Molecular Determinants of Malignant Brain Cancers: From Intracellular Alterations to Invasion Mediated by Extracellular Vesicles


Abstract
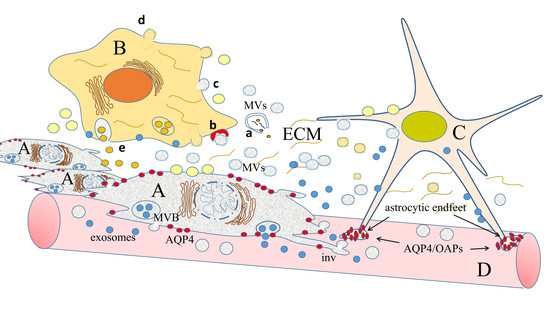
:
1. Introduction
2. Cellular and Molecular Bases of Glioma Growth and Invasion
2.1. The Extracellular Matrix (ECM)
2.2. The Cytoskeleton
2.3. Transcription Factors
2.4. Ion and Water Channels
2.5. Hypoxia, Metabolic Reprogramming and Angiogenesis
2.6. Non-Coding RNAs
3. The Pawns of Invasion: Extracellular Vesicles (EVs)
3.1. Extracellular Vesicles: Secretion by Producer Cells and Interaction with the Cell Environment
3.2. How EVs Can Both Directly and Indirectly Modify the Extracellular Matrix
3.3. EVs as Inducers of Gene Expression Modifications
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM17 | ADAM (a disintegrin and metalloprotease domain) metallopeptidase domain 17 |
| ADAMTS-AS2 | ADAM metallopeptidase with thrombospondin motif, antisense RNA 2 |
| ADD3 | Adducin 3 |
| AKT1 | Ak strain transforming (also known as protein-chinasi B o PKB) 1 |
| ASLNC | Anti-sense long non-coding RNA |
| ATM | Ataxia telangiectasia mutated |
| AURKA | Aurora kinase A |
| BCL-2 | B-cell lymphoma 2 |
| Bmi1 | B cell-specific Moloney murine leukemia virus integration site 1 (Polycomb complex protein BMI-1) |
| CASC 2 | Cancer susceptibility candidate 2 |
| CDC42 | Cell division control protein 42 homolog |
| CCNE1/CCNE2 | Cyclin E1/Ciclina E2 |
| CRNDE | Colorectal neoplasia differentially expressed |
| CYLD | Cylindromatosis (turban tumor syndrome) |
| DIXDC1 | Dixin; DIX domain-containing protein 1 |
| DNMT1 | DNA methyl transferase 1 |
| EGFR | Epidermal growth factor receptor |
| ESM-1 | Endothelial cell specific molecule 1 |
| FAK | Focal adhesion kinase |
| Fer1L4 | Fer (Feline Encephalitis Virus-Related) Kinase-1 Like Family Member 4 (pseudogene) |
| FoxM1 | Forkhead box M1 |
| GAS 5 | Growth arrest-specific 5 |
| HGS | Hepatocyte growth factor-regulated tyrosine kinase substrate |
| IGF | Insulin-like growth factor |
| IGFBP | Insulin-like growth factor-binding protein |
| IKK | IκB kinase |
| IRS 1/2 | Insulin Receptor Substrate 1/2 |
| HIF | Hypoxia-inducible factor |
| HOTAIR | HOX transcript antisense RNA |
| HOTTIP | HOXA transcript at the distal tip |
| HULC | Highly up-regulated in liver cancer |
| JAG-1 | Jagged-1 |
| KCNQ1OT1 | KCNQ1 (potassium voltage-gated channel subfamily Q member 1) opposite strand/antisense transcript 1 (non-protein coding) |
| Kir 4.1 | Inward-rectifier potassium ion channel 4.1 |
| KPNA4 | Karyopherin (Importin) Subunit Alpha 4 |
| LDH-A | Lactate dehydrogenase A |
| LINC | Long intergenic non-coding RNA |
| LINK-A | Long intergenic non-coding RNA for kinase activation |
| LKB1 | Liver Kinase B1 (also known as Serine/Threonine Kinase 11—STK11) |
| MALAT-1 | Metastasis associated lung adenocarcinoma transcript 1 |
| MBD2 | Methyl-CpG binding domain protein 2 |
| MEKK2 | Mitogen-Activated Protein Kinase Kinase Kinase 2 |
| MMP | Matrix metalloproteinase |
| mTOR | Mammalian target of rapamycin |
| NEAT | Nuclear enriched abundant transcript |
| NEDD9 | Neural precursor cell expressed developmentally down-regulated protein 9 |
| NOB1 | Nin1 (One) Binding protein 1 |
| Notch2 | Neurogenic locus notch homolog protein 2 |
| PAK4 | P21 (RAC1) Activated Kinase 4 |
| PBX3 | Pre-B-cell leukemia transcription factor 3 |
| PDCD4 | Programmed cell death protein 4 |
| PD-L1 | Programmed death-ligand 1 |
| PI3K | Phosphoinositide 3-kinase |
| PIWIL | Piwi-like RNA-mediated gene silencing |
| PKC | Protein kinase C |
| POU3F3 | POU Class 3 Homeobox 3 |
| PRC 1/2 | Polycomb repressor complex 1/2 |
| PLAC 2 | Placenta specific 2 |
| PTEN | Phosphatase and tensin homolog |
| Rac1 | Ras-related C3 botulinum toxin substrate 1, |
| RECK | Reversion-Inducing Cysteine-Rich Protein With Kazal Motifs |
| RhoC | Ras homolog gene family, member C |
| ROCK1 | Rho-associated, coiled-coil-containing protein kinase 1 |
| RTK | Receptor tyrosine kinase |
| SEMA3B | Semaphorin 3B |
| SIRT6 | Sirtuin 6 |
| SKA2 | Spindle and Kinetochore Associated protein 2 |
| SMAD | Small mother against decapentaplegic |
| SMO | Smoothened, Frizzled Class Receptor |
| SNORD47 | Small Nucleolar RNA, C/D Box 47 |
| Sox7/Sox9 | SRY-Box 7/SRY-Box 9 |
| STAT1/STAT5A | Signal transducer and activator of transcription 1/5A |
| TIMP-1/TIMP2 | Tissue inhibitor of metalloproteinase 1/2 |
| TSP 1 | Thrombospondin 1 |
| TSHZ3 | Teashirt zinc finger homeobox 3 |
| TUG1 | Taurine up-regulated gene |
| UCA1 | Urothelial cancer associated 1 (non-protein coding) |
| UPAR | Urokinase receptor |
| WIP | WAS/WASL interacting protein |
| Wnt | Wingless-related integration site |
| XIST | X-chromosome inactive specific transcript |
| YAP1 | Yes associated protein 1 |
References
- Jovčevska, I.; Kočevar, N.; Komel, R. Glioma and glioblastoma-how much do we (not) know? Mol. Clin. Oncol. 2013, 1, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Sampetrean, O.; Saya, H. Modeling phenotypes of malignant gliomas. Cancer Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Pan, Y.; Gutmann, D.H. The power of the few. Genes Dev. 2017, 31, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Fuller, G.N.; Scheithauer, B.W. The 2007 revised World Health Organization (WHO) classification of tumors of the central nervous system: Newly codified entities. Brain Pathol. 2007, 17, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumors of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, S.G.; Ahmad, M.; Toms, S.A. Mechanisms of Glioma Cell Invasion. In Neurooncology-Newer Developments; Agrawal, A., Ed.; InTech: London, UK, 2016; pp. 109–124. [Google Scholar] [CrossRef]
- Van den Bent, M.J. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: A clinician’s perspective. Acta Neuropathol. 2010, 120, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Leeper, H.E.; Caron, A.A.; Decker, P.A.; Jenkins, R.B.; Lachance, D.H.; Giannini, C. IDH mutation, 1p19q codeletion and ATRX loss in WHO grade II gliomas. Oncotarget 2015, 6, 30295–30305. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, A.; Godlewski, J.; Chiocca, E.A. Extracellular Vesicles and MicroRNAs: Their Role in Tumorigenicity and Therapy for Brain Tumors. Cell. Mol. Neurobiol. 2016, 36, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Cancer Genome Atlas Research Network: Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Rennert, R.C.; Hochberg, F.H.; Carter, B.S. ExRNA in Biofluids as Biomarkers for Brain Tumors. Cell. Mol. Neurobiol. 2016, 36, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Rooj, A.; Mineo, M.; Godlewski, J. MicroRNA and extracellular vesicles in glioblastoma: Small but powerful. Brain Tumor Pathol. 2016, 33, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, R.P.; Costello, J.F. Epigenetic mechanisms in glioblastoma multiforme. Semin. Cancer Biol. 2009, 19, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Nicolaidis, S. Biomarkers of glioblastoma multiforme. Metabolism 2015, 64 (Suppl. 1), S22–S27. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Pain, M.; Wang, H.; Herman, J.A.; Toledo, C.M.; DeLuca, J.G.; Yong, R.L.; Paddison, P.; Zhu, J. Sensitivity to BUB1B inhibition defines an alternative classification of glioblastoma. Cancer Res. 2017, 77, 5518–5529. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, T.; Gao, L.; Zheng, J.; Shao, A.; Zhang, J. Efficacy and safety of long-term therapy for high-grade glioma with temozolomide: A meta-analysis. Oncotarget 2017, 8, 51758–51765. [Google Scholar] [CrossRef] [PubMed]
- Tipping, M.; Eickhoff, J.; Ian Robins, H. Clinical outcomes in recurrent glioblastoma with bevacizumab therapy: An analysis of the literature. J. Clin. Neurosci. 2017, 44, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Murat, A.; Migliavacca, E.; Gorlia, T.; Lambiv, W.L.; Shay, T.; Hamou, M.F.; de Tribolet, N.; Regli, L.; Wick, W.; Kouwenhoven, M.C.; et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J. Clin. Oncol. 2008, 26, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Claes, A.; Idema, A.J.; Wesseling, P. Diffuse glioma growth: A guerrilla war. Acta Neuropathol. 2007, 114, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.F.; Xie, Q.; Su, Y.L.; Koeman, J.; Khoo, S.K.; Gustafson, M.; Knudsen, B.S.; Hay, R.; Shinomiya, N.; Vande Woude, G.F. Proliferation and invasion: Plasticity in tumor cells. Proc. Natl. Acad. Sci. USA 2005, 102, 10528–10533. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Mittal, S.; Berens, M.E. Targeting adaptive glioblastoma: An overview of proliferation and invasion. Neuro Oncol. 2014, 16, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Bellail, A.C.; Hunter, S.B.; Brat, D.J.; Van Meir, E.G. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell. Biol. 2004, 36, 1046–1069. [Google Scholar] [CrossRef] [PubMed]
- Iser, I.C.; Pereira, M.B.; Lenz, G.; Wink, M.R. The Epithelial-to-Mesenchymal Transition-Like Process in Glioblastoma: An Updated Systematic Review and In Silico Investigation. Med. Res. Rev. 2017, 37, 271–313. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, A.; Didier, S.; Fortin, S.; Chuang, Y.; White, T.; Berens, M.E.; Rushing, E.; Eschbacher, J.; Tran, N.L.; Chan, A.; et al. The small GTPase RhoG mediates glioblastoma cell invasion. Mol. Cancer 2012, 11, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortin Ensign, S.P.; Mathews, I.T.; Symons, M.H.; Berens, M.E.; Tran, N.L. Implications of Rho GTPase Signaling in Glioma Cell Invasion and Tumor Progression. Front. Oncol. 2013, 3, 241. [Google Scholar] [CrossRef] [PubMed]
- McFerrin, M.B.; Sontheimer, H. A role for ion channels in glioma cell invasion. Neuron Glia Biol. 2006, 2, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xie, J.; He, H.-Y.; Huang, E.-W.; Cao, Q.-H.; Luo, L.; Liao, Y.-S.; Guo, Y. Suppression of CLC-3 chloride channel reduces the aggressiveness of glioma through inhibiting nuclear factor-κB pathway. Oncotarget 2017, 8, 63788–63798. [Google Scholar] [PubMed]
- Maugeri, R.; Schiera, G.; Di Liegro, C.M.; Fricano, A.; Iacopino, D.G.; Di Liegro, I. Aquaporins and Brain Tumors. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, F.; Jiang, R.; Liu, H.; Hou, J.; Yi, Y.; Kang, L.; Liu, X.; Li, Y.; Yang, M. Down-Regulation of AQP4 Expression via p38 MAPK Signaling in Temozolomide-Induced Glioma Cells Growth Inhibition and Invasion Impairment. J. Cell. Biochem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.L.; Wang, X.; Lou, J.C.; Ma, X.C.; Zhang, B. The potential role of aquaporin 4 in malignant gliomas. Oncotarget 2017, 8, 32345–32355. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome–an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Tani, E.; Ametani, T. Extracellular distribution of ruthenium redpositive substance in the cerebral cortex. J. Ultrastruct. Res. 1971, 34, 1–14. [Google Scholar] [CrossRef]
