PD-1/PD-L1 Blockade Therapy for Tumors with Downregulated MHC Class I Expression


Abstract
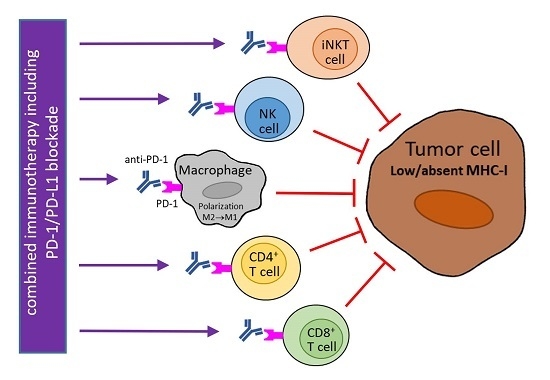
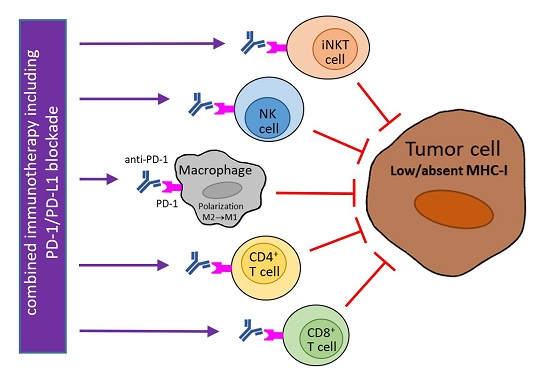
:
{kind=link}
{kind=link}
1. Introduction
2. PD-1/PD-L1 Signaling
3. Tumor Escape by MHC-I Downregulation
4. PD-1/PD-L1 Blockade and Predictive Biomarkers
5. PD-1/PD-L1 Blockade and MHC-I Expression
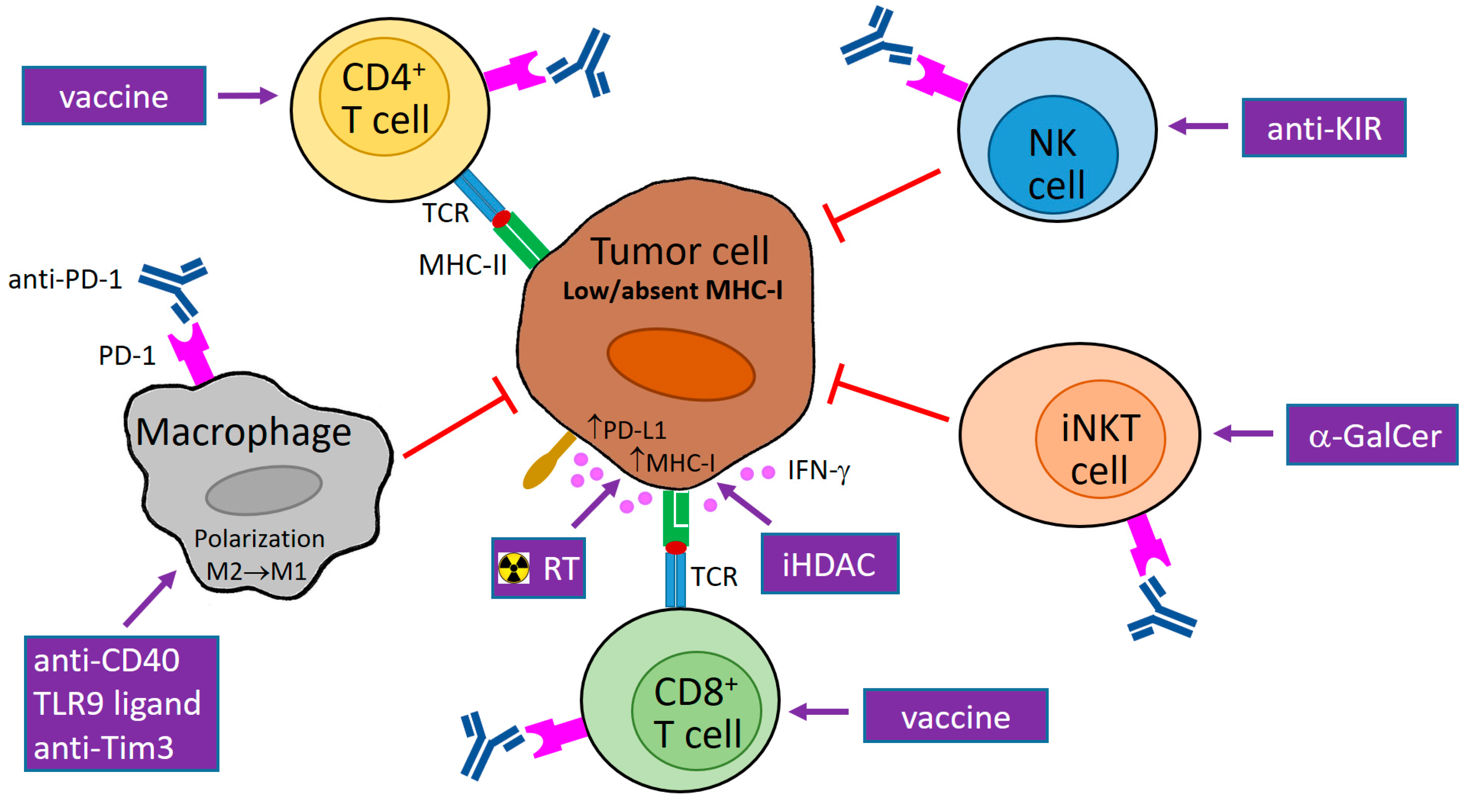
6. Utilization of PD-1/PD-L1 Blockade for Tumors with MHC-I Downregulation
7. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [PubMed]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Chikuma, S.; Terawaki, S.; Hayashi, T.; Nabeshima, R.; Yoshida, T.; Shibayama, S.; Okazaki, T.; Honjo, T. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J. Immunol. 2009, 182, 6682–6689. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Thompson, C.B. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. OncoTargets Ther. 2016, 9, 5023–5039. [Google Scholar] [CrossRef]
- Berry, S.; Taube, J.M. Innate vs. adaptive: PD-L1-mediated immune resistance by melanoma. OncoImmunology 2015, 4, e1029704. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Lastwika, K.J.; Wilson, W.; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Dorand, R.D.; Nthale, J.; Myers, J.T.; Barkauskas, D.S.; Avril, S.; Chirieleison, S.M.; Pareek, T.K.; Abbott, D.W.; Stearns, D.S.; Letterio, J.J.; et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science 2016, 353, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Shah, S.P.; Woolcock, B.W.; Rui, L.; Kawahara, M.; Farinha, P.; Johnson, N.A.; Zhao, Y.; Telenius, A.; Neriah, S.B.; et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011, 471, 377–381. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Bi, X.; Wang, H.; Zhang, W.; Wang, J.; Liu, W.; Xia, Z.; Huang, H.; Jiang, W.; Zhang, Y.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Shiraishi, Y.; Takeda, Y.; Sakata, S.; Matsumoto, M.; Nagano, S.; Maeda, T.; Nagata, Y.; Kitanaka, A.; Mizuno, S.; et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature 2016, 534, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef] [PubMed]
- Chikuma, S. Basics of PD-1 in self-tolerance, infection, and cancer immunity. Int. J. Clin. Oncol. 2016, 21, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Galon, J.; Fridman, W.-H.; Smyth, M.J. From mice to humans: Developments in cancer immunoediting. J. Clin. Investig. 2015, 125, 3338–3346. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Ruiz-Cabello, F.; Aptsiauri, N. Rejection versus escape: The tumor MHC dilemma. Cancer Immunol. Immunother. 2017, 66, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Shin, E.-C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Aust, S.; Felix, S.; Auer, K.; Bachmayr-Heyda, A.; Kenner, L.; Dekan, S.; Meier, S.M.; Gerner, C.; Grimm, C.; Pils, D. Absence of PD-L1 on tumor cells is associated with reduced MHC I expression and PD-L1 expression increases in recurrent serous ovarian cancer. Sci. Rep. 2017, 7, 42929. