Dietary Modulation of Oxidative Stress in Alzheimer’s Disease

{kind=link}
{kind=link}
Abstract
:1. Oxidative Stress and Aging Related Diseases
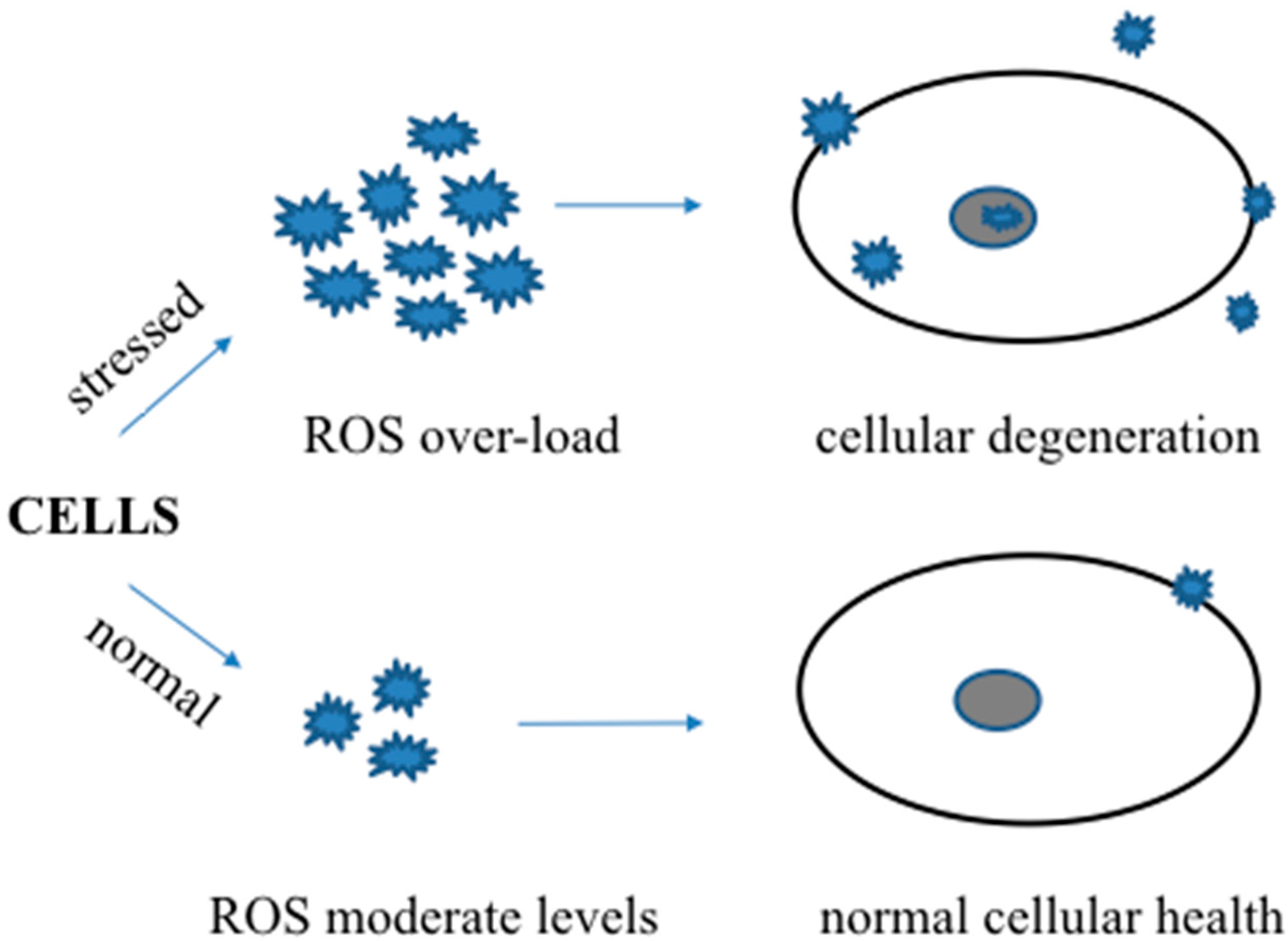
2. Oxidative Stress and Alzheimer’s Disease
3. Beneficial Effects of Oxidative Stress in Disease Resistance
4. Anti-Oxidative Defense in Alzheimer’s Disease
5. Natural Anti-Oxidative Therapeutics in Alzheimer’s Disease
6. Limitations of Anti-Oxidative Therapeutics in Alzheimer’s Disease
7. Dietary Approaches to Anti-Oxidative Therapeutics in Alzheimer’s Disease
8. Conclusions
Conflicts of Interest
References
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B. Understanding the odd science of aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Gemma, C.; Vila, J.; Bachstetter, A.; Bickford, P.C. Oxidative Stress and the Aging Brain: From Theory to Prevention. In Brain Aging: Models, Methods, and Mechanisms; Riddle, D.R., Ed.; CRC Press/Taylor & FrancisTaylor & Francis Group, LLC.: Boca Raton, FL, USA, 2007. [Google Scholar]
- Squier, T.C. Oxidative stress and protein aggregation during biological aging. Exp. Gerontol. 2001, 36, 1539–1550. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Agostinho, P.; Cunha, R.A.; Oliveira, C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2010, 16, 2766–2778. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J. Positive oxidative stress in aging and aging-related disease tolerance. Redox Biol. 2014, 2, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am. J. Med. 1991, 91, S14–S22. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, B.; Chakravarti, D.N. Oxidative modification of proteins: Age-related changes. Gerontology 2007, 53, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Vetrani, C.; Costabile, G.; Di Marino, L.; Rivellese, A.A. Nutrition and oxidative stress: A systematic review of human studies. Int. J. Food Sci. Nutr. 2013, 64, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative stress in Alzheimer’s disease: Why did antioxidant therapy fail? Oxid. Med. Cell. Longev. 2014, 2014, 427318. [Google Scholar] [CrossRef] [PubMed]
- Filipcik, P.; Cente, M.; Ferencik, M.; Hulin, I.; Novak, M. The role of oxidative stress in the pathogenesis of Alzheimer’s disease. Bratisl. Lekarsk. Listy 2006, 107, 384–394. [Google Scholar]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Dierckx, N.; Horvath, G.; van Gils, C.; Vertommen, J.; van de Vliet, J.; de Leeuw, I.; Manuel-y-Keenoy, B. Oxidative stress status in patients with diabetes mellitus: Relationship to diet. Eur. J. Clin. Nutr. 2003, 57, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A. Neuronal and vascular oxidative stress in Alzheimer’s disease. Curr. Neuropharmacol. 2011, 9, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Does the interdependence between oxidative stress and inflammation explain the antioxidant paradox? Oxid. Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [PubMed]
