
Inhibitory Effect of 2,3,5,6-Tetrafluoro-4-[4-(aryl)-1H-1,2,3-triazol-1-yl]benzenesulfonamide Derivatives on HIV Reverse Transcriptase Associated RNase H Activities

 , , , , , ,
, , , , , ,  , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
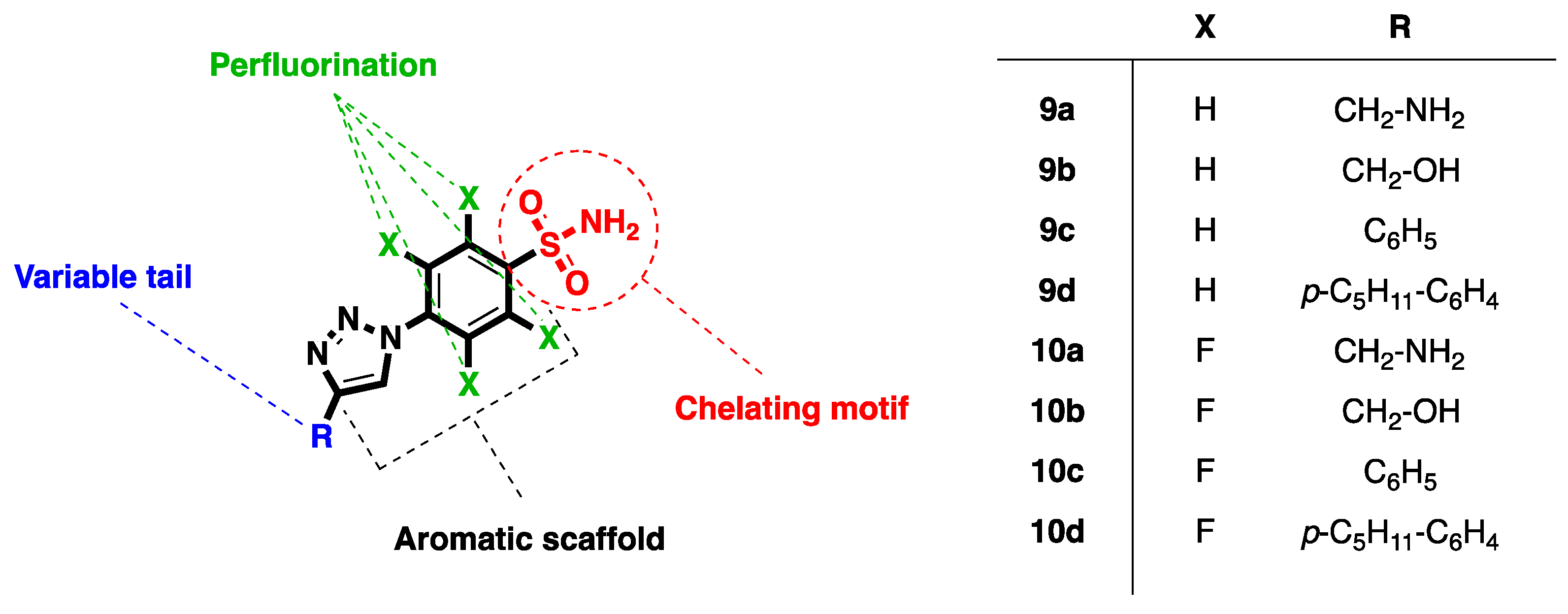
2.1. Compound Identification
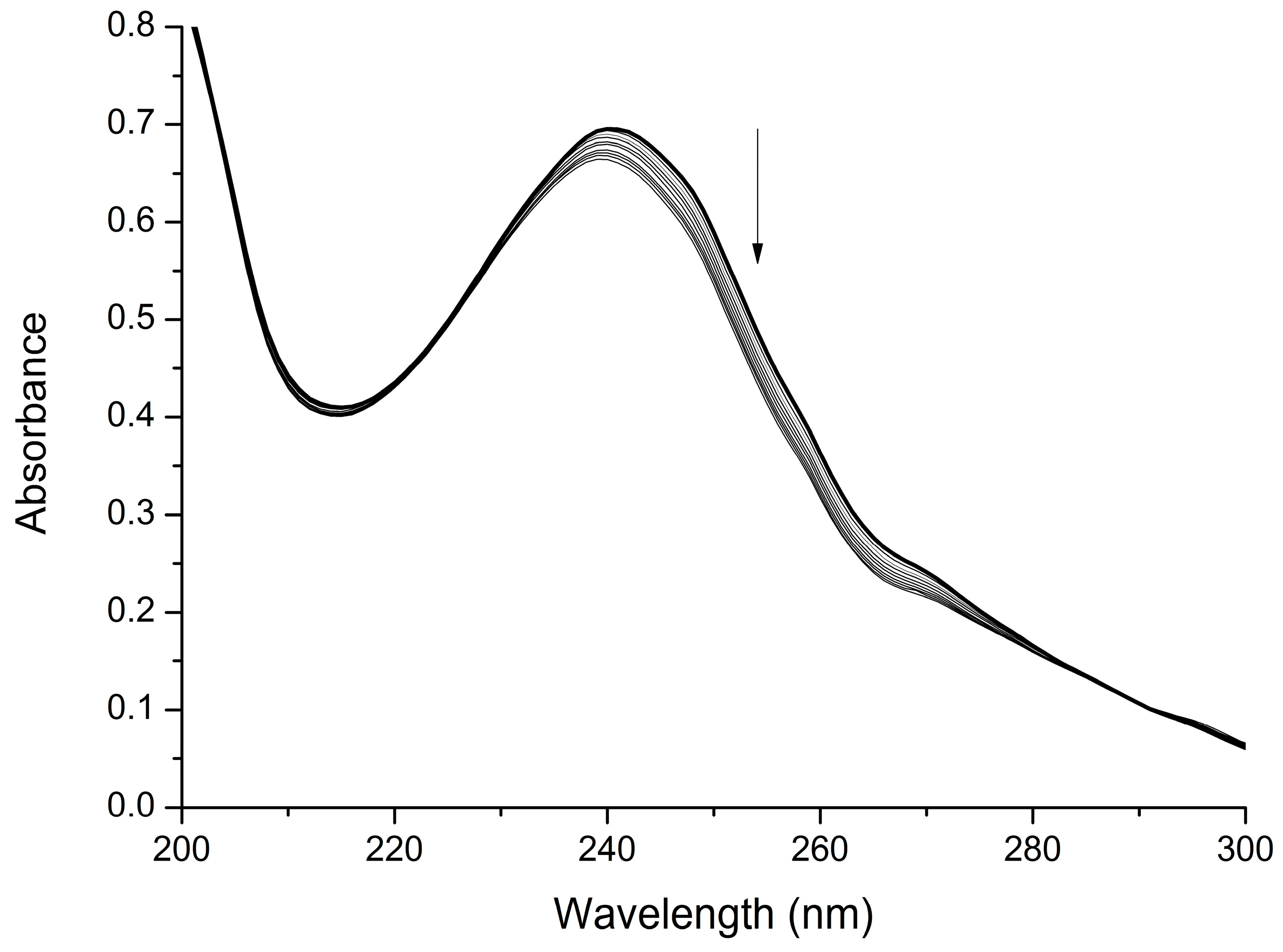
2.2. Interaction with Mg2+
2.3. Biology
2.4. Absorption, Distribution, Metabolism and Excretion Prediction
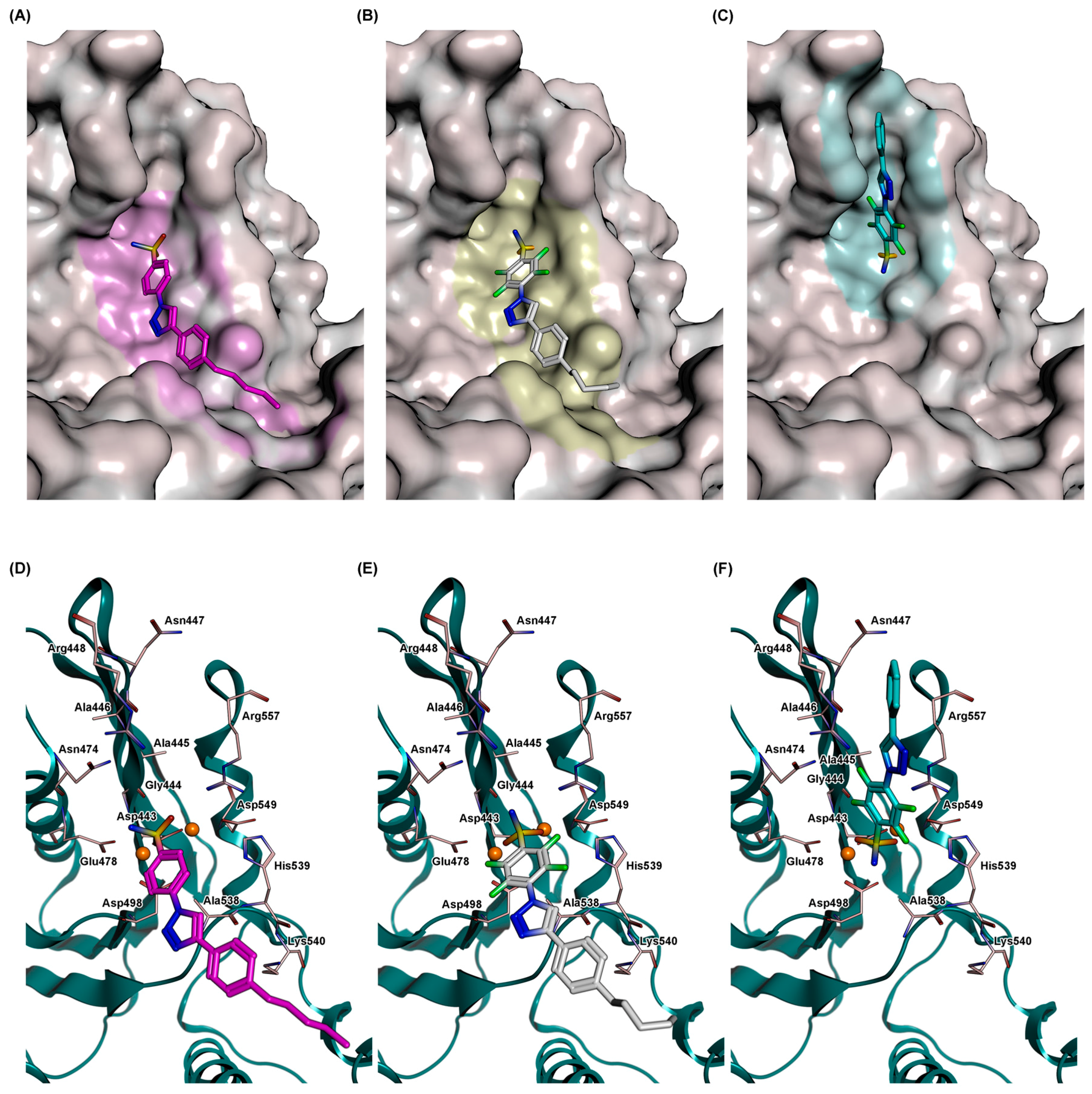
2.5. Docking
3. Materials and Methods
3.1. Chemistry
3.2. UV-Visible Titration
3.3. Biology
3.3.1. Protein Expression and Purification
3.3.2. HIV-1 RNase H Polymerase-Independent Cleavage Assay
3.3.3. HIV-1 RT-Associated RNA-Dependent DNA Polymerase Activity Determination
3.3.4. HIV-1 IN/LEDGF HTRF LEDGF-Dependent Assay
3.4. Molecular Modeling
3.4.1. Hardware Specifications
3.4.2. Protein Preparation
3.4.3. Ligands Preparation
3.4.4. Docking Procedures
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| RNase H | Ribonuclease H |
| RT | Reverse Transcriptase |
| RNA | Ribonucleic Acid |
| DNA | Deoxyribonucleic Acid |
| ADME | Absorption Distribution Metabolism Excretion |
| HIV-1 | Human Immunodeficiency Virus Type-1 |
| AIDS | Acquired Immune Deficiency Syndrome |
| HAART | Highly Active Antiretroviral Therapy |
| PR | Protease |
| IN | Integrase |
| vRNA | Viral RNA |
| vDNA | Viral DNA |
| NRTI | Nucleoside Reverse Transcriptase Inhibitor |
| NtRTI | Nucleotide Reverse Transcriptase Inhibitor |
| NNRTI | Non-Nucleoside Reverse Transcriptase Inhibitor |
| HID | 2-Hydroxyisoquinoline-1,3(2H,4H)-dione |
| CAIs | Carbonic Anhydrase Inhibitors |