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Nevot, M.A.; Esteban, F.; Ferron, A.; Gutierrez, J.; Oliva, M.R.; Romero, C.; Huelin, C.; Ruiz-Cabello, F.; Garrido, F. HLA class I gene expression on human primary tumours and autologous metastases: Demonstration of selective losses of HLA antigens on colorectal, gastric and laryngeal carcinomas. Br. J. Cancer 1989, 59, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Méndez, R.; Ruiz-Cabello, F.; Rodríguez, T.; Del Campo, A.; Paschen, A.; Schadendorf, D.; Garrido, F. Identification of different tumor escape mechanisms in several metastases from a melanoma patient undergoing immunotherapy. Cancer Immunol. Immunother. 2007, 56, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Carretero, R.; Romero, J.M.; Ruiz-Cabello, F.; Maleno, I.; Rodriguez, F.; Camacho, F.M.; Real, L.M.; Garrido, F.; Cabrera, T. Analysis of HLA class I expression in progressing and regressing metastatic melanoma lesions after immunotherapy. Immunogenetics 2008, 60, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Marincola, F.M.; Ferrone, S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol. Med. Today 1999, 5, 178–186. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhou, C. The past, present and future of immunotherapy against tumor. Transl. Lung Cancer Res. 2015, 4, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Gilligan, B.M.; Yuan, J.; Li, T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J. Hematol. Oncol. 2016, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.-J.; et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Capone, M.; Urba, W.J.; Bifulco, C.B.; Botti, G.; Lugli, A.; Marincola, F.M.; Ciliberto, G.; Galon, J.; Fox, B.A. The additional facet of immunoscore: Immunoprofiling as a possible predictive tool for cancer treatment. J. Transl. Med. 2013, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Champiat, S.; Ferté, C.; Lebel-Binay, S.; Eggermont, A.; Soria, J.C. Exomics and immunogenics: Bridging mutational load and immune checkpoints efficacy. OncoImmunology 2014, 3, e27817. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Park, J.-H.; Kiyotani, K.; Zewde, M.; Miyashita, A.; Jinnin, M.; Kiniwa, Y.; Okuyama, R.; Tanaka, R.; Fujisawa, Y.; et al. Intratumoral expression levels of PD-L1, GZMA, and HLA-A along with oligoclonal T cell expansion associate with response to nivolumab in metastatic melanoma. OncoImmunology 2016, 5, e1204507. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Hirano, F.; Kaneko, K.; Tamura, H.; Dong, H.; Wang, S.; Ichikawa, M.; Rietz, C.; Flies, D.B.; Lau, J.S.; Zhu, G.; et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005, 65, 1089–1096. [Google Scholar] [PubMed]
- Moreno, B.H.; Zaretsky, J.M.; Garcia-Diaz, A.; Tsoi, J.; Parisi, G.; Robert, L.; Meeth, K.; Ndoye, A.; Bosenberg, M.; Weeraratna, A.T.; et al. Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T cells. Cancer Immunol. Res. 2016, 4, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin lymphoma with reduced B2M/MHC class I expression is associated with Inferior outcome independent of 9p24.1 status. Cancer Immunol. Res. 2016, 4, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Bercovici, N.; Trautmann, A. Revisiting the role of T cells in tumor regression. OncoImmunology 2012, 1, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Thoreau, M.; Penny, H.L.; Tan, K.; Regnier, F.; Weiss, J.M.; Lee, B.; Johannes, L.; Dransart, E.; Bon, A.L.; Abastado, J.-P.; et al. Vaccine-induced tumor regression requires a dynamic cooperation between T cells and myeloid cells at the tumor site. Oncotarget 2015, 6, 27832–27846. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-Y.; Guarnieri, F.G.; Staveley-O’Carroll, K.F.; Levitsky, H.I.; August, J.T.; Pardoll, D.M.; Wu, T.-C. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996, 56, 21–26. [Google Scholar] [PubMed]