- McMillan, T.J.; Leatherman, E.; Ridley, A.; Shorrocks, J.; Tobi, S.E.; Whiteside, J.R. Cellular effects of long wavelength UV light (UVA) in mammalian cells. J. Pharm. Pharmacol. 2008, 60, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Isik, B.; Ceylan, A.; Isik, R. Oxidative stress in smokers and non-smokers. Inhal. Toxicol. 2007, 19, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Hanbauer, I.; Grisham, M.B.; Laval, F.; Nims, R.W.; Laval, J.; Cook, J.; Pacelli, R.; Liebmann, J.; Krishna, M.; et al. Chemical biology of nitric oxide: Regulation and protective and toxic mechanisms. Curr. Top. Cell. Regul. 1996, 34, 159–187. [Google Scholar] [PubMed]
- Ak, T.; Gulcin, I. Antioxidant and radical scavenging properties of curcumin. Chem. Biol. Interact. 2008, 174, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Thapa, A.; Chi, E.Y. Biflavonoids as potential small molecule therapeutics for Alzheimer’s disease. Adv. Exp. Med. Biol. 2015, 863, 55–77. [Google Scholar] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative stress, prooxidants, and antioxidants: The interplay. BioMed Res. Int. 2014, 2014, 761264. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [PubMed]
- Hamley, I.W. The amyloid β peptide: A chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [PubMed]
- Pollard, H.B.; Arispe, N.; Rojas, E. Ion channel hypothesis for Alzheimer amyloid peptide neurotoxicity. Cell. Mol. Neurobiol. 1995, 15, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [PubMed]
- Gu, F.; Zhu, M.; Shi, J.; Hu, Y.; Zhao, Z. Enhanced oxidative stress is an early event during development of Alzheimer-like pathologies in presenilin conditional knock-out mice. Neurosci. Lett. 2008, 440, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.; Chauhan, A. Oxidative stress in Alzheimer’s disease. Pathophysiology 2006, 13, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Sultana, R. Methionine-35 of β(1–42): Importance for oxidative stress in Alzheimer disease. J. Amino Acids 2011, 2011, 198430. [Google Scholar] [CrossRef] [PubMed]
- Hardas, S.S.; Sultana, R.; Clark, A.M.; Beckett, T.L.; Szweda, L.I.; Murphy, M.P.; Butterfield, D.A. Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol. 2013, 1, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-peptide (1–42)-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid β-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Smith, D.G.; Cappai, R.; Barnham, K.J. The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim. Biophys. Acta 2007, 1768, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Allan Butterfield, D. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Moslemnezhad, A.; Mahjoub, S.; Moghadasi, M. Altered plasma marker of oxidative DNA damage and total antioxidant capacity in patients with Alzheimer’s disease. Casp. J. Intern. Med. 2016, 7, 88–92. [Google Scholar]
- Mao, P.; Reddy, P.H. Aging and amyloid β-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta 2011, 1812, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. DNA damage and repair: Relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, G.; Chirumbolo, S. Role of oxidative stress and antioxidants in daily nutrition and human health. Nutrition 2017, 33, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N.; Chandra, M.; Tuteja, R.; Misra, M.K. Nitric oxide as a unique bioactive signaling messenger in physiology and pathophysiology. J. Biomed. Biotechnol. 