- Zimmermann, D.R.; Dours-Zimmermann, M.T. Extracellular matrix of the central nervous system: From neglect to challenge. Histochem. Cell Biol. 2008, 130, 635–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, T.A.; de Juan Pardo, E.M.; Kumar, S. The mechanical rigidity of the extracellular matrix regulates the structure, motility and proliferation of glioma cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef] [PubMed]
- Wildeboer, D.; Naus, S.; Amy Sang, Q.X.; Bartsch, J.W.; Pagenstecher, A. Metalloproteinase disintegrins ADAM8 and ADAM19 are highly regulated in human primary brain tumors and their expression levels and activities are associated with invasiveness. J. Neuropathol. Exp. Neurol. 2006, 65, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Siney, E.J.; Holden, A.; Casselden, E.; Bulstrode, H.; Thomas, G.J.; Willaime-Morawek, S. Metalloproteinases ADAM10 and ADAM17 Mediate Migration and Differentiation in Glioblastoma Sphere-Forming Cells. Mol. Neurobiol. 2017, 54, 3893–3905. [Google Scholar] [CrossRef] [PubMed]
- D’Abaco, G.M.; Ng, K.; Paradiso, L.; Godde, N.J.; Kaye, A.; Novak, U. ADAM22, expressed in normal brain but not in high-grade gliomas, inhibits cellular proliferation via the disintegrin domain. Neurosurgery 2006, 58, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, S.J.; Seo, H.H.; Shin, S.P.; Kim, D.; Park, C.S.; Kim, K.T.; Kim, Y.H.; Jeong, J.S.; Kim, I.H. Over-expression of miR-145 enhances the effectiveness of HSVtk gene therapy for malignant glioma. Cancer Lett. 2012, 320, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Stanton, H.; Melrose, J.; Little, C.B.; Fosang, A.F. Proteoglycan degradation by the ADAMTS family of proteinases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 1616–1629. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D. Secretion of cathepsin B by human gliomas in vitro. Neuropathol. Appl. Neurobiol. 1993, 19, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Rempel, S.A.; Rosenblum, M.L.; Mikkelsen, T.; Yan, P.-S.; Ellis, K.D.; Golembieski, W.A.; Sameni, M.; Rozhin, J.; Ziegler, G.; Sloane, B.F. Cathepsin B expression and localization in glioma progression and invasion. Cancer Res. 1994, 54, 6027–6031. [Google Scholar] [PubMed]
- Demchik, L.L.; Sameni, M.; Nelson, K.; Mikkelsen, T.; Sloane, B.F. Cathepsin B and glioma invasion. Int. J. Dev. Neurosci. 1999, 17, 483–494. [Google Scholar] [CrossRef]
- Pei, J.; Moon, K.S.; Pan, S.; Lee, K.H.; Ryu, H.H.; Jung, T.Y.; Kim, I.Y.; Jang, W.Y.; Jung, C.H.; Jung, S. Proteomic Analysis between U87MG and U343MG-A Cell Lines: Searching for Candidate Proteins for Glioma Invasion. Brain Tumor Res. Treat. 2014, 2, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Momeny, M.; Moghaddaskho, F.; Gortany, N.K.; Yousefi, H.; Sabourinejad, Z.; Zarrinrad, G.; Mirshahvaladi, S.; Eyvani, H.; Barghi, F.; Ahmadinia, L.; et al. Blockade of vascular endothelial growth factor receptors by tivozanib has potential anti-tumor effects on human glioblastoma cells. Sci. Rep. 2017, 7, 44075. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ye, L.; Owen, S.; Weeks, H.P.; Zhang, Z.; Jiang, W.G. Emerging role of CCN family proteins in tumorigenesis and cancer metastasis (Review). Int. J. Mol. Med. 2015, 36, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Thorne, A.H.; Meisen, W.H.; Russell, L.; Yoo, J.Y.; Bolyard, C.M.; Lathia, J.D.; Rich, J.; Puduvalli, V.K.; Mao, H.; Yu, J.; et al. Role of cysteine-rich 61 protein (CCN1) in macrophage-mediated oncolytic herpes simplex virus clearance. Mol. Ther. 2014, 22, 1678–1687. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Oguchi, M.; Nakashima, Y.; Yamabe, H. Distribution of collagen type IV in brain tumors: An immunohistochemical study. J. Neuro Oncol. 1989, 7, 357–366. [Google Scholar] [CrossRef]
- Mammoto, T.; Jiang, A.; Jiang, E.; Panigrahy, D.; Kieran, M.W.; Mammoto, A. Role of collagen matrix in tumor angiogenesis and glioblastoma multiforme progression. Am. J. Pathol. 2013, 183, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Delpech, B.; Maingonnat, C.; Girard, N.; Chauzy, C.; Maunoury, R.; Olivier, A.; Tayot, J.; Creissard, P. Hyaluronan and hyaluronectin in the extracellular matrix of human brain tumor stroma. Eur. J. Cancer 1993, 29, 1012–1017. [Google Scholar] [CrossRef]
- Martinez-Quintanilla, J.; He, D.; Wakimoto, H.; Alemany, R.; Shah, K. Encapsulated Stem Cells Loaded with Hyaluronidase-expressing Oncolytic Virus for Brain Tumor Therapy. Mol. Ther. 2015, 23, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Stojic, J.; Hagemann, C.; Haas, S.; Herbold, C.; Kühnel, S.; Gerngras, S.; Roggendorf, W.; Roosen, K.; Vince, G.H. Expression of matrix metalloproteinases MMP-1, MMP-11 and MMP-19 is correlated with the WHO-grading of human malignant gliomas. Neurosci. Res. 2008, 60, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H.; Zhu, J.; Ding, K.; Xu, J. FTY720 reduces migration and invasion of human glioblastoma cell lines via inhibiting the PI3K/AKT/mTOR/p70S6K signaling pathway. Tumor Biol. 2014, 35, 10707–10714. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Inbar, O.; Zaaroor, M. Glioblastoma multiforme targeted therapy: The Chlorotoxin story. J. Clin. Neurosci. 2016, 33, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, Q.; Tan, Y.; Liu, B.; Liu, C. Ellagic acid inhibits human glioblastoma growth in vitro and in vivo. Oncol. Rep. 2017, 37, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zheng, J.; Zhang, Y.; Wang, Z.; Yang, Y.; Bai, M.; Dai, Y. Fucoxanthin Activates Apoptosis via Inhibition of PI3K/Akt/mTOR Pathway and Suppresses Invasion and Migration by Restriction of p38-MMP-2/9 Pathway in Human Glioblastoma Cells. Neurochem. Res. 2016, 41, 2728–2751. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Tu, Y.; Liu, Q.; Ouyang, Y.; He, M.; Luo, M.; Chen, J.; Pi, R.; Liu, A. PT93, a novel caffeic acid amide derivative, suppresses glioblastoma cells migration, proliferation and MMP-2/-9 expression. Oncol. Lett. 2017, 13, 1990–1996. [Google Scholar] [CrossRef] [PubMed]
- Cesarini, V.; Martini, M.; Vitiani, L.R.; Gravina, G.L.; Di Agostino, S.; Graziani, G.; D’Alessandris, Q.G.; Pallini, R.; Larocca, L.M.; Rossi, P.; et al. Type 5 phosphodiesterase regulates glioblastoma multiforme aggressiveness and clinical outcome. Oncotarget 2017, 8, 13223–13239. [Google Scholar] [CrossRef] [PubMed]
- Kegelman, T.P.; Wu, B.; Das, S.K.; Talukdar, S.; Beckta, J.M.; Hu, B.; Emdad, L.; Valerie, K.; Sarkar, D.; Furnari, F.B.; et al. Inhibition of radiation-induced glioblastoma invasion by genetic and pharmacological targeting of MDA-9/Syntenin. Proc. Natl. Acad. Sci. USA 2017, 114, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Imanishi, Y.; Cackowski, F.C.; Jarzynka, M.J.; Tao, H.Q.; Nishikawa, R.; Hirose, T.; Hu, B.; Cheng, S.Y. Up-regulation of angiopoietin-2, matrix metalloprotease-2, membrane type 1 metalloprotease and laminin 5 gamma 2 correlates with the invasiveness of human glioma. Am. J. Pathol. 2005, 166, 877–890. [Google Scholar] [CrossRef]
- Wang, M.; Wang, T.; Liu, S.; Yoshida, D.; Teramoto, A. The expression of matrix metalloproteinase-2 and -9 in human gliomas of different pathological grades. Brain Tumor Pathol. 2003, 20, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, P.A.; Wong, H.; Laing, T.D.; Rewcastle, N.B.; Morris, D.G.; Muzik, H.; Leco, K.J.; Johnston, R.N.; Brasher, P.M.; Sutherland, G.; et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br. J. Cancer 1999, 79, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.J.; Butler, G.S.; Rodriguez, D.; Overall, C.M. Matrix metalloproteinase proteomics: Substrates, targets and therapy. Curr. Opin. Cell Biol. 2009, 21, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Rome, C.; Arsaut, J.; Taris, C.; Couillaud, F.; Loiseau, H. MMP-7 (matrilysin) expression in human brain tumors. Mol. Carcinog. 2007, 46, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Nuttall, R.K.; Liu, S.; Edwards, D.R.; Yong, V.W. Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res. 2006, 66, 11771–11780. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, W.; Li, Y.; Yang, H.; Xu, H.; Liang, S.; Zhang, L.; Li, Y. Expression of Matrix Metalloproteinase-26 promotes human glioma U251 cell invasion in vitro and in vivo. Oncol. Rep. 2010, 23, 69–78. [Google Scholar] [PubMed]
- Sato, H.; Takino, T. Coordinate action of membrane-type matrix metalloproteinase-1 (MT1-MMP) and MMP-2 enhances pericellular proteolysis and invasion. Cancer Sci. 2010, 101, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Mahesparan, R.; Read, T.-A.; Lund-Johansen, M.; Skaftnesmo, K.; Bjerkvig, R.; Engebraaten, O. Expression of extracellular matrix components in a highly infiltrative in vivo glioma model. Acta Neuropathol. 2003, 105, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Brösicke, N.; Faissner, A. Role of tenascins in the ECM of gliomas. Cell Adhes. Migr. 2015, 9, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Kim, O.R.; Choi, Y.S.; Lee, H.; Park, K.; Lee, C.T.; Kang, K.W.; Jeong, S. Selection and characterization of tenascin C targeting peptide. Mol. Cells 2012, 33, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Aaberg-Jessen, C.; Christensen, K.; Offenberg, H.; Bartels, A.; Dreehsen, T.; Hansen, S.; Schrøder, H.D.; Brünner, N.; Kristensen, B.W. Low expression of tissue inhibitor of metalloproteinases-1 (TIMP-1) in glioblastoma predicts longer patient survival. J. Neuro Oncol. 2009, 95, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.C.; Volpert, O.V.; Steck, P.A.; Mikkelsen, T.; Polverini, P.J.; Rao, S.; Chou, P.; Bouck, N.P. Inhibition of angiogenesis in human glioblastomas by chromosome 10 induction of thrombospondin-1. Cancer Res. 1996, 56, 5684–5691. [Google Scholar] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Katsetos, C.D.; Dráberová, E.; Smejkalová, B.; Reddy, G.; Bertrand, L.; de Chadarévian, J.P.; Legido, A.; Nissanov, J.; Baas, P.W.; Dráber, P. Class III beta-tubulin and gamma-tubulin are co-expressed and form complexes in human glioblastoma cells. Neurochem. Res. 2007, 32, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Katsetos, C.D.; Dráberová, E.; Legido, A.; Dumontet, C.; Dráber, P. Tubulin targets in the pathobiology and therapy of glioblastoma multiforme. I. Class III beta-tubulin. J. Cell. Physiol. 2009, 221, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Katsetos, C.D.; Dráberová, E.; Legido, A.; Dráber, P. Tubulin targets in the pathobiology and therapy of glioblastoma multiforme. II. gamma-Tubulin. J. Cell. Physiol. 2009, 221, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Ludueña, R.F.; Banerjee, A. The isotypes of tubulin: Distribution and functional significance. In Cancer Drug Discovery and Development: The Role of Microtubules in Cell Biology, Neurobiology and Oncology; Fojo, T., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 123–175. [Google Scholar]
