
| Transcriptional Regulator Genes | ||||||||||
| 33 | Ets1 | ETS1 | 1.694 | 0.001 | 2.076 | 0.016 | 2.111 | 2.97 × 10−10 | −1.114 | 1 |
| 34 | Ets2 | ETS2 | 1.78 | 1.76 × 10−6 | 1.761 | 0.012 | −1.102 | 0.908 | −1.218 | 0.618 |
| 35 | ID1 | ID1 | 1.356 | 0.015 | 1.375 | 0.278 | 1.21 | 0.025 | 1.242 | 0.317 |
| 36 | ID2 | ID2 | 1.765 | 1.79 × 10−4 | 2.022 | 0.258 | 1.066 | 0.193 | 1.038 | 0.362 |
| 37 | ID3 | ID3 | 3.023 | 2.14 × 10−4 | n.a. | n.a. | 1.072 | 0.208 | 1.043 | 0.447 |
| 38 | YB1 | YXB1 | 1.421 | 0.001 | 1.644 | 6.26 × 10−4 | −1.362 | 0.998 | 1.637 | 0.001 |
| 39 | SP1 | SP1 | 1.292 | 0.95 | −1.382 | 0.717 | −1.232 | 1 | 1.008 | 0.372 |
| 40 | HBP1 | PBRM1 | −1.131 | 0.716 | n.a. | n.a. | −1.303 | 1.00 | 1.039 | 0.137 |
| 41 | b-myb | MYB | 1.003 | 0.495 | 1.414 | 0.13 | −1.842 | 1 | 1.134 | 0.229 |
| 42 | JUNB | JUNB | 1.38 | 0.006 | 1.252 | 0.093 | 1.003 | 0.485 | −1.029 | 0.552 |
| 43 | c-Jun | C-JUN | 1.549 | 1.13 × 10−5 | 1.18 | 0.083 | 1.318 | 0.008 | −1.093 | 0.508 |
| 44 | JunD | JUND | −1.048 | 0.549 | 1.176 | 0.05 | −1.227 | 0.999 | 1.045 | 0.086 |
| 45 | c-Fos | C-FOS | 13.503 | 2.88 × 10−5 | 1.481 | 0.212 | −1.415 | 0.956 | 1.225 | 0.365 |
| 46 | FosB | FOSB | 3.016 | 0.005 | 1.385 | 0.021 | −1.042 | 0.527 | −1.881 | 1 |
| 47 | FRA1 | FOSL1 | 1.663 | 0.076 | −1.194 | 0.922 | 1.2 | 0.045 | −3.719 | 0.946 |
| 48 | FRA2 | FOSL2 | −1.355 | 0.997 | 1.13 | 0.205 | −2.032 | 1 | −1.149 | 0.909 |
| 49 | PPARalpha | PPARA | −1.043 | 0.406 | −1.134 | 0.585 | −1.079 | 0.984 | 1.021 | 0.104 |
| 50 | ANRIL | CDKN2BAS | n.a. | n.a. | n.a. | n.a. | 1.802 | 8.85 × 10−12 | n.a. | n.a. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Arcangelo, D.; Tinaburri, L.; Dellambra, E. The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer. Int. J. Mol. Sci. 2017, 18, 1591. https://doi.org/10.3390/ijms18071591
D’Arcangelo D, Tinaburri L, Dellambra E. The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer. International Journal of Molecular Sciences. 2017; 18(7):1591. https://doi.org/10.3390/ijms18071591
Chicago/Turabian StyleD’Arcangelo, Daniela, Lavinia Tinaburri, and Elena Dellambra. 2017. "The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer" International Journal of Molecular Sciences 18, no. 7: 1591. https://doi.org/10.3390/ijms18071591