Control of Nucleotide Metabolism Enables Mutant p53’s Oncogenic Gain-of-Function Activity

{kind=link}
{kind=link}
Abstract
:1. Mutant p53’s Oncogenic Gain-of-Function
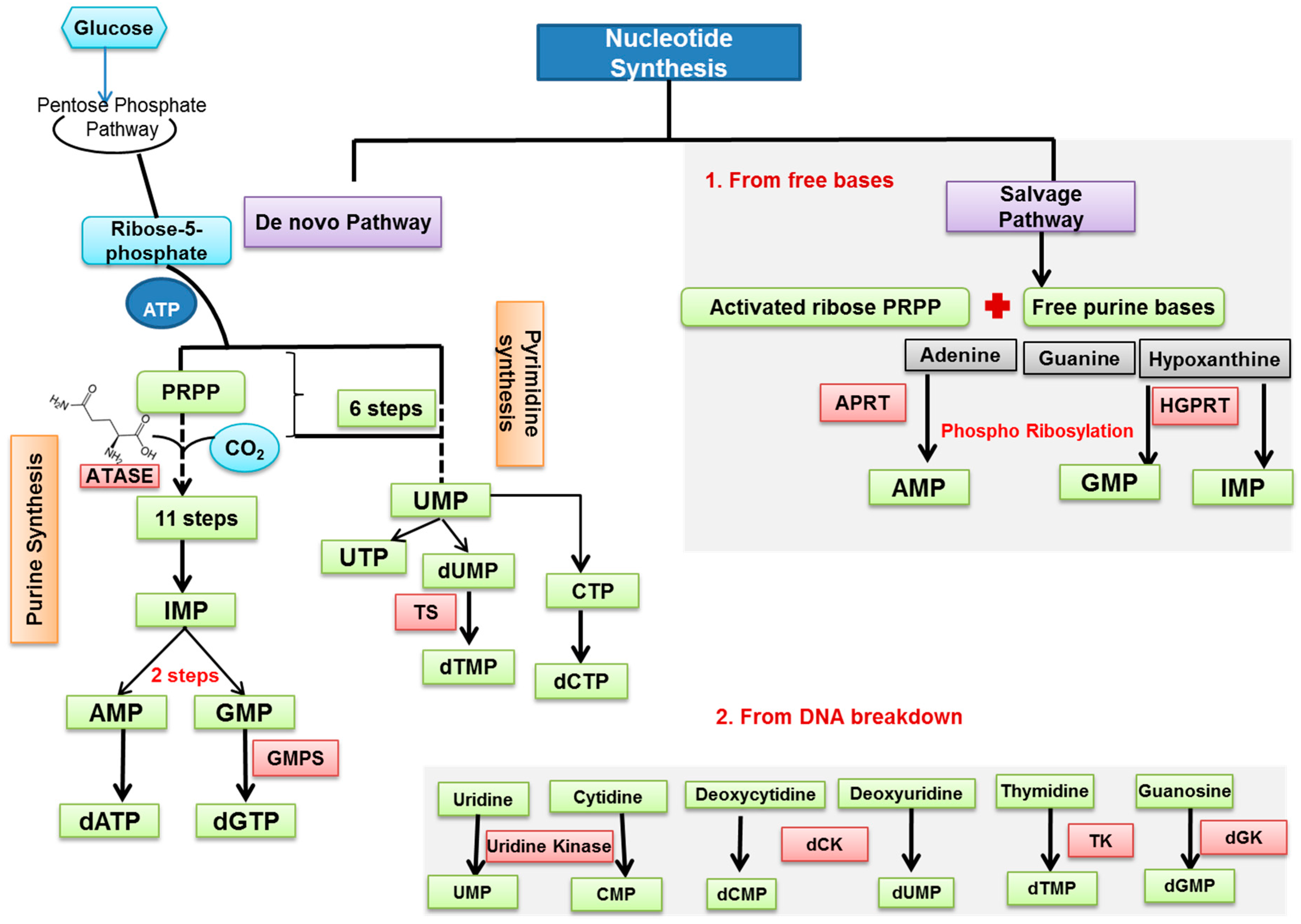
2. Nucleotide Metabolism and Its Regulation
3. Control of Nucleotide Metabolism by Mtp53
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | APC, WNT signaling pathway regulator |
| RB1 | RB transcriptional corepressor 1 |
| PDGFRβ | Platelet-derived growth factor receptor β |
| NF-Y | Trimeric complex composed of NFYA, NFYB, NFYC |
| SP1 | SP1 transcription factor |
| SREBP | Sterol regulatory element binding protein |
| VDR | Vitamin D receptor |
| ETS1 | ETS proto-oncogene 1, transcription factor |
| ETS2 | ETS proto-oncogene 2, transcription factor |
| GLUT1 | SLC2A1, solute carrier 2, member 1 |
| AMPK | PRKAA1, protein kinase AMP-activated catalytic subunit alpha 1 |
| cGAMP | cyclic GMP-AMP |
| ENTPD5 | Ectonucleoside triphosphate diphosphohydrolase 5 |
References
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Humpton, T.J.; Vousden, K.H. Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Li, F.P.; Fraumeni, J.F., Jr. Rhabdomyosarcoma in children: Epidemiologic study and identification of a familial cancer syndrome. J. Natl. Cancer Inst. 1969, 43, 1365–1373. [Google Scholar] [PubMed]
- Li, F.P.; Fraumeni, J.F., Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann. Int. Med. 1969, 71, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Adv. Cancer Res. 2011, 110, 107–139. [Google Scholar] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell. Biol. 1999, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Zhang, Y.; Lozano, G. Mutant p53: Multiple Mechanisms Define Biologic Activity in Cancer. Front. Oncol. 2015, 5, 249. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Deppert, W. Interactions of mutant p53 with DNA: Guilt by association. Oncogene 2007, 26, 2185–2190. [Google Scholar] [CrossRef] [PubMed]
- Adamik, M.; Kejnovska, I.; Bazantova, P.; Petr, M.; Renciuk, D.; Vorlickova, M.; Brazdova, M. p53 binds human telomeric G-quadruplex in vitro. Biochimie 2016, 128–129, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Brazdova, M.; Navratilova, L.; Tichy, V.; Nemcova, K.; Lexa, M.; Hrstka, R.; Pecinka, P.; Adamik, M.; Vojtesek, B.; Palecek, E.; et al. Preferential binding of hot spot mutant p53 proteins to supercoiled DNA in vitro and in cells. PLoS ONE 2013, 8, e59567. [Google Scholar] [CrossRef] [PubMed]
- Brazdova, M.; Tichy, V.; Helma, R.; Bazantova, P.; Polaskova, A.; Krejci, A.; Petr, M.; Navratilova, L.; Ticha, O.; Nejedly, K.; et al. p53 Specifically Binds Triplex DNA In Vitro and in Cells. PLoS ONE 2016, 11, e0167439. [Google Scholar] [CrossRef] [PubMed]