- Song, Y.; Mu, L.; Han, X.; Li, Q.; Dong, B.; Li, H.; Liu, X. MicroRNA-9 inhibits vasculogenic mimicry of glioma cell lines by suppressing Stathmin expression. J. Neurooncol. 2013, 115, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Zohrabian, V.M.; Forzani, B.; Chau, Z.; Murali, R.; Jhanwar-Uniyal, M. Rho/ROCK and MAPK signaling pathways are involved in glioblastoma cell migration and proliferation. Anticancer Res. 2009, 29, 119–123. [Google Scholar] [PubMed]
- Stylli, S.S.; Kaye, A.H.; Lock, P. Invadopodia: At the cutting edge of tumor invasion. J. Clin. Neurosci. 2008, 15, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Boguski, M.S.; McCormick, F. Proteins regulating Ras and its relatives. Nature 1993, 366, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Tcherkezian, J.; Lamarche-Vane, N. Current knowledge of the large RhoGAP family of proteins. Biol. Cell 2007, 99, 67–86. [Google Scholar] [CrossRef] [PubMed]
- Tseliou, M.; Al-Qahtani, A.; Alarifi, S.; Alkahtani, S.H.; Stournaras, C.; Sourvinos, G. The Role of RhoA, RhoB and RhoC GTPases in Cell Morphology, Proliferation and Migration in Human Cytomegalovirus (HCMV) Infected Glioblastoma Cells. Cell. Physiol. Biochem. 2016, 38, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Drake, K.L.; Nakada, S.; Niska, J.A.; Berens, M.E. Ephrin-B3 ligand promotes glioma invasion through activation of Rac1. Cancer Res. 2006, 66, 8492–8500. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Hu, B.; Vuori, K.; Sarkaria, J.N.; Furnari, F.B.; Cavenee, W.K.; Cheng, S.Y. EGFRvIII stimulates glioma growth and invasion through PKA-dependent serine phospho-rylation of Dock180. Oncogene 2014, 33, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Hu, B.; Liu, K.W.; Li, Y.; Lu, X.; Cheng, T.; Yiin, J.J.; Lu, S.; Keezer, S.; Fenton, T.; et al. Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRα-stimulated glioma tumorigenesis in mice and humans. J. Clin. Investig. 2011, 121, 4670–4684. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Song, X.; Mouneimne, G.; Sidani, M.; Lawrence, D.S.; Condeelis, J.S. Cofilin promotes actin polymerization and defines the direction of cell motility. Science 2004, 304, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Hotulainen, P.; Paunola, E.; Vartiainen, M.K.; Lappalainen, P. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol. Biol. Cell 2005, 16, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.T.; Simpson, T.I.; Pratt, T.; Price, D.J.; Maciver, S.K. The motility of glioblastoma tumor cells is modulated by intracellular cofilin expression in a concentration-dependent manner. Cytoskeleton 2005, 60, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Schiapparelli, P.; Guerrero-Cazares, H.; Magaña-Maldonado, R.; Hamilla, S.M.; Ganaha, S.; Goulin Lippi Fernandes, E.; Huang, C.H.; Aranda-Espinoza, H.; Devreotes, P.; Quinones-Hinojosa, A. NKCC1 Regulates Migration Ability of Glioblastoma Cells by Modulation of Actin Dynamics and Interacting with Cofilin. EBioMedicine 2017, 21, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Geiger, K.D.; Stoldt, P.; Schlote, W.; Derouiche, A. Ezrin immunoreactivity is associated with increasing malignancy of astrocytic tumors but is absent in oligodendrogliomas. Am. J. Pathol. 2000, 157, 1785–1793. [Google Scholar] [CrossRef]
- Roma, A.A.; Prayson, R.A. Fascin expression in 90 patients with glioblastoma multiforme. Ann. Diagn. Pathol. 2005, 9, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Quick, Q.; Skalli, O. Alpha-actinin 1 and alpha-actinin 4: Contrasting roles in the survival, motility and RhoA signaling of astrocytoma cells. Exp. Cell Res. 2010, 316, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Finniss, S.; Cazacu, S.; Xiang, C.; Poisson, L.M.; Blumberg, P.M.; Brodie, C. RasGRP3 regulates the migration of glioma cells via interaction with Arp3. Oncotarget 2015, 6, 1850–1864. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Ikota, H.; Sugawara, K.; Nobusawa, S.; Hirato, J.; Nakazato, Y. Nestin expression in brain tumors: Its utility for pathological diagnosis and correlation with the prognosis of high-grade gliomas. Brain Tumor Pathol. 2012, 29, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Lépinoux-Chambaud, C.; Eyer, J. Review on intermediate filaments of the nervous system and their pathological alterations. Histochem. Cell Biol. 2013, 140, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quick, Q.; Paul, M.; Skalli, O. Roles and potential clinical applications of intermediate filament proteins in brain tumors. Semin. Pediatr. Neurol. 2015, 22, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Brommeland, T.; Rosengren, L.; Fridlund, S.; Hennig, R.; Isaksen, V. Serum levels of glial fibrillary acidic protein correlate to tumor volume of high-grade gliomas. Acta Neurol. Scand. 2007, 116, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.S.; Foerch, C.; Schanzer, A.; Heck, A.; Plate, K.H.; Seifert, V.; Steinmetz, H.; Raabe, A.; Sitzer, M. Serum GFAP is a diagnostic marker for glioblastoma multiforme. Brain 2007, 130, 3336–3341. [Google Scholar] [CrossRef] [PubMed]
- Vietheer, J.M.; Rieger, J.; Wagner, M.; Senft, C.; Tichy, J.; Foerch, C. Serum concentrations of glial fibrillary acidic protein (GFAP) do not indicate tumor recurrence in patients with glioblastoma. J. Neuro Oncol. 2017, 135, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Denysenko, T.; Annovazzi, L.; Cassoni, P.; Melcarne, A.; Mellai, M.; Schiffer, D. WNT/β-catenin Signaling Pathway and Downstream Modulators in Low- and High-grade Glioma. Cancer Genom. Proteom. 2016, 13, 31–45. [Google Scholar]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Gong, A.; Huang, S. Foxm1 and Wnt/Beta-catenin signaling in glioma stem cells. Cancer Res. 2012, 72, 5658–5662. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.X.; Tu, Y.; Zhang, S. FoxM1 promotes glioma cells progression by upregulating Anxa1 expression. PLoS ONE 2013, 8, e72376. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.T.; Lee, H.T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y.; et al. Foxm1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, S.; Siu, T.L.; Huang, S. Glioblastoma Multiforme Formation and EMT: Role of FoxM1 Transcription Factor. Curr. Pharm. Des. 2015, 21, 1268–1271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lv, Q.L.; Huang, Y.T.; Zhang, L.H.; Zhou, H.H. Akt/FoxM1 signaling pathway-mediated upregulation of MYBL2 promotes progression of human glioma. J. Exp. Clin. Cancer Res. 2017, 36, 105. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Liu, X.; Li, Y.; Cheng, M.; Wang, W.; Tian, K.; Mu, L.; Zeng, T.; Liu, Y.; Jiang, X.; et al. HMGA2 sustains self-renewal and invasiveness of glioma-initiating cells. Oncotarget 2016, 7, 44365–44380. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillò, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Yang, H.; Tian, Y.; Xie, Q.; Lu, Y.; Wang, Y.; Su, N.; Dong, B.; Liu, X.; Wang, C.; et al. SOX7 is associated with the suppression of human glioma by HMG-box dependent regulation of Wnt/β-catenin signaling. Cancer Lett. 2016, 375, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Cahill, K.E.; Moshead, R.A.; Yamini, B. NF-κB in glioblastoma: Insights into regulators and targeted therapy. Neuro Oncol. 2016, 18, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Chandran, U.R.; Luthra, S.; Santana-Santos, L.; Mao, P.; Kim, S.H.; Minata, M.; Li, J.; Benos, P.V.; DeWang, M.; Hu, B.; et al. Gene expression profiling distinguishes proneural glioma stem cells from mesenchymal glioma stem cells. Genom. Data 2015, 5, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Romashkova, J.A.; Makarov, S.S. NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Inda, M.M.; Vandenberg, S.; Cheng, S.Y.; Nagane, M.; Hadwiger, P.; Tan, P.; Sah, D.W.; Cavenee, W.K.; Furnari, F.B. EGFRvIII promotes glioma angiogenesis and growth through the NF-kB, interleukin-8 pathway. Oncogene 2012, 31, 4054–4066. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B.; Baritaki, S. The novel role of Yin Yang 1 in the regulation of epithelial to mesenchymal transition in cancer via the dysregulated NF-kB/Snail/YY1/RKIP/PTEN circuitry. Crit. Rev. Oncog. 2011, 16, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.L.; McDonough, W.S.; Savitch, B.A.; Fortin, S.P.; Winkles, J.A.; Symons, M.; Nakada, M.; Cunliffe, H.E.; Hostetter, G.; Hoelzinger, D.B.; et al. Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and NF-κB and correlate with poor patient outcome. Cancer Res. 2006, 66, 9535–9542. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jin, X.; Sohn, X.; Jeon, H.Y.; Kim, E.J.; Ham, S.W.; Jeon, H.M.; Chang, S.Y.; Oh, S.Y.; Yin, J.; et al. Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer Lett. 2014, 353, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; He, X.X.; Ma, C.; Wu, X.M.; Wan, X.L.; Xing, Z.K.; Pei, Q.Q.; Dong, X.P.; Liu, D.X.; Xiong, W.C.; et al. Netrin-1 promotes glioma growth by activating NF-κB via UNC5A. Sci. Rep. 2017, 7, 5454. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tang, C.; Jin, D.; Guan, L.; Wu, Y.; Liu, X.; Wu, X.; Wu, Q.Y.; Gao, D. CUEDC2 suppresses glioma tumorigenicity by inhibiting the activation of STAT3 and NF-κB signaling pathway. Int. J. Oncol. 2017, 51, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Westermarck, J.; Kahari, V.M. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999, 13, 781–792. [Google Scholar] [PubMed]