| SAR | Structure-Activity Relationships |
| CuAAC | Copper-Catalyzed Azide-Alkyne Cycloaddition |
| IPTG | β-d-1-thio Galactopyranoside |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| DTT | Dithiothreitol |
| EDTA | Ethylenediaminetetraacetic Acid |
References
- Zhang, J.; Crumpacker, C. Eradication of HIV and cure of AIDS, now and how? Front. Immunol. 2013, 4, 337. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. HIV-1 eradication strategies: Design and assessment. Curr. Opin. HIV AIDS 2013, 8, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Siliciano, R.F. Targeting HIV latency: Pharmacologic strategies toward eradication. Drug Discov. Today 2013, 18, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Su, H.P.; Yan, Y.; Prasad, G.S.; Smith, R.F.; Daniels, C.L.; Abeywickrema, P.D.; Reid, J.C.; Loughran, H.M.; Kornienko, M.; Sharma, S.; et al. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J. Virol. 2010, 84, 7625–7633. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.; Masaoka, T.; Tocco, G.; Tramontano, E.; Le Grice, S.F. Active site and allosteric inhibitors of the ribonuclease H activity of HIV reverse transcriptase. Future Med. Chem. 2013, 5, 2127–2139. [Google Scholar] [CrossRef] [PubMed]
- Distinto, S.; Maccioni, E.; Meleddu, R.; Corona, A.; Alcaro, S.; Tramontano, E. Molecular aspects of the RT/Drug interactions. perspective of dual inhibitors. Curr. Pharm. Des. 2013, 19, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Tramontano, E. Past and future. Current drugs targeting HIV-1 integrase and reverse transcriptase-associated ribonuclease H activity: Single and dual active site inhibitors. Antivir. Chem. Chemother. 