- Moynihan, K.D.; Opel, C.F.; Szeto, G.L.; Tzeng, A.; Zhu, E.F.; Engreitz, J.M.; Williams, R.T.; Rakhra, K.; Zhang, M.H.; Rothschilds, A.M.; et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med. 2016, 22, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Beldi-Ferchiou, A.; Lambert, M.; Dogniaux, S.; Vely, F.; Vivier, E.; Olive, D.; Dupuy, S.; Levasseur, F.; Zucman, D.; Lebbe, C.; et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 2016, 7, 72961–72977. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Kamata, T.; Suzuki, A.; Mise, N.; Ihara, F.; Takami, M.; Makita, Y.; Horinaka, A.; Harada, K.; Kunii, N.; Yoshida, S.; et al. Blockade of programmed death-1/programmed death ligand pathway enhances the antitumor immunity of human invariant natural killer T cells. Cancer Immunol. Immunother. 2016, 65, 1477–1489. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-S.; Kim, J.-Y.; Kim, Y.-J.; Kim, Y.-S.; Lee, J.-M.; Azuma, M.; Yagita, H.; Kang, C.-Y. Cutting edge: Programmed death-1/programmed death ligand 1 interaction regulates the induction and maintenance of invariant NKT cell anergy. J. Immunol. 2008, 181, 6707–6710. [Google Scholar] [CrossRef] [PubMed]
- Parekh, V.V.; Lalani, S.; Kim, S.; Halder, R.; Azuma, M.; Yagita, H.; Kumar, V.; Wu, L.; Kaer, L.V. PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J. Immunol. 2009, 182, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Durgan, K.; Ali, M.; Warner, P.; Latchman, Y.E. Targeting NKT cells and PD-L1 pathway results in augmented anti-tumor responses in a melanoma model. Cancer Immunol. Immunother. 2011, 60, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Van de Berg, P.J.; van Leeuwen, E.M.; ten Berge, I.J.; van Lier, R. Cytotoxic human CD4+ T cells. Curr. Opin. Immunol. 2008, 20, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Peggs, K.S. Tumor-reactive CD4+ cells: Plasticity beyond helper and regulatory activities. Immunotherapy 2011, 3, 915–917. [Google Scholar] [CrossRef] [PubMed]
- Perez-Diez, A.; Joncker, N.T.; Choi, K.; Chan, W.F.N.; Anderson, C.C.; Lantz, O.; Matzinger, P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 2007, 109, 5346–5354. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Hou, X.; Li, T.; Zhao, L.; Yuan, X.; Fu, H.; Zhu, R. CD4+ T cell-mediated cytotoxicity eliminates primary tumor cells in metastatic melanoma through high MHC class II expression and can be enhanced by inhibitory receptor blockade. Tumor Biol. 2016, 15949–15958. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Kleinovink, J.W.; Marijt, K.A.; Schoonderwoerd, M.J.A.; van Hall, T.; Ossendorp, F.; Fransen, M.F. PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. OncoImmunology 2017, 6, e1294299. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.J.; Ni, L.; Zhang, Y.; Zhang, C.L.; Wu, X.Y.; Atia, A.N.; Thayer, P.; Moorman, J.P.; Yao, Z.Q. PD-1 negatively regulates interleukin-12 expression by limiting STAT-1 phosphorylation in monocytes/macrophages during chronic hepatitis C virus infection. Immunology 2011, 132, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Yao, A.; Liu, F.; Chen, K.; Tang, L.; Liu, L.; Zhang, K.; Yu, C.; Bian, G.; Guo, H.; Zheng, J.; et al. Programmed death 1 deficiency induces the polarization of macrophages/microglia to the M1 phenotype after spinal cord injury in mice. Neurotherapeutics 2014, 11, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Merlino, G. The two faces of interferon-γ in cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Hamanishi, J.; Abiko, K.; Matsumura, N.; Baba, T.; Konishi, I. Dual faces of IFNγ in cancer progression: A role of PD-L1 induction in the determination of pro- and antitumor immunity. Clin. Cancer Res. 2016, 22, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Terawaki, S.; Chikuma, S.; Shibayama, S.; Hayashi, T.; Yoshida, T.; Okazaki, T.; Honjo, T. IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J. Immunol. 2011, 186, 2772–2779. [Google Scholar] [CrossRef] [PubMed]
- Kakizaki, A.; Fujimura, T.; Furudate, S.; Kambayashi, Y.; Yamauchi, T.; Yagita, H.; Aiba, S. Immunomodulatory effect of peritumorally administered interferon-beta on melanoma through tumor-associated macrophages. OncoImmunology 2015, 4, e1047584. [Google Scholar] [CrossRef] [PubMed]
- Swart, M.; Verbrugge, I.; Beltman, J.B. Combination approaches with immune-checkpoint blockade in cancer therapy. Front. Oncol. 2016, 6, 233. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W.L. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2015, 13, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Buhtoiarov, I.N.; Sondel, P.M.; Wigginton, J.M.; Buhtoiarova, T.N.; Yanke, E.M.; Mahvi, D.A.; Rakhmilevich, A.L. Anti-tumour synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumour-associated macrophages. Immunology 2011, 132, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Magner, W.J.; Kazim, A.L.; Stewart, C.; Romano, M.A.; Catalano, G.; Grande, C.; Keiser, N.; Santaniello, F.; Tomasi, T.B. Activation of MHC Class I, II, and CD40 gene expression by histone deacetylase inhibitors. J. Immunol. 2000, 165, 7017–7024. [Google Scholar] [CrossRef] [PubMed]
- Licciardi, P.V.; Karagiannis, T.C. Regulation of immune responses by histone deacetylase inhibitors. Int. Sch. Res. Not. 2012, 2012, e690901. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.M.; Sodré, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhao, W.; Yan, C.; Watson, C.C.; Massengill, M.; Xie, M.; Massengill, C.; Noyes, D.R.; Martinez, G.V.; Afzal, R.; et al. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin. Cancer Res. 2016, 22, 4119–4132. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Schoenhals, J.E.; Li, A.; Valdecanas, D.R.; Ye, H.; Zhang, F.; Tang, C.; Tang, M.; Liu, C.-G.; Liu, X.; et al. Suppression of type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Res. 2017, 77, 839–850. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šmahel, M. PD-1/PD-L1 Blockade Therapy for Tumors with Downregulated MHC Class I Expression. Int. J. Mol. Sci. 2017, 18, 1331. https://doi.org/10.3390/ijms18061331
Šmahel M. PD-1/PD-L1 Blockade Therapy for Tumors with Downregulated MHC Class I Expression. International Journal of Molecular Sciences. 2017; 18(6):1331. https://doi.org/10.3390/ijms18061331
Chicago/Turabian StyleŠmahel, Michal. 2017. "PD-1/PD-L1 Blockade Therapy for Tumors with Downregulated MHC Class I Expression" International Journal of Molecular Sciences 18, no. 6: 1331. https://doi.org/10.3390/ijms18061331