- Guan, H.; Cai, J.; Zhang, N.; Wu, J.; Yuan, J.; Li, J.; Li, M. Sp1 is upregulated in human glioma, promotes MMP-2-mediated cell invasion and predicts poor clinical outcome. Int. J. Cancer 2012, 130, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Yang, K.; Zhang, M.; Zou, M.; Bai, E.; Ma, Q.; Xu, R. Specific protein 1 depletion attenuates glucose uptake and proliferation of human glioma cells by regulating GLUT3 expression. Oncol. Lett. 2016, 12, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Kaufhold, S.; Bonavida, B. Central role of Snail1 in the regulation of EMT and resistance in cancer: A target for therapeutic intervention. J. Exp. Clin. Cancer Res. 2014, 33, 62. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.K.; Choi, S.A.; Kim, S.K.; Wang, K.C.; Park, S.H. Snail plays an oncogenic role in glioblastoma by promoting epithelial mesenchymal transition. Int. J. Clin. Exp. Pathol. 2014, 7, 1977–1987. [Google Scholar] [PubMed]
- Méndez, O.; Zavadil, J.; Esencay, M.; Lukyanov, Y.; Santovasi, D.; Wang, S.C.; Newcomb, E.W.; Zagzag, D. Knock down of HIF-1α in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol. Cancer 2010, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Bertout, J.A.; Hu, C.J.; Diehl, J.A.; Simon, M.C. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.E.; McGuire, M.F. Oncogenesis recapitulates embryogenesis via the hypoxia pathway: Morphoproteomics and biomedical analytics provide proof of concept and therapeutic options. Ann. Clin. Lab. Sci. 2012, 42, 243–257. [Google Scholar] [PubMed]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Valsesia-Wittmann, S.; Magdeleine, M.; Dupasquier, S.; Garin, E.; Jallas, A.C.; Combaret, V.; Krause, A.; Leissner, P.; Puisieux, A. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 2004, 6, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Ehtesham, M.; Winston, J.A.; Kabos, P.; Thompson, R.C. CXCR4 expression mediates glioma cell invasiveness. Oncogene 2006, 25, 2801–2806. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Kress, S.; Stein, A.; Maurer, P.; Weber, B.; Reichert, J.; Buchmann, A.; Huppert, P.; Schwarz, M. Expression of hypoxia-inducible genes in tumor cells. J. Cancer Res. Clin. Oncol. 1998, 124, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed]
- Mikheeva, S.A.; Mikheev, A.M.; Petit, A.; Beyer, R.; Oxford, R.G.; Khorasani, L.; Maxwell, J.P.; Glackin, C.A.; Wakimoto, H.; González-Herrero, I.; et al. Twist1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer 2010, 9, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.P.; Kim, J.H.; Han, M.E.; Sim, H.E.; Kim, K.S.; Yoon, S.; Baek, S.Y.; Kim, B.S.; Oh, S.O. Snai1 is involved in the proliferation and migration of glioblastoma cells. Cell. Mol. Neurobiol. 2011, 31, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Menon, L.G.; Black, P.M.; Carroll, R.S.; Johnson, M.D. Snai2/Slug promotes growth and invasion in human gliomas. BMC Cancer 2010, 10, 301. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Seol, H.J.; Kim, E.H.; Rheey, J.; Jin, H.J.; Lee, Y.; Joo, K.M.; Lee, J.; Nam, D.H. WNT/β-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. Neuro Oncol. 2013, 15, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Saito, N.; Miyazawa, K.; Miyazono, K. Glioma-initiating cells retain their tumorigenicity through integration of the Sox axis and Oct4 protein. J. Biol. Chem. 2011, 286, 41434–41441. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G. How Sox2 maintains neural stem cell identity. Biochem. J. 2013, 450, e1–e2. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Temme, A.; Senner, V.; Ebner, R.; Schwind, S.; Stevanovic, S.; Wehner, R.; Schackert, G.; Schackert, H.K.; Fussel, M.; et al. Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. Br. J. Cancer 2007, 96, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.M.; Diez-Valle, R.; Manterola, L.; Rubio, A.; Liu, D.; Cortes-Santiago, N.; Urquiza, L.; Jauregi, P.; Lopez de Munain, A.; Sampron, N.; et al. Genetic and epigenetic modifications of Sox2 contribute to the invasive phenotype of malignant gliomas. PLoS ONE 2011, 6, e26740. [Google Scholar] [CrossRef] [PubMed]
- Garros-Regulez, L.; Garcia, I.; Carrasco-Garcia, E.; Lantero, A.; Aldaz, P.; Moreno-Cugnon, L.; Arrizabalaga, O.; Undabeitia, J.; Torres-Bayona, S.; Villanua, J.; et al. Targeting SOX2 as a Therapeutic Strategy in Glioblastoma. Front. Oncol. 2016, 6, 222. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.-C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Fiorelli, R.; Kupp, R.; Rajan, S.; Szeto, E.; Lo Cascio, C.; Maire, C.L.; Sun, Y.; Alberta, J.A.; Eschbacher, J.M.; et al. Post-translational Modifications of OLIG2 Regulate Glioma Invasion through the TGF-β Pathway. Cell Rep. 2016, 16, 950–966. [Google Scholar] [CrossRef] [PubMed]
- Homma, J.; Yamanaka, R.; Yajima, N.; Tsuchiya, N.; Genkai, N.; Sano, M.; Tanaka, R. Increased expression of CCAAT/enhancer binding protein beta correlates with prognosis in glioma patients. Oncol. Rep. 2006, 15, 595–601. [Google Scholar] [PubMed]
- Aguilar-Morante, D.; Cortes-Canteli, M.; Sanz-Sancristobal, M.; Santos, A.; Perez-Castillo, A. Decreased CCAAT/enhancer binding protein β expression inhibits the growth of glioblastoma cells. Neuroscience 2011, 176, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soroceanu, L.; Manning, T.J., Jr.; Sontheimer, H. Modulation of glioma cell migration and invasion using Cl(−) and K(+) ion channel blockers. J. Neurosci. 1999, 19, 5942–5954. [Google Scholar] [PubMed]
- Watkins, S.; Sontheimer, H. Hydrodynamic Cellular Volume Changes Enable Glioma Cell Invasion. J. Neurosci. 2011, 31, 17250–17259. [Google Scholar] [CrossRef] [PubMed]
- Setti, M.; Savalli, N.; Osti, D.; Richichi, C.; Angelini, M.; Brescia, P.; Fornasari, L.; Carro, M.S.; Mazzanti, M.; Pelicci, G. Functional role of CLIC1 ion channel in glioblastoma-derived stem/progenitor cells. J. Natl. Cancer Inst. 2013, 105, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Setti, M.; Osti, D.; Richichi, C.; Ortensi, B.; Del Bene, M.; Fornasari, L.; Beznoussenko, G.; Mironov, A.; Rappa, G.; Cuomo, A.; et al. Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget 2015, 6, 31413–31427. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ding, X. TRPC Channels and Glioma. Adv. Exp. Med. Biol. 2017, 976, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Higashimori, H.; Sontheimer, H. Role of Kir4.1 channels in growth control of glia. Glia 2007, 55, 1668–1679. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Chanteloup, G.; Boucher, J.; Pernet, N.; Boudesco, C.; Jego, G.; Chatelier, A.; Bois, P.; Gobbo, J.; Cronier, L.; et al. Modulation of the inwardly rectifying potassium channel Kir4.1 by the pro-invasive miR-5096 in glioblastoma cells. Oncotarget 2017, 8, 37681–37693. [Google Scholar] [CrossRef] [PubMed]
- Loo, D.D.; Zeuthen, T.; Chandy, G.; Wright, E.M. Cotransport of water by the Na+/glucose cotransporter. Proc. Natl. Acad. Sci. USA 1996, 93, 13367–13370. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Busch, G.L.; Ritter, M.; Völkl, H.; Waldegger, S.; Gulbins, E.; Häussinger, D. Functional significance of cell volume regulatory mechanisms. Physiol. Rev. 1998, 78, 247–306. [Google Scholar] [PubMed]
- Day, R.E.; Kitchen, P.; Owen, D.S.; Bland, C.; Marshall, L.; Conner, A.C.; Bill, R.M.; Conner, M.T. Human aquaporins: Regulators of transcellular water flow. Biochim. Biophys. Acta 2014, 1840, 1492–1506. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Edwards, N.A.; Proescholdt, M.A.; Oldfield, E.H.; Merrill, M.J. Regulation and function of aquaporin-1 in glioma cells. Neoplasia 2007, 9, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized membrane domains for water transport in glial cells: High-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 1997, 17, 171–180. [Google Scholar] [PubMed]
- Nagelhus, E.A.; Mathiisen, T.M.; Ottersen, O.P. Aquaporin-4 in the central nervous system: Cellular and subcellular distribution and coexpression with Kir4.1. Neuroscience 2004, 129, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Frigeri, A.; Nicchia, G.P.; Quondamatteo, F.; Herken, R.; Errede, M.; Ribatti, D.; Svelto, M.; Roncali, L. Role of aquaporin-4 water channel in the development and integrity of the blood-brain barrier. J. Cell Sci. 2001, 114, 1297–1307. [Google Scholar] [PubMed]
- Wolburg, H. Orthogonal arrays of intramembranous particles. A review with special reference to astrocytes. J. Brain Res. 1995, 36, 239–258. [Google Scholar]
- Yang, B.; Brown, D.; Verkman, A.S. The mercurial insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J. Biol. Chem. 1996, 271, 4577–4580. [Google Scholar] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Amiry-Moghaddam, M.; Otsuka, T.; Hurn, P.D.; Traystman, R.J.; Haug, F.-M.; Froehner, S.C.; Adams, M.E.; Neely, J.D.; Agre, P.; Ottersen, O.P.; et al. An α-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc. Natl. Acad. Sci. USA 2003, 100, 2106–2111. [Google Scholar] [CrossRef] [PubMed]
- Noell, S.; Wolburg-Buchholz, K.; Mack, A.F.; Beedle, A.M.; Satz, J.S.; Campbell, K.P.; Wolburg, H.; Fallier-Becker, P. Evidence for a role of dystroglycan regulating the membrane architecture of astroglial endfeet. Eur. J. Neurosci. 2011, 33, 2179–2186. [Google Scholar] [CrossRef] [PubMed]
- Fossdal, G.; Vik-Mo, E.O.; Sandberg, C.; Varghese, M.; Kaarbø, M.; Telmo, E.; Langmoen, I.A.; Murrell, W. Aqp 9 and brain tumor stem cells. Sci. World J. 2012, 915176. [Google Scholar] [CrossRef]
- McCoy, E.; Sontheimer, H. Expression and function of water channels (aquaporins) in migrating malignant astrocytes. Glia 2007, 55, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- McCoy, E.S.; Haas, B.R.; Sontheimer, H. Water permeability through aquaporin-4 is regulated by protein kinase C and becomes rate-limiting for glioma invasion. Neuroscience 2010, 168, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Engelhorn, T.; Savaskan, N.E.; Schwarz, M.A.; Kreutzer, J.; Meyer, E.P.; Hahnen, E.; Ganslandt, O.; Dörfler, A.; Nimsky, C.; Buchfelder, M.; et al. Cellular characterization of the peritumoral edema zone in malignant brain tumors. Cancer Sci. 2009, 100, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Jain, R.K.; Witwer, B.; Brown, D. Water channel (aquaporin 1) expression and distribution in mammary carcinomas and glioblastomas. Microvasc. Res. 1999, 58, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Warth, A.; Kröger, S.; Wolburg, H. Redistribution of aquaporin-4 in human glioblastoma correlates with loss of agrin immunoreactivity from brain capillary basal laminae. Acta Neuropathol. 2004, 107, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Warth, A.; Mittelbronn, M.; Wolburg, H. Redistribution of the water channel protein aquaporin-4 and the K+ channel protein Kir4.1 differs in low- and high-grade human brain tumors. Acta Neuropathol. 2005, 109, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.L. Hypoxia in the tumorigenesis of gliomas and as a potential target for therapeutic measures. Neurosurg. Focus 2006, 20, E24. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Radermacher, J.; Mayer, J.; Mehraein, Y.; Meese, E. Tumor hypoxia: Impact on gene amplification in glioblastoma. Int. J. Oncol. 2008, 33, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Irshad, K.; Mohapatra, S.K.; Srivastava, C.; Garg, H.; Mishra, S.; Dikshit, B.; Sarkar, C.; Gupta, D.; Chandra, P.S.; Chattopadhyay, P.; et al. A combined gene signature of hypoxia and notch pathway in human glioblastoma and its prognostic relevance. PLoS ONE 2015, 10, E0118201. [Google Scholar] [CrossRef] [PubMed]