2004, 2004, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Okabe, A.; Urano, Y.; Itoh, S.; Suda, N.; Kotani, R.; Nishimura, Y.; Saito, Y.; Noguchi, N. Adaptive responses induced by 24 S-hydroxycholesterol through liver X receptor pathway reduce 7-ketocholesterol-caused neuronal cell death. Redox Biol. 2013, 2, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Kang, I.; Marchant, R.E.; Zagorski, M.G. Methionine 35 oxidation reduces fibril assembly of the amyloid β-(1–42) peptide of Alzheimer’s disease. J. Biol. Chem. 2002, 277, 40173–40176. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Long, C.; Reaney, S.H.; Di Monte, D.A.; Fink, A.L.; Uversky, V.N. Methionine oxidation stabilizes non-toxic oligomers of α-synuclein through strengthening the auto-inhibitory intra-molecular long-range interactions. Biochim. Biophys. Acta 2010, 1802, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Niki, E. Antioxidants: Basic principles, emerging concepts, and problems. Biomed. J. 2014, 37, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.W.; Lu, S.; Su, Y.J.; Xue, D.; Yu, X.L.; Wang, S.W.; Zhang, H.; Xu, P.X.; Xie, X.X.; Liu, R.T. Decreasing oxidative stress and neuroinflammation with a multifunctional peptide rescues memory deficits in mice with Alzheimer’s disease. Free Radic. Biol. Med. 2014, 74, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Mett, J.; Hartmann, T. The impact of vitamin E and other fat-Soluble vitamins on Alzheimer’s disease. Int. J. Mol. Sci. 2016, 17, 1785. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.R.; Roberts, B.R.; Bush, A.I.; Hare, D.J. Selenium, selenoproteins and neurodegenerative diseases. Metallomics 2015, 7, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef] [PubMed]
- Taheri, E.; Djalali, M.; Saedisomeolia, A.; Moghadam, A.M.; Djazayeri, A.; Qorbani, M. The relationship between the activates of antioxidant enzymes in red blood cells and body mass index in Iranian type 2 diabetes and healthy subjects. J. Diabetes Metab. Disord. 2012, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Conti, V.; Izzo, V.; Corbi, G.; Russomanno, G.; Manzo, V.; de Lise, F.; di Donato, A.; Filippelli, A. Antioxidant supplementation in the treatment of aging-associated diseases. Front. Pharmacol. 2016, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Fusco, D.; Colloca, G.; Lo Monaco, M.R.; Cesari, M. Effects of antioxidant supplementation on the aging process. Clin. Interv. Aging 2007, 2, 377–387. [Google Scholar] [PubMed]
- Franceschelli, S.; Pesce, M.; Ferrone, A.; De Lutiis, M.A.; Patruno, A.; Grilli, A.; Felaco, M.; Speranza, L. Astaxanthin treatment confers protection against oxidative stress in U937 cells stimulated with lipopolysaccharide reducing O2-production. PLoS ONE 2014, 9, e88359. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R., 3rd; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Inter. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef]
- Poljsak, B.; Milisav, I. The neglected significance of “antioxidative stress”. Oxid. Med. Cell. Longev. 2012, 2012, 480895. [Google Scholar] [CrossRef] [PubMed]
- Thapa, A.; Woo, E.R.; Chi, E.Y.; Sharoar, M.G.; Jin, H.G.; Shin, S.Y.; Park, I.S. Biflavonoids are superior to monoflavonoids in inhibiting amyloid-β toxicity and fibrillogenesis via accumulation of nontoxic oligomer-like structures. Biochemistry 2011, 50, 2445–2455. [Google Scholar] [CrossRef] [PubMed]