- Petr, M.; Helma, R.; Polaskova, A.; Krejci, A.; Dvorakova, Z.; Kejnovska, I.; Navratilova, L.; Adamik, M.; Vorlickova, M.; Brazdova, M. Wild-type p53 binds to MYC promoter G-quadruplex. Biosci. Rep. 2016, 36, e00397. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Coufal, J. Recognition of Local DNA Structures by p53 Protein. Int. J. Mol. Sci. 2017, 18, 375. [Google Scholar] [CrossRef] [PubMed]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.T.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Tu, H.; Kollareddy, M.; Pant, V.; Li, Q.; Zhang, Y.; Jackson, J.G.; Suh, Y.A.; Elizondo-Fraire, A.C.; Yang, P.; et al. Pla2g16 phospholipase mediates gain-of-function activities of mutant p53. Proc. Natl. Acad. Sci. USA 2014, 111, 11145–11150. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Chicas, A.; Molina, P.; Bargonetti, J. Mutant p53 forms a complex with Sp1 on HIV-LTR DNA. Biochem. Biophys. Res. Commun. 2000, 279, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Stambolsky, P.; Tabach, Y.; Fontemaggi, G.; Weisz, L.; Maor-Aloni, R.; Siegfried, Z.; Shiff, I.; Kogan, I.; Shay, M.; Kalo, E.; et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 2010, 17, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Sampath, J.; Sun, D.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Gunther, W.; Yoshizato, K.; Meissner, H.; Zapf, S.; Nusing, R.M.; Yamamoto, H.; Van Meir, E.G.; Deppert, W.; Giese, A. Tumor suppressor p53 inhibits transcriptional activation of invasion gene thromboxane synthase mediated by the proto-oncogenic factor ets-1. Oncogene 2003, 22, 7716–7727. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Marchenko, N.D. Mutant p53—Heat Shock Response Oncogenic Cooperation: A New Mechanism of Cancer Cell Survival. Front. Endocrinol. 2015, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Do, P.M.; Varanasi, L.; Fan, S.; Li, C.; Kubacka, I.; Newman, V.; Chauhan, K.; Daniels, S.R.; Boccetta, M.; Garrett, M.R.; et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012, 26, 830–845. [Google Scholar]
- Carrero, Z.I.; Kollareddy, M.; Chauhan, K.M.; Ramakrishnan, G.; Martinez, L.A. Mutant p53 protects ETS2 from non-canonical COP1/DET1 dependent degradation. Oncotarget 2016, 7, 12554–12567. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.A. Mutant p53 and ETS2, a Tale of Reciprocity. Front. Oncol. 2016, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, W.; Huang, P.; Xu, Y.Z.; Heinemann, V.; Grunewald, R.; Gandhi, V. Gemcitabine: Metabolism, mechanisms of action, and self-potentiation. Semin. Oncol. 1995, 22, 3–10. [Google Scholar] [PubMed]
- Evans, W.E. Pharmacogenetics of thiopurine S-methyltransferase and thiopurine therapy. Ther. Drug Monit. 2004, 26, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Reichard, P. Interactions between deoxyribonucleotide and DNA synthesis. Ann. Rev. Biochem. 1988, 57, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.; Eriksson, S. Mammalian deoxyribonucleoside kinases. Pharmacol. Ther. 1995, 67, 155–186. [Google Scholar] [CrossRef]