2014, 23, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, M.; Yang, W. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 2006, 25, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Ilina, T.; Labarge, K.; Sarafianos, S.G.; Ishima, R.; Parniak, M.A. Inhibitors of HIV-1 reverse transcriptase-associated ribonuclease H activity. Biology 2012, 1, 521–541. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Dal Peraro, M.; Klein, M.L. Phosphodiester cleavage in ribonuclease H occurs via an associative two-metal-aided catalytic mechanism. J. Am. Chem. Soc. 2008, 130, 10955–10962. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A.; Steitz, J.A. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6498–6502. [Google Scholar] [CrossRef] [PubMed]
- Rogolino, D.; Carcelli, M.; Sechi, M.; Neamati, N. Viral enzymes containing magnesium: Metal binding as successful strategy in drug design. Coord. Chem. Rev. 2012, 256, 3063–3086. [Google Scholar] [CrossRef]
- Tramontano, E.; Esposito, F.; Badas, R.; di Santo, R.; Costi, R.; La Colla, P. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antivir. Res. 2005, 65, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.; Di Leva, F.S.; Thierry, S.; Pescatori, L.; Cuzzucoli Crucitti, G.; Subra, F.; Delelis, O.; Esposito, F.; Rigogliuso, G.; Costi, R.; et al. Identification of highly conserved residues involved in inhibition of HIV-1 RNase H function by diketo acid derivatives. Antimicrob. Agents Chemother. 2014, 58, 6101–6110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costi, R.; Métifiot, M.; Esposito, F.; Cuzzucoli Crucitti, G.; Pescatori, L.; Messore, A.; Scipione, L.; Tortorella, S.; Zinzula, L.; Marchand, C.; et al. 6-(1-Benzyl-1H-pyrrol-2-yl)-2,4-dioxo-5-hexenoic acids as dual inhibitors of recombinant HIV-1 integrase and ribonuclease H, synthesized by a parallel synthesis approach. J. Med. Chem. 2013, 56, 8588–8598. [Google Scholar] [CrossRef] [PubMed]