- Lakey-Beitia, J.; Berrocal, R.; Rao, K.S.; Durant, A.A. Polyphenols as therapeutic molecules in Alzheimer’s disease through modulating amyloid pathways. Mol. Neurobiol. 2015, 51, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-β aggregation pathway. Am. J. Pathol. 2009, 175, 2557–2565. [Google Scholar] [CrossRef] [PubMed]
- Ramassamy, C. Emerging role of polyphenolic compounds in the treatment of neurodegenerative diseases: A review of their intracellular targets. Eur. J. Pharmacol. 2006, 545, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hamaguchi, T.; Naiki, H.; Yamada, M. Anti-amyloidogenic effects of antioxidants: Implications for the prevention and therapeutics of Alzheimer’s disease. Biochim. Biophys. Acta 2006, 1762, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Frautschy, S.A.; Cole, G.M.; Masterman, D.L.; Cummings, J.L. A potential role of the curry spice curcumin in Alzheimer's disease. Curr. Alzheimer Res. 2005, 2, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [PubMed]
- Assis, R.P.; Arcaro, C.A.; Gutierres, V.O.; Oliveira, J.O.; Costa, P.I.; Baviera, A.M.; Brunetti, I.L. Combined effects of curcumin and lycopene or Bixin in yoghurt on inhibition of LDL oxidation and increases in HDL and paraoxonase levels in streptozotocin-diabetic rats. Int. J. Mol. Sci. 2017, 18, 332. [Google Scholar] [CrossRef] [PubMed]
- Canevelli, M.; Adali, N.; Kelaiditi, E.; Cantet, C.; Ousset, P.J.; Cesari, M. Effects of Gingko biloba supplementation in Alzheimer’s disease patients receiving cholinesterase inhibitors: Data from the ICTUS study. Phytomedicine 2014, 21, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Smith, J.V.; Paramasivam, V.; Burdick, A.; Curry, K.J.; Buford, J.P.; Khan, I.; Netzer, W.J.; Xu, H.; Butko, P. Inhibition of amyloid-β aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proc. Natl. Acad. Sci. USA 2002, 99, 12197–12202. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Williamson, J.D.; Fitzpatrick, A.L.; Kronmal, R.A.; Ives, D.G.; Saxton, J.A.; Lopez, O.L.; Burke, G.; Carlson, M.C.; Fried, L.P. Ginkgo biloba for prevention of dementia: A randomized controlled trial. JAMA 2008, 300, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Geng, Z.; Lau, B.H. Ginkgo biloba attenuates oxidative stress in macrophages and endothelial cells. Free Radic. Biol. Med. 1996, 20, 121–127. [Google Scholar] [CrossRef]
- Niki, E.; Noguchi, N.; Tsuchihashi, H.; Gotoh, N. Interaction among vitamin C, vitamin E, and β-carotene. Am. J. Clin. Nutr. 1995, 62, 1322s–1326s. [Google Scholar] [PubMed]
- Noguchi, N.; Gotoh, N.; Niki, E. Action of vitamin E as antioxidant against oxidative modification of low density lipoprotein. BioFactors 1998, 7, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Tangirala, R.K.; Rader, D.J.; Rokach, J.; FitzGerald, G.A. Vitamin E suppresses isoprostane generation in vivo and reduces atherosclerosis in ApoE-deficient mice. Nat. Med. 1998, 4, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.; Yao, Y.; Uryu, K.; Yang, H.; Lee, V.M.; Trojanowski, J.Q.; Pratico, D. Early vitamin E supplementation in young but not aged mice reduces Aβ levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J 2004, 18, 323–325. [Google Scholar] [PubMed]