- Ho, I.A.W.; Shim, W.S.N. Contribution of the Microenvironmental Niche to Glioblastoma Heterogeneity. Biomed. Res. Int. 2017, 2017, 9634172. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Colombo, S.L.; Palacios-Callender, M.; Frakich, N.; De Leon, J.; Schmitt, C.A.; Boorn, L.; Davis, N.; Moncada, S. Anaphase-promoting complex/cyclosome-Cdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 18868–18873. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Gonçalves, V.; Granja, S.; Martinho, O.; Honavar, M.; Pojo, M.; Costa, B.M.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; et al. Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget 2016, 7, 46335–46353. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Pinto, A.; Martherus, R.; Santiago de Jesus, J.P.; Polet, F.; Feron, O. Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab. 2016, 24, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Proia, P.; Di Liegro, C.M.; Schiera, G.; Fricano, A.; Di Liegro, I. Lactate as a Metabolite and a Regulator in the Central Nervous System. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newsholme, E.A.; Crabtree, B.; Ardawi, M.S. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci. Rep. 1985, 5, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, G.; Zhao, L.; Ma, Z.; Chen, H. Metabolic reprogramming in cancer cells: Glycolysis, glutaminolysis and Bcl-2 proteins as novel therapeutic targets for cancer. World J. Surg. Oncol. 2016, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Ríos, M.; Foretz, M.; Viollet, B.; Prieto, A.; Fraga, M.; García-Caballero, T.; Costoya, J.A.; Señarís, R. Lipoprotein internalization induced by oncogenic AMPK activation is essential to maintain glioblastoma cell growth. Eur. J. Cancer 2014, 50, 3187–3197. [Google Scholar] [CrossRef] [PubMed]
- Kathagen-Buhmann, A.; Schulte, A.; Weller, J.; Holz, M.; Herold-Mende, C.; Glass, R.; Lamszus, K. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro Oncol. 2016, 18, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Fischer, I.; Gagner, J.P.; Law, M.; Newcomb, E.W.; Zagzag, D. Angiogenesis in gliomas: Biology and molecular pathophysiology. Brain Pathol. 2005, 15, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Guillevin, R.; Vallée, J.N. Vasculogenesis and angiogenesis initiation under normoxic conditions through Wnt/β-catenin pathway in gliomas. Rev. Neurosci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Brooks, L.J.; Parrinello, S. Vascular regulation of glioma stem-like cells: A balancing act. Curr. Opin. Neurobiol. 2017, 47, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Anido, J.; Sáez-Borderías, A.; Gonzàlez-Juncà, A.; Rodón, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martínez-Sáez, E.; Prudkin, L.; et al. TGF-β Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Borovski, T.; Medema, J.P. Endothelial cells induce cancer stem cell features in differentiated glioblastoma cells via bFGF. Mol. Cancer 2015, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Debruyne, D.N.; Turchi, L.; Burel-Vandenbos, F.; Fareh, M.; Almairac, F.; Virolle, V.; Figarella-Branger, D. DOCK4 promotes loss of proliferation in glioblastoma progenitor cells through nuclear beta-catenin accumulation and subsequent miR-302-367 cluster expression. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. Shining a light on the genome’s ‘dark matter’. Science 2010, 330, 1614. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Di Liegro, C.M.; Schiera, G.; Di Liegro, I. Extracellular Vesicle-Associated RNA as a Carrier of Epigenetic Information. Genes (Basel) 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, A.; Wang, Y.; Nowicki, M.O.; Peruzzi, P.; Ansari, K.; Ogawa, D.; Balaj, L.; De Rienzo, G.; Mineo, M.; Nakano, I.; et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014, 74, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Kumari Singh, D.; Panda, S.; Shiras, A. Extracellular Vesicles as Modulators of Tumor Microenvironment and Disease Progression in Glioma. Front. Oncol. 2017, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- Moller, H.G.; Rasmussen, A.P.; Andersen, H.H.; Johnsen, K.B.; Henriksen, M.; Duroux, M. A systematic review of microRNA in glioblastoma multiforme: Micro-modulators in the mesenchymal mode of migration and invasion. Mol. Neurobiol. 2013, 47, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Beyer, S.; Fleming, J.; Meng, W.; Singh, R.; Haque, S.J.; Chakravarti, A. The Role of miRNAs in Angiogenesis, Invasion and Metabolism and Their Therapeutic Implications in Gliomas. Cancers (Basel) 2017, 9. [Google Scholar] [CrossRef]
- Baroni, S.; Romero-Cordoba, S.; Plantamura, I.; Dugo, M.; D’Ippolito, E.; Cataldo, A.; Cosentino, G.; Angeloni, V.; Rossini, A.; Daidone, M.G.; et al. Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis. 2016, 7, e2312. [Google Scholar] [CrossRef] [PubMed]
- Sasayama, T.; Nishihara, M.; Kondoh, T.; Hosoda, K.; Kohmura, E. MicroRNA-10b is overexpressed in malignant glioma and associated with tumor invasive factors, UPAR and RHOC. Int. J. Cancer 2009, 125, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.Q.; Lu, X.J.; Wu, T.F.; Ding, D.D.; Zhao, Z.H.; Chen, G.L.; Xie, X.S.; Li, B.; Wei, Y.X.; Guo, L.C.; et al. MicroRNA-16 inhibits glioma cell growth and invasion through suppression of BCL2 and the nuclear factor-κB1/MMP9 signaling pathway. Cancer Sci. 2014, 105, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.H.; Xu, Q.Y.; Tian, R.; Yan, H.; Zhang, M.; Wu, J.; Wang, W.; He, J. MicroRNA16 regulates glioma cell proliferation, apoptosis and invasion by targeting Wip1-ATM-p53 feedback loop. Oncotarget 2017, 8, 54788–54798. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumor growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Gabriely, G.; Wurdinger, T.; Kesari, S.; Esau, C.C.; Burchard, J.; Linsley, P.S.; Krichevsky, A.M. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol. Cell. Biol. 2008, 28, 5369–5380. [Google Scholar] [CrossRef] [PubMed]
- Hermansen, S.K.; Nielsen, B.S.; Aaberg-Jessen, C.; Kristensen, B.W. miR-21 is Linked to Glioma Angiogenesis: A Co-Localization Study. J. Histochem. Cytochem. 2016, 64, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Luo, W.; Sun, X.; Lin, J.; Wang, M.; Zhang, Y.; Luo, W.; Zhang, Y. MicroRNA-21 promotes migration and invasion of glioma cells via activation of Sox2 and β-catenin signaling. Mol. Med. Rep. 2017, 15, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Li, C.C.Y.; Eaton, S.A.; Young, P.E.; Lee, M.; Shuttleworth, R.; Humphreys, D.T.; Grau, G.E.; Combes, V.; Bebawy, M.; Gong, J.; et al. Glioma microvesicles carry selectively packaged coding and non-coding RNAs which alter gene expression in recipient cells. RNA Biol. 2013, 10, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Krichevsky, A.M.; Johnson, M.D.; Chiocca, E.A.; Bronisz, A. Belonging to a network–microRNAs, extracellular vesicles and the glioblastoma microenvironment. Neuro Oncol. 2015, 17, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Dai, W.M.; Huang, Q.; Jie, Y.Q.; Yu, G.F.; Fan, X.F.; Wu, A.; Mao, D.D. Effects of microRNA-26b on proliferation and invasion of glioma cells and related mechanisms. Mol. Med. Rep. 2017, 16, 4165–4170. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Sun, J.; Shi, C.; Sun, C.; Yu, L.; Wen, Y.; Zhao, S.; Liu, J.; Xu, J.; Li, H.; et al. miR-29s inhibit the malignant behavior of U87MG glioblastoma cell line by targeting DNMT3A and 3B. Neurosci. Lett. 2015, 590, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, P.; Ma, J.; Liu, Y.H.; Li, Z.; Li, Z.Q.; Wang, Z.H.; Chen, L.Y.; Xue, Y.X. MiR-34a regulates blood-tumor barrier function by targeting protein kinase Cε. Mol. Biol. Cell 2015, 26, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L. miR-34a attenuates glioma cells progression and chemoresistance via targeting PD-L1. Biotechnol. Lett. 2017, 39, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Deng, Z.; Shatseva, T.; Yang, J.; Peng, C.; Du, W.W.; Yee, A.J.; Ang, L.C.; He, C.; Shan, S.W.; et al. MicroRNA miR-93 promotes tumor growth and angiogenesis by targeting integrin-β8. Oncogene 2011, 30, 806–821. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, E.; Brognara, E.; Montagner, G.; Ghimenton, C.; Eccher, A.; Cantù, C.; Khalil, S.; Bezzerri, V.; Provezza, L.; Bianchi, N.; et al. Regulation of IL-8 gene expression in gliomas by microRNA miR-93. BMC Cancer 2015, 15, 661. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.H.; Ye, M.H.; Wu, L.; Lv, S.G.; Wu, M.J.; Xiao, B.; Liao, C.C.; Ji, Q.K.; Chai, Y.; Zhu, X.G. Overexpression of miR-98 inhibits cell invasion in glioma cell lines via downregulation of IKKε. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3593–3604. [Google Scholar] [PubMed]
- Qiao, W.; Guo, B.; Zhou, H.; Xu, W.; Chen, Y.; Liang, Y.; Dong, B. miR-124 suppresses glioblastoma growth and potentiates chemosensitivity by inhibiting AURKA. Biochem. Biophys. Res. Commun. 2017, 486, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gong, Q.; Li, M.; Xu, J.; Zheng, Y.; Ge, P.; Chi, G. MicroRNA-124 inhibits the proliferation of C6 glioma cells by targeting Smad4. Int. J. Mol. Med. 2017, 40, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Fu, W.; Wo, L.; Shu, X.; Liu, F.; Li, C. miR-128 and its target genes in tumorigenesis and metastasis. Exp. Cell Res. 2013, 319, 3059–3064. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.Q.; Yin, T.K.; Li, Y.X.; Zhang, J.H.; Gu, J.J. miR-130b regulates the proliferation, invasion and apoptosis of glioma cells via targeting of CYLD. Oncol. Rep. 2017, 38, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Cannistraci, A.; Federici, G.; Addario, A.; Di Pace, A.L.; Grassi, L.; Muto, G.; Collura, D.; Signore, M.; De Salvo, L.; Sentinelli, S.; et al. C-Met/miR-130b axis as novel mechanism and biomarker for castration resistance state acquisition. Oncogene 2017, 36, 3718–3728. [Google Scholar] [CrossRef] [PubMed]