- Cuzzucoli Crucitti, G.; Métifiot, M.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Saccoliti, F.; Scipione, L.; Tortorella, S.; Esposito, F.; et al. Structure-activity relationship of pyrrolyl diketo acid derivatives as dual inhibitors of HIV-1 integrase and reverse transcriptase ribonuclease H domain. J. Med. Chem. 2015, 58, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Costi, R.; Métifiot, M.; Chung, S.; Cuzzucoli Crucitti, G.; Maddali, K.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Scipione, L.; et al. Basic quinolinonyl diketo acid derivatives as inhibitors of HIV integrase and their activity against RNase H function of reverse transcriptase. J. Med. Chem. 2014, 57, 3223–3234. [Google Scholar] [CrossRef] [PubMed]
- Himmel, D.M.; Sarafianos, S.G.; Dharmasena, S.; Hossain, M.M.; McCoy-Simandle, K.; Ilina, T.; Clark, A.D., Jr.; Knight, J.L.; Julias, J.G.; Clark, P.K.; et al. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem. Biol. 2006, 1, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, K.; Hang, J.Q.; Rajendran, S.; Yang, Y.; Derosier, A.; Wong Kai In, P.; Overton, H.; Parkes, K.E.; Cammack, N.; Martin, J.A. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. 2003, 31, 6852–6859. [Google Scholar] [CrossRef] [PubMed]