- Ahmed, T.; Enam, S.A.; Gilani, A.H. Curcuminoids enhance memory in an amyloid-infused rat model of Alzheimer’s disease. Neuroscience 2010, 169, 1296–1306. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Badia, M.C.; Mora, N.J.; Pallardo, F.V.; Alonso, M.D.; Vina, J. Vitamin E paradox in Alzheimer’s disease: It does not prevent loss of cognition and may even be detrimental. J. Alzheimers Dis. 2009, 17, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Why vitamin E therapy fails for treatment of Alzheimer’s disease. J. Alzheimers Dis 2010, 19, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Garrido, M.; Terron, M.P.; Rodriguez, A.B. Chrononutrition against oxidative stress in aging. Oxid. Med. Cell. Longev. 2013, 2013, 729804. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Naiki, H.; Yamada, M. The development of preventives and therapeutics for Alzheimer’s disease that inhibit the formation of β-amyloid fibrils (fAβ), as well as destabilize preformed fAβ. Curr. Pharm. Des. 2006, 12, 4357–4375. [Google Scholar] [CrossRef] [PubMed]
- Kirsh, V.A.; Hayes, R.B.; Mayne, S.T.; Chatterjee, N.; Subar, A.F.; Dixon, L.B.; Albanes, D.; Andriole, G.L.; Urban, D.A.; Peters, U.; et al. Supplemental and dietary vitamin E, β-carotene, and vitamin C intakes and prostate cancer risk. J. Natl. Cancer Inst. 2006, 98, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Nie, Q.; Du, X.G.; Geng, M.Y. Small molecule inhibitors of amyloid β peptide aggregation as a potential therapeutic strategy for Alzheimer’s disease. Acta Pharmacol. Sin. 2011, 32, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Bae, Y.C.; Kim-Han, J.S.; Lee, J.H.; Choi, I.Y.; Son, K.H.; Kang, S.S.; Kim, W.K.; Han, B.H. Polyphenol amentoflavone affords neuroprotection against neonatal hypoxic-ischemic brain damage via multiple mechanisms. J. Neurochem. 2006, 96, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Hadden, M.K.; Blagg, B.S. Dimeric approaches to anti-cancer chemotherapeutics. Anticancer Agents Med. Chem. 2008, 8, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Gil, B.; Sanz, M.J.; Terencio, M.C.; Gunasegaran, R.; Paya, M.; Alcaraz, M.J. Morelloflavone, a novel biflavonoid inhibitor of human secretory phospholipase A2 with anti-inflammatory activity. Biochem. Pharmacol. 1997, 53, 733–740. [Google Scholar] [CrossRef]
- Chan, K.F.; Zhao, Y.; Burkett, B.A.; Wong, I.L.; Chow, L.M.; Chan, T.H. Flavonoid dimers as bivalent modulators for P-glycoprotein-based multidrug resistance: Synthetic apigenin homodimers linked with defined-length poly(ethylene glycol) spacers increase drug retention and enhance chemosensitivity in resistant cancer cells. J. Med. Chem. 2006, 49, 6742–6759. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Rezai-Zadeh, K.; Koyama, N.; Arendash, G.W.; Yamaguchi, H.; Kakuda, N.; Horikoshi-Sakuraba, Y.; Tan, J.; Town, T. Tannic acid is a natural β-secretase inhibitor that prevents cognitive impairment and mitigates Alzheimer-like pathology in transgenic mice. J. Biol. Chem. 2012, 287, 6912–6927. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Sunagawa, T.; Kanda, T.; Tagashira, M.; Shirasawa, T.; Shimizu, T. Apple procyanidins suppress amyloid β-protein aggregation. Biochem. Res. Int. 2011, 2011, 784698. [Google Scholar] [CrossRef] [PubMed]