- Bian, E.B.; Ma, C.C.; He, X.J.; Wang, C.; Zong, G.; Wang, H.L.; Zhao, B. Epigenetic modification of miR-141 regulates SKA2 by an endogenous ‘sponge’ HOTAIR in glioma. Oncotarget 2016, 7, 30610–30625. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Rong, X.; Dong, J.; Yu, C.; Yang, J. miR-142 inhibits the migration and invasion of glioma by targeting Rac1. Oncol. Rep. 2017, 38, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Qi, Y.; Ng, S.S.; Chen, X.; Li, D.; Chen, S.; Ge, R.; Jiang, S.; Li, G.; Chen, Y.; et al. microRNA-146b inhibits glioma cell migration and invasion by targeting MMPs. Brain Res. 2009, 1269, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Manterola, L.; Guruceaga, E.; Gállego Pérez-Larraya, J.; González-Huarriz, M.; Jauregui, P.; Tejada, S.; Diez-Valle, R.; Segura, V.; Samprón, N.; Barrena, C.; et al. A small noncoding RNA signature found in exosomes of GBM patient serum as a diagnostic tool. Neuro Oncol. 2014, 16, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Tian, X.; Zhang, J.; Huang, Y.; Lin, X.; Chen, L.; Zhang, S. Regulation of human glioma cell apoptosis and invasion by miR-152–3p through targeting DNMT1 and regulating NF2: MiR-152–3p regulate glioma cell apoptosis and invasion. J. Exp. Clin. Cancer Res. 2017, 36, 100. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhu, W.; Shi, D.; Lv, L.; Zhang, C.; Liu, P.; Hu, W. MicroRNA-181a sensitizes human malignant glioma U87MG cells to radiation by targeting Bcl-2. Oncol. Rep. 2010, 23, 997–1003. [Google Scholar] [PubMed]
- Wang, H.; Tao, T.; Yan, W.; Feng, Y.; Wang, Y.; Cai, J.; You, Y.; Jiang, T.; Jiang, C. Upregulation of miR-181s reverses mesenchymal transition by targeting KPNA4 in glioblastoma. Sci. Rep. 2015, 5, 13072. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Cho, B.J.; Kim, D.H.; Park, J.M.; Choi, E.J.; Kim, H.H.; Lee, D.J.; Kim, I.A. MicroRNA-200c increases radiosensitivity of human cancer cells with activated EGFR-associated signaling. Oncotarget 2017, 8, 65457–65468. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Huang, Q.; Chen, K.; Liu, L.; Lin, C.; Dai, T.; Yu, C.; Wu, Z.; Li, J. miR-218 inhibits the invasive ability of glioma cells by direct downregulation of IKK-beta. Biochem. Biophys. Res. Commun. 2010, 402, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Gao, X.; Li, G.; Fu, H.; Cui, D.; Liu, H.; Jin, W.; Zhang, Y. MicroRNA-218 inhibits glioma invasion, migration, proliferation and cancer stem-like cell self-renewal by targeting the polycomb group gene Bmi1. Cancer Res. 2013, 73, 6046–6055. [Google Scholar] [CrossRef] [PubMed]
- Mathew, L.K.; Skuli, N.; Mucaj, V.; Lee, S.S.; Zinn, P.O.; Sathyan, P.; Imtiyaz, H.Z.; Zhang, Z.; Davuluri, R.V.; Rao, S.; et al. miR-218 opposes a critical RTK-HIF pathway in mesenchymal glioblastoma. Proc. Natl. Acad. Sci. USA 2014, 111, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, J.; Hao, J.; Shi, Z.; Wang, Y.; Han, L.; Yu, S.; You, Y.; Jiang, T.; Wang, J.; et al. High level of miR-221/222 confers increased cell invasion and poor prognosis in glioma. J. Transl. Med. 2012, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Qiao, S.; Chen, K. Suppression of miR-221 inhibits glioma cells proliferation and invasion via targeting SEMA3B. Biol. Res. 2015, 48, 37. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, W.; Zhou, C.; Xi, W.; Yuan, L.; Chen, X.; Li, Y.; Yang, A.; Zhang, J.; Wang, T. MiR-221/222 promote human glioma cell invasion and angiogenesis by targeting TIMP2. Tumor Biol. 2015, 36, 3763–3773. [Google Scholar] [CrossRef] [PubMed]
- Würdinger, T.; Tannous, B.A.; Saydam, O.; Skog, J.; Grau, S.; Soutschek, J.; Weissleder, R.; Breakefield, X.O.; Krichevsky, A.M. miR-296 regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell 2008, 14, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hwang, S.J.; Kim, H.R.; Shin, C.H.; Choi, K.H.; Joung, J.G.; Kim, H.H. Neurofibromatosis 2 (NF2) controls the invasiveness of glioblastoma through YAP-dependent expression of CYR61/CCN1 and miR-296–3p. Biochim. Biophys. Acta 2016, 1859, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Liu, X.; Chen, L.; Dou, Z.; Lei, X.; Chang, L.; Cai, J.; Cui, Y.; Yang, D.; Sun, Y.; et al. Targeting the SMO oncogene by miR-326 inhibits glioma biological behaviors and stemness. Neuro Oncol. 2015, 17, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, Z.; Patil, V.; Paul, Y.; Hegde, A.S.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. PI3 kinase pathway regulated miRNome in glioblastoma: Identification of miR-326 as a tumor suppressor miRNA. Mol. Cancer 2016, 15, 74. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wu, T.; Li, Y.; Xu, Z.; Zhang, S.; Liu, B.; Chen, Q.; Tian, D. MicroRNA-370-3p inhibits human glioma cell proliferation and induces cell cycle arrest by directly targeting β-catenin. Brain Res. 2016, 1644, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Zheng, J.; Liu, X.; Liu, Y.; Guo, J.; Gao, Y.; Tao, W.; Chen, J.; Li, Z.; Ma, J.; et al. Knockdown of Long Non-Coding RNA KCNQ1OT1 Restrained Glioma Cells’ Malignancy by Activating miR-370/CCNE2 Axis. Front. Cell. Neurosci. 2017, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Han, L.; Zhang, A.; Wang, G.; Jia, Z.; Yang, Y.; Yue, X.; Pu, P.; Zhong, Y.; Kang, C. MiRNA-451 plays a role as tumor suppressor in human glioma cells. Brain Res. 2010, 1359, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Bronisz, A.; Nowicki, M.O.; Chiocca, E.A.; Lawler, S. microRNA-451: A conditional switch controlling glioma cell proliferation and migration. Cell Cycle 2010, 9, 2742–2748. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; de Lay, M.; van Brocklyn, J.; Ostrowski, M.C.; Chiocca, E.A.; Lawler, S.E. microRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 2010, 37, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Chekhonin, V.P. Extracellular vesicles shed by glioma cells: Pathogenic role and clinical value. Tumor Biol. 2014, 35, 8425–8438. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Zhou, L.; Qi, H.; Wang, G.; Luan, Y.; Zuo, L. MiR-592 functions as a tumor suppressor in glioma by targeting IGFBP2. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Hodges, T.R.; Song, R.; Gong, Y.; Calin, G.A.; Heimberger, A.B.; Zhao, H. Serum HOTAIR and GAS5 levels as predictors of survival in patients with glioblastoma. Mol. Carcinog. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Han, L.; Bao, Z.S.; Wang, Y.Y.; Chen, L.Y.; Yan, W.; Yu, S.Z.; Pu, P.Y.; Liu, N.; You, Y.P.; et al. Chinese Glioma Cooperative Group. HOTAIR, a cell cycle-associated long noncoding RNA and a strong predictor of survival, is preferentially expressed in classical and mesenchymal glioma. Neuro Oncol. 2013, 15, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhou, B.; Zhang, J.; Geng, P.; Liu, K.; Zhu, Y.; Zhu, W. A new tumor suppressor LncRNA ADAMTS9-AS2 is regulated by DNMT1 and inhibits migration of glioma cells. Tumor Biol. 2014, 35, 7935–7944. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, P.; Liu, J.; Zheng, J.; Liu, Y.; Chen, J.; Xue, Y. Gas5 Exerts Tumor-suppressive Functions in Human Glioma Cells by Targeting miR-222. Mol. Ther. 2015, 23, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, Y.; Zheng, J.; Liu, X.; Chen, J.; Liu, L.; Wang, P.; Xue, Y. GAS5 suppresses malignancy of human glioma stem cells via a miR-196a-5p/FOXO1 feedback loop. Biochim. Biophys. Acta 2017, 1864, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, Y.H.; Yao, Y.L.; Li, Z.; Li, Z.Q.; Ma, J.; Xue, Y.X. Long non-coding RNA CASC2 suppresses malignancy in human gliomas by miR-21. Cell. Signal. 2015, 27, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Li, X.D.; Wang, P.; Liu, X.B.; Xue, Y.X.; Hu, Y.; Li, Z.; Li, Z.Q.; Wang, Z.H.; Liu, Y.H. CRNDE affects the malignant biological characteristics of human glioma stem cells by negatively regulating miR-186. Oncotarget 2015, 6, 25339–25355. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Liu, X.; Wang, P.; Xue, Y.; Ma, J.; Qu, C.; Liu, Y. CRNDE Promotes Malignant Progression of Glioma by Attenuating miR-384/PIWIL4/STAT3 Axis. Mol. Ther. 2016, 24, 1199–1215. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Luan, W.; Wang, P.; Tao, T.; Zhang, J.; Qian, J.; Liu, N.; You, Y. Long non-coding RNA H19 promotes glioma cell invasion by deriving miR-675. PLoS ONE 2014, 9, e86295. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Yan, Y.; Hu, M.; Chen, X.; Wang, Y.; Dai, Y.; Wu, D.; Wang, Y.; Zhuang, Z.; Xia, H. Increased level of H19 long noncoding RNA promotes invasion, angiogenesis and stemness of glioblastoma cells. J. Neurosurg. 2016, 2016, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Hu, Q.; Nie, E.; Yu, T.; Wu, Y.; Zhi, T.; Jiang, K.; Shen, F.; Wang, Y.; Zhang, J.; et al. Hypoxia induces H19 expression through direct and indirect Hif-1α activity, promoting oncogenic effects in glioblastoma. Sci. Rep. 2017, 7, 45029. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Ye, M.H.; Lv, S.G.; Wang, Q.X.; Wu, M.J.; Xiao, B.; Kang, C.S.; Zhu, X.G. SNORD47, a box C/D snoRNA, suppresses tumorigenesis in glioblastoma. Oncotarget 2017, 8, 43953–43966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, W.; Liu, G.; Xie, S.; Li, Q.; Li, Y.; Lin, Z. Long non-coding RNA HOTTIP promotes hypoxia-induced epithelial-mesenchymal transition of malignant glioma by regulating the miR-101/ZEB1 axis. Biomed. Pharmacother. 2017, 95, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, X.; Qi, L.; Cai, Y.; Yang, P.; Xuan, G.; Jiang, Y. HULC long noncoding RNA silencing suppresses angiogenesis by regulating ESM-1 via the PI3K/Akt/mTOR signaling pathway in human gliomas. Oncotarget 2016, 7, 14429–14440. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Dai, J.; Liao, Y.; Ma, J.; Zhou, W. Knockdown of Long Noncoding RNA LINC0000125 Suppresses Cellular Proliferation and Invasion in Glioma Cells by Regulating MiR-4775. Oncol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wu, L.; Yang, Q.; Ye, M.; Zhu, X. Functional linc-POU3F3 is overexpressed and contributes to tumorigenesis in glioma. Gene 2015, 554, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.L.; Hu, G.W.; Chen, Y.; Liu, Y.; Tu, W.; Lu, Y.M.; Wu, L.; Xu, G.H. Glioma cells promote angiogenesis through the release of exosomes containing long non-coding RNA POU3F3. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 959–972. [Google Scholar] [PubMed]