- Toy, G.; Austin, W.R.; Liao, H.I.; Cheng, D.; Singh, A.; Campbell, D.O.; Ishikawa, T.O.; Lehmann, L.W.; Satyamurthy, N.; Phelps, M.E.; et al. Requirement for deoxycytidine kinase in T and B lymphocyte development. Proc. Natl. Acad. Sci. USA 2010, 107, 5551–5556. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Voit, E.O. Identification of cancer mechanisms through computational systems modeling. Trans. Cancer Res. 2014, 3, 233–242. [Google Scholar]
- Zauri, M.; Berridge, G.; Thezenas, M.L.; Pugh, K.M.; Goldin, R.; Kessler, B.M.; Kriaucionis, S. CDA directs metabolism of epigenetic nucleosides revealing a therapeutic window in cancer. Nature 2015, 524, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, P.; Reichard, P. Ribonucleotide reductases. Ann. Rev. Biochem. 2006, 75, 681–706. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, Y.; Eriksson, S.; Jildevik, I.; Skog, S.; Thelander, L.; Tribukait, B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J. Biol. Chem. 1985, 260, 9114–9116. [Google Scholar] [PubMed]
- Hakansson, P.; Hofer, A.; Thelander, L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J. Biol. Chem. 2006, 281, 7834–7841. [Google Scholar] [CrossRef] [PubMed]
- Aird, K.M.; Zhang, R. Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett. 2015, 356, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Jones, T.L.; Martin, S.E.; Caplen, N.J.; Pommier, Y. Implication of checkpoint kinase-dependent up-regulation of ribonucleotide reductase R2 in DNA damage response. J. Biol. Chem. 2009, 284, 18085–18095. [Google Scholar] [CrossRef] [PubMed]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, O.; Chicas, A.; Zeng, T.; Zhao, Z.; McCurrach, M.; Wang, X.; Lowe, S.W. The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence. Genes Dev. 2012, 26, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Aird, K.M.; Li, H.; Xin, F.; Konstantinopoulos, P.A.; Zhang, R. Identification of ribonucleotide reductase M2 as a potential target for pro-senescence therapy in epithelial ovarian cancer. Cell Cycle 2014, 13, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Mannava, S.; Moparthy, K.C.; Wheeler, L.J.; Leonova, K.I.; Wawrzyniak, J.A.; Bianchi-Smiraglia, A.; Berman, A.E.; Flanagan, S.; Shewach, D.S.; Zeitouni, N.C.; et al. Ribonucleotide reductase and thymidylate synthase or exogenous deoxyribonucleosides reduce DNA damage and senescence caused by C-MYC depletion. Aging 2012, 4, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Mannava, S.; Moparthy, K.C.; Wheeler, L.J.; Natarajan, V.; Zucker, S.N.; Fink, E.E.; Im, M.; Flanagan, S.; Burhans, W.C.; Zeitouni, N.C.; et al. Depletion of deoxyribonucleotide pools is an endogenous source of DNA damage in cells undergoing oncogene-induced senescence. Am. J. Pathol. 2013, 182, 142–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Lamm, N.; Maoz, K.; Bester, A.C.; Im, M.M.; Shewach, D.S.; Karni, R.; Kerem, B. Folate levels modulate oncogene-induced replication stress and tumorigenicity. EMBO Mol. Med. 2015, 7, 1138–1152. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Moreno, M.V.; Lodi, A.; Ronen, S.M.; Ruggero, D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 2014, 157, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Angus, S.P.; Wheeler, L.J.; Ranmal, S.A.; Zhang, X.; Markey, M.P.; Mathews, C.K.; Knudsen, E.S. Retinoblastoma tumor suppressor targets dNTP metabolism to regulate DNA replication. J. Biol. Chem. 2002, 277, 44376–44384. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Li, F.; Handler, J.; Huang, C.R.; Xiang, Y.; Neretti, N.; Sedivy, J.M.; Zeller, K.I.; Dang, C.V. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS ONE 2008, 3, e2722. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Fan, T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucl. Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Balint, E.; Ashcroft, M.; Vousden, K.H. A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene 2000, 19, 4283–4289. [Google Scholar]
- Kollareddy, M.; Dimitrova, E.; Vallabhaneni, K.C.; Chan, A.; Le, T.; Chauhan, K.M.; Carrero, Z.I.; Ramakrishnan, G.; Watabe, K.; Haupt, Y.; et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat. Commun. 2015, 6, 7389. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.N.; Ivanov, A.V.; Kravchenko, J.E.; Kopnin, B.P.; Levine, A.J.; Chumakov, P.M. Novel gain of function activity of p53 mutants: Activation of the dUTPase gene expression leading to resistance to 5-fluorouracil. Oncogene 2002, 21, 4595–4600. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ramirez, A.S.; Diaz-Munoz, M.; Butanda-Ochoa, A.; Vazquez-Cuevas, F.G. Nucleotides and nucleoside signaling in the regulation of the epithelium to mesenchymal transition (EMT). Purinergic Signal. 2017, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De Andrade Mello, P.; Coutinho-Silva, R.; Savio, L.E.B. Multifaceted Effects of Extracellular Adenosine Triphosphate and Adenosine in the Tumor-Host Interaction and Therapeutic Perspectives. Front. Immunol. 2017, 8, 1526. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ramirez, A.S.; Garay, E.; Garcia-Carranca, A.; Vazquez-Cuevas, F.G. The P2RY2 Receptor Induces Carcinoma Cell Migration and EMT Through Cross-Talk With Epidermal Growth Factor Receptor. J. Cell. Biochem. 2016, 117, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Liu, Y.; Yang, H.; Zhang, H.Q.; Tian, X.X.; Fang, W.G. ATP-P2Y2-β-catenin axis promotes cell invasion in breast cancer cells. Cancer Sci. 2017, 108, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Beavis, P.A.; Darcy, P.K.; Stagg, J. Immunosuppressive activities of adenosine in cancer. Curr. Opin. Pharmacol. 2016, 29, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [PubMed]
- Robson, S.C.; Sevigny, J.; Zimmermann, H. The E-NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signal. 2006, 2, 409–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogiatzi, F.; Brandt, D.T.; Schneikert, J.; Fuchs, J.; Grikscheit, K.; Wanzel, M.; Pavlakis, E.; Charles, J.P.; Timofeev, O.; Nist, A.; et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc. Natl. Acad. Sci. USA 2016, 113, E8433–E8442. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, E.S.; Helenius, A. Glycoprotein reglucosylation and nucleotide sugar utilization in the secretory pathway: Identification of a nucleoside diphosphatase in the endoplasmic reticulum. EMBO J. 1999, 18, 3282–3292. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Shen, Z.; Huang, S.; Zhao, L.; Chen, S.; Mak, T.W.; Wang, X. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 2010, 143, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Khramtsova, S.; Stromskaya, T.; Potapova, G.; Chumakov, P.; Kopnin, B. Human p53, mutated at codon 273, causes distinct effects on nucleotide biosynthesis salvage pathway key enzymes in Rat-1 cells and in their derivatives expressing activated ras oncogene. Biochem. Biophys. Res. Commun. 1993, 194, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Frazier, M.W.; He, X.; Wang, J.; Gu, Z.; Cleveland, J.L.; Zambetti, G.P. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol. Cell. Biol. 1998, 18, 3735–3743. [Google Scholar] [CrossRef] [PubMed]
- Wawrzyniak, J.A.; Bianchi-Smiraglia, A.; Bshara, W.; Mannava, S.; Ackroyd, J.; Bagati, A.; Omilian, A.R.; Im, M.; Fedtsova, N.; Miecznikowski, J.C.; et al. A purine nucleotide biosynthesis enzyme guanosine monophosphate reductase is a suppressor of melanoma invasion. Cell Rep. 2013, 5, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Bar-Sagi, D.; Hall, A. Ras and Rho GTPases: A family reunion. Cell 2000, 103, 227–238. [Google Scholar] [CrossRef]
- Bossi, G.; Marampon, F.; Maor-Aloni, R.; Zani, B.; Rotter, V.; Oren, M.; Strano, S.; Blandino, G.; Sacchi, A. Conditional RNA interference in vivo to study mutant p53 oncogenic gain of function on tumor malignancy. Cell Cycle 2008, 7, 1870–1879. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.V.; Parrales, A.; Begani, P.; Narkar, A.; Adhikari, A.S.; Martinez, L.A.; Iwakuma, T. Allele-specific silencing of mutant p53 attenuates dominant-negative and gain-of-function activities. Oncotarget 2016, 7, 5401–5415. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.A.; Naguibneva, I.; Lehrmann, H.; Vervisch, A.; Tchenio, T.; Lozano, G.; Harel-Bellan, A. Synthetic small inhibiting RNAs: Efficient tools to inactivate oncogenic mutations and restore p53 pathways. Proc. Natl. Acad. Sci. USA 2002, 99, 14849–14854. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, V.; Nagar, R.; Martinez, L.A. Control of Nucleotide Metabolism Enables Mutant p53’s Oncogenic Gain-of-Function Activity. Int. J. Mol. Sci. 2017, 18, 2759. https://doi.org/10.3390/ijms18122759
Schmidt V, Nagar R, Martinez LA. Control of Nucleotide Metabolism Enables Mutant p53’s Oncogenic Gain-of-Function Activity. International Journal of Molecular Sciences. 2017; 18(12):2759. https://doi.org/10.3390/ijms18122759
Chicago/Turabian StyleSchmidt, Valentina, Rachana Nagar, and Luis A. Martinez. 2017. "Control of Nucleotide Metabolism Enables Mutant p53’s Oncogenic Gain-of-Function Activity" International Journal of Molecular Sciences 18, no. 12: 2759. https://doi.org/10.3390/ijms18122759