- Picone, P.; Nuzzo, D.; di Carlo, M. Ferulic acid: A natural antioxidant against oxidative stress induced by oligomeric Aβ on sea urchin embryo. Biol. Bull. 2013, 224, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, D.P.; Kiyota, T.; Dong, Y.; Ikezu, T.; Vennerstrom, J.L. Phenolic bis-styrylbenzenes as β-amyloid binding ligands and free radical scavengers. J. Med. Chem. 2010, 53, 7992–7999. [Google Scholar] [CrossRef] [PubMed]
- Ritter, E. Vitamin supplements in oncology—Necessary, useful or superfluous? MMW Fortschr. Med. 2002, 144, 33–37. [Google Scholar] [PubMed]
- Karunadharma, P.P.; Nordgaard, C.L.; Olsen, T.W.; Ferrington, D.A. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5470–5479. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Zimbron, L.F.; Rivas-Arancibia, S. Oxidative stress caused by ozone exposure induces β-amyloid 1–42 overproduction and mitochondrial accumulation by activating the amyloidogenic pathway. Neuroscience 2015, 304, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, X.; Tung, Y.C.; Xie, S.; Liu, F.; Iqbal, K. Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimers Dement. 2016, 12, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, X.; Morris, M.E. Combined effects of multiple flavonoids on breast cancer resistance protein (ABCG2)-mediated transport. Pharm. Res. 2004, 21, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Maiti, P.; Ma, Q.; Zuo, X.; Jones, M.R.; Cole, G.M.; Frautschy, S.A. Clinical development of curcumin in neurodegenerative disease. Expert Rev. Neurother. 2015, 15, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.P.; Chiam, P.C.; Lee, T.; Chua, H.C.; Lim, L.; Kua, E.H. Curry consumption and cognitive function in the elderly. Am. J. Epidemiol. 2006, 164, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; Polidori, M.C. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, R.; Balaraman, R.; Sen, A.K.; Shukla, D.; Seth, A. Effect of concomitant administration of coenzyme Q10 with sitagliptin on experimentally induced diabetic nephropathy in rats. Ren. Fail. 2017, 39, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.; Liu, R.H. Apple phytochemicals and their health benefits. Nutr. J. 2004, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.H.; Lin, Y.S.; Huang, Y.W.; Fang, S.U.; Lin, S.Y.; Hou, W.C. Protective effects of minor components of curcuminoids on hydrogen peroxide-treated human HaCaT keratinocytes. J. Agric. Food Chem. 2016, 64, 3598–3608. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thapa, A.; Carroll, N.J. Dietary Modulation of Oxidative Stress in Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1583. https://doi.org/10.3390/ijms18071583
Thapa A, Carroll NJ. Dietary Modulation of Oxidative Stress in Alzheimer’s Disease. International Journal of Molecular Sciences. 2017; 18(7):1583. https://doi.org/10.3390/ijms18071583
Chicago/Turabian StyleThapa, Arjun, and Nick J. Carroll. 2017. "Dietary Modulation of Oxidative Stress in Alzheimer’s Disease" International Journal of Molecular Sciences 18, no. 7: 1583. https://doi.org/10.3390/ijms18071583