- Wu, D.; Zhao, B.; Cao, X.; Wan, J. Long non-coding RNA LINK-A promotes glioma cell growth and invasion via lactate dehydrogenase A. Oncol. Rep. 2017, 38, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Luo, W.; Wang, J.; Peng, T.; Sun, G.; Shi, J.; Li, Z.; Zhang, B. Malat1 activates autophagy and promotes cell proliferation by sponging miR-101 and upregulating STMN1, RAB5A and ATG4D expression in glioma. Biochem. Biophys. Res. Commun. 2017, 492, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.W.; Kang, C.M.; Zhao, J.J.; Nie, Y.; Zheng, L.; Li, H.X.; Li, X.; Wang, Q.; Qiu, Y.R. LncRNA PLAC2 down-regulates RPL36 expression and blocks cell cycle progression in glioma through a mechanism involving STAT1. J. Cell. Mol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; An, G.; Zhang, M.; Ma, Q. Long non-coding RNA TUG1 acts as a miR-26a sponge in human glioma cells. Biochem. Biophys. Res. Commun. 2016, 477, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Xue, Y.; Wang, P.; Wang, Z.; Li, Z.; Hu, Y.; Li, Z.; Shang, X.; Liu, Y. The long noncoding RNA TUG1 regulates blood-tumor barrier permeability by targeting miR-144. Oncotarget 2015, 6, 19759–19779. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Liu, X.; Zheng, J.; Xue, Y.; Ma, J.; Li, Z.; Xi, Z.; Li, Z.; Bao, M.; Liu, Y. Long non-coding RNA taurine upregulated 1 enhances tumor-induced angiogenesis through inhibiting microRNA-299 in human glioblastoma. Oncogene 2017, 36, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, Y.; Zheng, Y. MiR-122/Wnt/β-catenin regulatory circuitry sustains glioma progression. Tumor Biol. 2014, 35, 8565–8572. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jin, J.G.; Mi, W.Y.; Wu, H.; Zhang, S.R.; Meng, Q.; Zhang, S.T. Long Non-coding RNA UCA1 Targets miR-122 to Promote Proliferation, Migration and Invasion of Glioma Cells. Oncol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhao, H.; Peng, R.; Liu, Q.; Yuan, J.; Peng, G.; Liao, Y. LncRNA-XIST interacts with miR-29c to modulate the chemoresistance of glioma cell to TMZ through DNA mismatch repair pathway. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xue, Y.; Wang, P.; Liu, X.; Ma, J.; Zheng, J.; Li, Z.; Li, Z.; Cai, H.; Liu, Y. Knockdown of long non-coding RNA XIST increases blood-tumor barrier permeability and inhibits glioma angiogenesis by targeting miR-137. Oncogenesis 2017, 6, e303. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Ma, J.; Xue, Y.; Wang, P.; Li, Z.; Liu, J.; Chen, L.; Xi, Z.; Teng, H.; Wang, Z.; et al. Knockdown of long non-coding RNA XIST exerts tumor-suppressive functions in human glioblastoma stem cells by up-regulating miR-152. Cancer Lett. 2015, 359, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Mashburn-Warren, L.M.; Whiteley, M. Special delivery: Vesicle trafficking in prokaryotes. Mol. Microbiol. 2006, 61, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Schiera, G.; Di Liegro, C.M.; Di Liegro, I. Extracellular Membrane Vesicles as Vehicles for Brain Cell-to-Cell Interactions in Physiological as well as Pathological Conditions. Biomed. Res. Int. 2015, 2015, 152926. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Mateescu, B.; Kowal, E.J.; van Balkom, B.W.; Bartel, S.; Bhattacharyya, S.N.; Buzás, E.I.; Buck, A.H.; de Candia, P.; Chow, F.W.; Das, S.; et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA-an ISEV position paper. J. Extracell. Vesicles 2017, 6, 1286095. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [PubMed]
- Blanc, L.; De Gassart, A.; Géminard, C.; Bette-Bobillo, P.; Vidal, M. Exosome release by reticulocytes–an integral part of the red blood cell differentiation system. Blood Cells Mol. Dis. 2005, 35, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Sharples, R.A.; Nisbet, R.M.; Cappai, R.; Hill, A.F. The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur. Biophys. J. 2008, 37, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Schiera, G.; Di Liegro, C.M.; Saladino, P.; Pitti, R.; Savettieri, G.; Proia, P.; Di Liegro, I. Oligodendroglioma cells synthesize the differentiation-specific linker histone H1° and release it into the extracellular environment through shed vesicles. Int. J. Oncol. 2013, 43, 1771–1776. [Google Scholar] [CrossRef] [PubMed]
- Lo Cicero, A.; Majkowska, I.; Nagase, H.; Di Liegro, I.; Troeberg, L. Microvesicles shed by oligodendroglioma cells and rheumatoid synovial fibroblasts contain aggrecanase activity. Matrix Biol. 2012, 31, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Di Liegro, C.M.; Schiera, G.; Di Liegro, I. Regulation of mRNA transport, localization and translation in the nervous system of mammals. Int. J. Mol. Med. 2014, 33, 747–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irion, U.; St. Johnston, D. Bicoid RNA localization requires specific binding of an endosomal sorting complex. Nature 2007, 445, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Christianson, H.C.; Svensson, K.J.; van Kuppevelt, T.H.; Li, J.P.; Belting, M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [PubMed]
- Penfornis, P.; Vallabhaneni, K.C.; Whitt, J.; Pochampally, R. Extracellular vesicles as carriers of microRNA, proteins and lipids in tumor microenvironment. Int. J. Cancer 2016, 138, 14–21. [Google Scholar] [CrossRef] [PubMed]
- D’Asti, E.; Gamier, D.; Lee, T.H.; Montermini, L.; Meehan, B.; Rak, J. Oncogenic extracellular vesicles in brain tumor progression. Front. Physiol. 2012, 3, 294. [Google Scholar] [CrossRef] [PubMed]
- D’Asti, E.; Chennakrishnaiah, S.; Lee, T.H.; Rak, J. Extracellular Vesicles in Brain Tumor Progression. Cell. Mol. Neurobiol. 2016, 36, 383–407. [Google Scholar] [CrossRef] [PubMed]
- Nakano, I.; Garnier, D.; Minata, M.; Rak, J. Extracellular vesicles in the biology of brain tumor stem cells—Implications for inter-cellular communication, therapy and biomarker development. Semin. Cell Dev. Biol. 2015, 40, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Giusti, I.; Delle Monache, S.; Di Francesco, M.; Sanità, P.; D’Ascenzo, S.; Gravina, G.L.; Festuccia, C.; Dolo, V. From glioblastoma to endothelial cells through extracellular vesicles: Messages for angiogenesis. Tumor Biol. 2016, 37, 12743–12753. [Google Scholar] [CrossRef] [PubMed]
- Giusti, I.; Di Francesco, M.; Dolo, V. Extracellular Vesicles in Glioblastoma: Role in Biological Processes and in Therapeutic Applications. Curr. Cancer Drug Targets 2017, 17, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Gourlay, J.; Morokoff, A.P.; Luwor, R.B.; Zhu, H.-J.; Kaye, A.H.; Stylli, S.S. The emergent role of exosomes in glioma. J. Clin. Neurosci. 2017, 35, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Mallawaaratchy, D.M.; Hallal, S.; Russell, B.; Ly, L.; Ebrahimkhani, S.; Wei, H.; Christopherson, R.I.; Buckland, M.E.; Kaufman, K.L. Comprehensive proteome profiling of glioblastoma-derived extracellular vesicles identifies markers for more aggressive disease. J. Neuro Oncol. 2017, 131, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Ahn, S.; Yeom, C.H.; Park, S. Exosome Proteome of U-87MG Glioblastoma Cells. Biol. (Basel) 2016, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.E.; Iwadate, Y.; Machida, T.; Hiwasa, T.; Nimura, Y.; Nagai, Y.; Takiguchi, M.; Tanzawa, H.; Yamaura, A.; Seki, N. Cathepsin D is a potential serum marker for poor prognosis in glioma patients. Cancer Res. 2005, 65, 5190–5194. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, C.; Cavallini, L.; Spinelli, C.; Yang, J.; Reis-Sobreiro, M.; de Candia, P.; Minciacchi, V.R.; Di Vizio, D. Focus on Extracellular Vesicles: New Frontiers of Cell-to-Cell Communication in Cancer. Int. J. Mol. Sci. 2016, 17, 175. [Google Scholar] [CrossRef] [PubMed]
- Antonyak, M.A.; Li, B.; Boroughs, L.K.; Johnson, J.L.; Druso, J.E.; Bryant, K.L.; Holowka, D.A.; Cerione, R.A. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4852–4857. [Google Scholar] [CrossRef] [PubMed]
- Trylcova, J.; Busek, P.; Smetana, K., Jr.; Balaziova, E.; Dvorankova, B.; Mifkova, A.; Sedo, A. Effect of cancer-associated fibroblasts on the migration of glioma cells in vitro. Tumor Biol. 2015, 36, 5873–5879. [Google Scholar] [CrossRef] [PubMed]
- De Vrij, J.; Maas, S.L.; Kwappenberg, K.M.; Schnoor, R.; Kleijn, A.; Dekker, L.; Luider, T.M.; de Witte, L.D.; Litjens, M.; van Strien, M.E.; et al. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int. J. Cancer 2015, 137, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Treps, L.; Edmond, S.; Harford-Wright, E.; Galan-Moya, E.M.; Schmitt, A.; Azzi, S.; Citerne, A.; Bidère, N.; Ricard, D.; Gavard, J. Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene 2016, 19, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringnér, M.; Mörgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef] [PubMed]
- Pen, A.; Moreno, M.J.; Martin, J.; Stanimirovic, D.B. Molecular markers of extracellular matrix remodeling in glioblastoma vessels: Microarray study of laser-captured glioblastoma vessels. Glia 2007, 55, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Arscott, W.T.; Tandle, A.T.; Zhao, S.; Shabason, J.E.; Gordon, I.K.; Schlaff, C.D.; Zhang, G.; Tofilon, P.J.; Camphausen, K.A. Ionizing radiation and glioblastoma exosomes: Implications in tumor biology and cell migration. Transl. Oncol. 2013, 6, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Baulch, J.E.; Geidzinski, E.; Tran, K.K.; Yu, L.; Zhou, Y.H.; Limoli, C.L. Irradiation of primary human gliomas triggers dynamic and aggressive survival responses involving microvesicle signaling. Environ. Mol. Mutagen. 2016, 57, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Luhtala, N.; Aslanian, A.; Yates, J.R., 3rd; Hunter, T. Secreted Glioblastoma Nanovesicles Contain Intracellular Signaling Proteins and Active Ras Incorporated in a Farnesylation-dependent Manner. J. Biol. Chem. 2017, 292, 611–628. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.; Clippinger, A.K.; Talledge, N.; Skowyra, M.L.; Saunders, M.G.; Naismith, T.V.; Colf, L.A.; Afonine, P.; Arthur, C.; Sundquist, W.I.; et al. Structure and membrane remodeling activity of ESCRT-III helical polymers. Science 2015, 350, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Costa Verdera, H.; Gitz-Francois, J.J.; Schiffelers, R.M.; Vader, P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J. Control. Release 2017, 266, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zheng, H.; Wang, C.; Lu, X.; Zhao, X.; Li, X. Insight into HOTAIR structural features and functions as landing pads for transcription regulation proteins. Biochem. Biophys. Res. Commun. 2017, 485, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Fareh, M.; Almairac, F.; Turchi, L.; Burel-Vandenbos, F.; Paquis, P.; Fontaine, D.; Lacas-Gervais, S.; Junier, M.P.; Chneiweiss, H.; Virolle, T. Cell-based therapy using miR-302–367 expressing cells represses glioblastoma growth. Cell Death Dis. 2017, 8, e2713. [Google Scholar] [CrossRef] [PubMed]
- Van der Vos, K.E.; Balaj, L.; Skog, J.; Breakefield, X.O. Brain tumor microvesicles: Insights into intercellular communication in the nervous system. Cell. Mol. Neurobiol. 2011, 31, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef] [PubMed]
- Van der Vos, K.E.; Abels, E.R.; Zhang, X.; Lai, C.; Carrizosa, E.; Oakley, D.; Prabhakar, S.; Mardini, O.; Crommentuijn, M.H.; Skog, J.; et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol. 2016, 18, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Schiera, G.; Di Liegro, C.M.; Puleo, V.; Colletta, O.; Fricano, A.; Cancemi, P.; Di Cara, G.; Di Liegro, I. Extracellular vesicles shed by melanoma cells contain a modified form of H1.0 linker histone and H1.0 mRNA-binding proteins. Int. J. Oncol. 2016, 49, 1807–1814. [Google Scholar] [CrossRef] [PubMed]


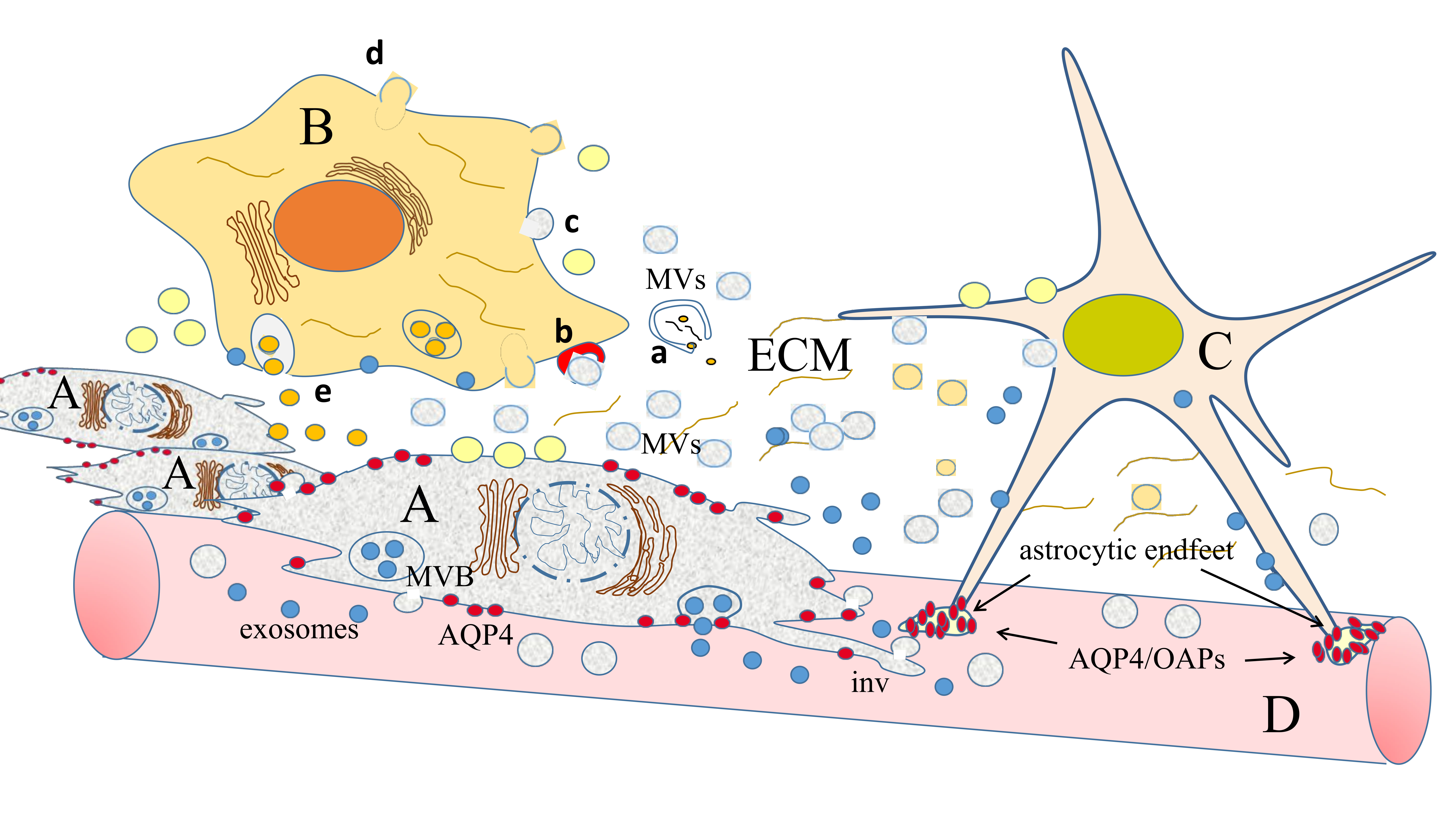{kind=link}
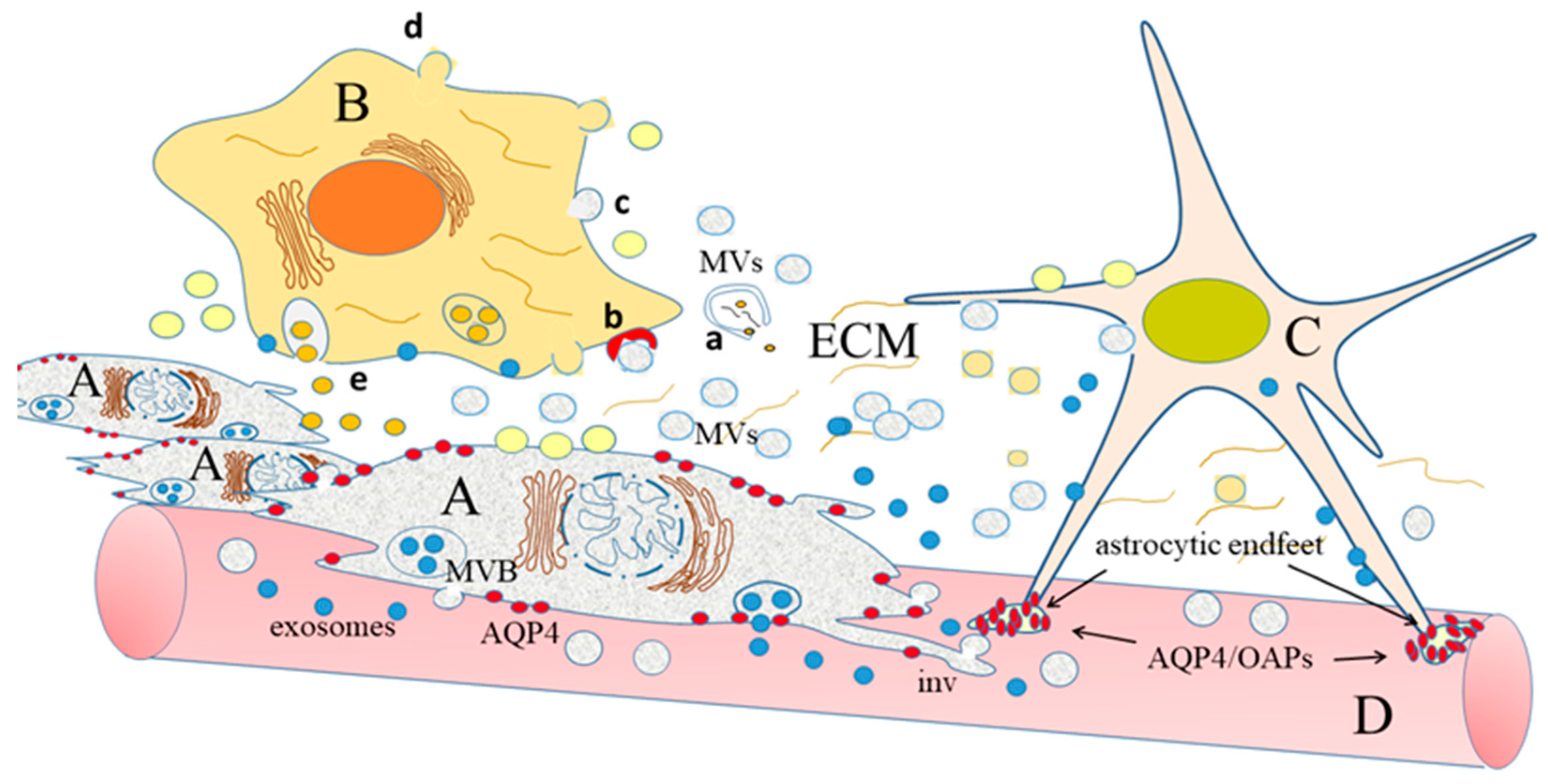{kind=link}
| Factor | Function | Up/Down Regulated in Glioma [References] | Therapies Targeting These Proteins [References] |
|---|---|---|---|
| ADAMs 8, 9, 10, 17, 19 | extracellular disintegrin and metalloproteases | up-regulated [39] | ADAM 10 and 17 [40] |
| ADAM-22 | inhibitor of astrocyte proliferation | downregulated in high-grade gliomas [41] | over-expression of miR-145 targets, among other genes, also ADAM-22 [42] |
| ADAMTS-4 and ADAMTS-5 | degrade lectican and small leucine-rich repeat families of proteoglycans | expression correlates with glioma invasiveness [43] | no example of specific targeting found |
| Cathepsin B and D | extracellular proteases | upregulated in high-grade gliomas [44,45,46,47] | tivozanib diminished glioblastoma multiforme (GBM) cell invasion by impairing the proteolytic cascade of cathepsin B/urokinase-type plasminogen activator (uPA)/matrix metalloproteinase-2 (MMP-2) [48] |
| CCN1 | heparin-binding protein; interacts with the integrins α-v β-3 and α-6β-1 and increases the migration of glioma cells | highly up-regulated in primary gliomas and invasive glioblastoma cell lines [49] | potential therapy based on oncolytic HSV1 (OV) [50] |
| Collagen Type IV | the major structural component of basement membranes | up-regulated [51] | the lysyl oxidase inhibitor β-aminopropionitrile disrupts collagen structure in the tumor and inhibits tumor angiogenesis and glioblastoma multiforme growth in a mouse orthotopic brain tumor model [52] |
| Hyaluronan | the major component of the brain ECM | up-regulated in primary brain tumors [53] It stimulates secretion of matrix metalloproteases | hyaluronidase can improve penetration of therapeutic agents into brain tumors [54] |
| Matrix metalloproteinase (MMP)-1 | interstitial collagenase | expression increases with WHO grade [55] | a collection of new drugs targeting matrix metalloproteases have been tested in vitro. Among them: 2-Amino-2-[2-(4-octylphenyl)]-1,3-propanediol hydrochloride (FTY720) [56], chlorotoxin [57], ellagic acid [58], fucoxanthin [59], caffeic acid [60]. Moreover, silencing of specific genes appears as a promising tool for inhibiting growth and invasiveness of glioma cells, by reducing expression of matrix metalloproteases [61,62] |
| MMP-2 | gelatinase activity | highly upregulated, secreted, activated [63,64,65] | |
| MMP-3 | broad substrate specifity | highly upregulated, secreted, activated [66] | |
| MMP-7 | broad substrate specifity | highly upregulated, secreted, activated [67] | |
| MMP-9 | gelatinase activity | highly upregulated, secreted, activated [64,65] | |
| MMP-11 | does not degrade laminin, fibronectin and elastin; has a strong activity on serine protease inhibitor α1-antitrypsin and insulin-like growth factor binding protein-1 (IGFBP-1) | expression increases with WHO grade [55] | |
| MMP-12 | degrades soluble and insoluble elastin, type IV collagen, fibronectin, fibrillin-1, laminin, vitronectin, chondroitin sulfate and heparin sulfate proteoglycans, MMP2/3 activation | elevation of MMP-12 by tenascin-C in glioma [68] | |
| MMP-19 | degrades various ECM components including collagen type IV, nidogen-1, fibronectin, tenascin-C isoform, aggrecan and laminin-5-gamma-2-chain | expression increases with WHO grade [55] | |
| MMP-26 | degrades type IV collagen, fibronectin, vitronectin, alpha 1-antitrypsin (A1AT), insulin-like growth factor-binding protein 1 (IGFBP) and activates MMP9 | significantly up-regulated [69] | |
| (MT1)-MMP/MMP-14 | involved in the maturation of active MMP-2 | highly upregulated, secreted, activated [70] | no example of specific targeting found |
| Tenascin-C | plays a crucial role in angiogenesis, proliferation and cell migration | up-regulated [71,72] | a peptide that bound to tenascin C has been isolated by phage display peptide library. The selected peptide specifically recognized tenascin C protein in xenograft mouse tissue [73] |
| Tenascin-R | influences cell adhesion, neural cell migration, cell-matrix interaction and axon outgrowth | increasingly down-regulated with glioma progression: (in grade IV glioblastoma only a weak TN-R expression is detected [72] | no example of specific targeting found |
| TIMP-1 | natural inhibitor of MMPs | higher levels in GBM compared to lower grade glioma [74] | 2-Amino-2-[2-(4-octylphenyl)]-1,3-propanediol hydrochloride (FTY720) [56] |
| Thrombospondin 1 (TSP-1) | Implicated in cancer cell, adhesion, migration, invasion, inhibition of angiogenesis | may decrease with tumor grade [75] | no example of specific induction found |
| miRNA | Proposed Mode of Action | Some Proposed Targets [References] | Presence in EVs [References] |
|---|---|---|---|
| miR-1 | tumor suppressor | Annexin A2 [211] | [10,212] |
| miR-7 | tumor suppressor | EGFR, FAK, IRS1/2 [213,214] | - |
| miR-9 | oncogenic | Stathmin [81] | Found in EVs from breast cancer cell lines [215] |
| miR-10b | oncogenic | UPAR, RhoC [216] | [10] |
| miR-16 | tumor suppressor | BCL2, WIP1-ATM-p53 pathway [217,218] | [219] |
| miR-21 | oncogenic | TIMP3, RECK4, PDCD4, β-catenin [220,221,222] | [13,212,223] |
| miR-26a | oncogenic | PTEN, Rb, MEKK2 [224] | [212] |
| miR-26b | tumor suppressor | BCL2 [225] | [212] |
| miR-29 | tumor suppressor | DNMT3A and 3B. [226] | - |
| miR-29a | oncogenic | PTEN [223] | [223] |
| miR-30e | oncogenic | NFkB, VEGF-C, MMPs [223] | [223] |
| miR-34a | tumor suppressor | PKCε, PD-L1 [227,228] | - |
| miR-93 | oncogenic | Integrin β8 [229,230] | [212] |
| miR-98 | tumor suppressor | IKK-ε [231] | - |
| miR-124 | tumor suppressor | AURKA, Smad4 [232,233] | [224] |
| miR-128 | tumor suppressor | EGFR, PDGFRA, EphB2, p70S6K PRC1, PRC2 (reduces levels of phospho-Akt and derepresses p21 expression) [224,234] | [10,13,224] |
| miR-130b | oncogenic | CYLD [235] | Found in EVs from prostate cancers [236] |
| miR-141 | tumor suppressor | SKA2 [237] | - |
| miR-142 | tumor suppressor | Rac1 [238] | - |
| miR-146b | tumor suppressor | MMPs [239,240] | [10,241] |
| miR-152-3p | tumor suppressor | DNMT1 [242] | - |
| miR-181 | tumor suppressor | Bcl-2, KPNA4 [243,244] | - |
| miR-200c | tumor suppressor | EGFR, AKT [245] | - |
| miR-210 | oncogenic | Glycerol-3-phosphate dehydrogenase 1-like; increased levels of HIF3A and of VEGF [13] | [13] |
| miR-218 | tumor suppressor | IKK-β, Bmi1, RTK-HIF pathway [246,247,248] | - |
| miR-221/222 | oncogenic | TIMP2, SEMA3B [249,250,251] | [13,223] |
| miR-296 | oncogenic | HGS, STAT5A [252,253] | - |
| miR-320 | oncogenic | [241] | |
| miR-326 | tumor suppressor | SMO, Notch2, NOB1 [254,255] | - |
| miR-370 | tumor suppressor | beta-catenin, CCNE2 [256,257] | - |
| miR-451 | tumor suppressor | Akt1, CyclinD1, MMP-2, MMP-9 and Bcl-2, LKB1 [258,259,260] | [13,261] |
| miR-592 | tumor suppressor | IGFBP2 [262] | - |
| miR-5096 | oncogenic | Kir4.1 [160] | [223] |
| LncRNA | Proposed Mode of Action | Some Proposed Targets |
|---|---|---|
| ADAMTS-AS2 | tumor suppressor | DNMT1 [265] |
| CASC 2 | tumor suppressor | miR-21 [268] |
| CRNDE | oncogenic | miR-186, miR-384/PIWIL4 [269,270] |
| GAS 5 | tumor suppressor | miR-196a, miR-222 [263,266,267] |
| H19 | oncogenic (generates miR-675) | Cadherin 13 (CDH13) [271,272,273] |
| HOTAIR | oncogenic | PDCD4, miR-141, SNORD47 [237,263,264,274] |
| HOTTIP | oncogenic | miR-101 [275] |
| HULC | oncogenic | ESM-1; PI3K/AKT/mTOR [276] |
| KCNQ1OT1 | oncogenic | miR-370 [257] |
| LINC0000125 | oncogenic | miR-4775 [277] |
| LINC-POU3F3 | oncogenic | POU3F3; bFGF, bFGFR, VEGFA [278,279] |
| LINK-A | oncogenic | LDH-A [280] |
| MALAT-1(NEAT-2) | oncogenic | miR-101 [281] |
| PLAC 2 | tumor suppressor | ribosomal protein (RP)L36, STAT1 [282] |
| TUG1 | tumor suppressor | miR-26a, miR-144, miR-299 [283,284,285] |
| UCA1 | oncogenic | miR-122 [286,287] |
| XIST | oncogenic | miR-29c, miR-137, miR-152 [288,289,290] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiera, G.; Di Liegro, C.M.; Di Liegro, I. Molecular Determinants of Malignant Brain Cancers: From Intracellular Alterations to Invasion Mediated by Extracellular Vesicles. Int. J. Mol. Sci. 2017, 18, 2774. https://doi.org/10.3390/ijms18122774
Schiera G, Di Liegro CM, Di Liegro I. Molecular Determinants of Malignant Brain Cancers: From Intracellular Alterations to Invasion Mediated by Extracellular Vesicles. International Journal of Molecular Sciences. 2017; 18(12):2774. https://doi.org/10.3390/ijms18122774
Chicago/Turabian StyleSchiera, Gabriella, Carlo Maria Di Liegro, and Italia Di Liegro. 2017. "Molecular Determinants of Malignant Brain Cancers: From Intracellular Alterations to Invasion Mediated by Extracellular Vesicles" International Journal of Molecular Sciences 18, no. 12: 2774. https://doi.org/10.3390/ijms18122774