- Lansdon, E.B.; Liu, Q.; Leavitt, S.A.; Balakrishnan, M.; Perry, J.K.; Lancaster-Moyer, C.; Kutty, N.; Liu, X.; Squires, N.H.; Watkins, W.J.; et al. Structural and binding analysis of pyrimidinol carboxylic acid and N-hydroxy quinazolinedione HIV-1 RNase H inhibitors. Antimicrob. Agents Chemother. 2011, 55, 2905–2915. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.D.; Staas, D.D.; Venkatraman, S.; Loughran, H.M.; Ruzek, R.D.; Booth, T.M.; Lyle, T.A.; Wai, J.S.; Vacca, J.P.; Feuston, B.P.; et al. Potent and selective HIV-1 ribonuclease H inhibitors based on a 1-hydroxy-1,8-naphthyridin-2(1H)-one scaffold. Bioorg. Med. Chem. Lett. 2010, 20, 6754–6757. [Google Scholar] [CrossRef] [PubMed]
- Beilhartz, G.L.; Ngure, M.; Johns, B.A.; DeAnda, F.; Gerondelis, P.; Gotte, M. Inhibition of the ribonuclease H activity of HIV-1 reverse transcriptase by GSK5750 correlates with slow enzyme-inhibitor dissociation. J. Biol. Chem. 2014, 289, 16270–16277. [Google Scholar] [CrossRef] [PubMed]
- Suchaud, V.; Bailly, F.; Lion, C.; Tramontano, E.; Esposito, F.; Corona, A.; Christ, F.; Debyser, Z.; Cotelle, P. Development of a series of 3-hydroxyquinolin-2(1H)-ones as selective inhibitors of HIV-1 reverse transcriptase associated RNase H activity. Bioorg. Med. Chem. Lett. 2012, 22, 3988–3992. [Google Scholar] [CrossRef] [PubMed]
- Himmel, D.M.; Myshakina, N.S.; Ilina, T.; Van Ry, A.; Ho, W.C.; Parniak, M.A.; Arnold, E. Structure of a dihydroxycoumarin active-site inhibitor in complex with the RNase H domain of HIV-1 reverse transcriptase and structure-activity analysis of inhibitor analogs. J. Mol. Biol. 2014, 426, 2617–2631. [Google Scholar] [CrossRef] [PubMed]
- Rogolino, D.; Bacchi, A.; de Luca, L.; Rispoli, G.; Sechi, M.; Stevaert, A.; Naesens, L.; Carcelli, M. Investigation of the salicylaldehyde thiosemicarbazone scaffold for inhibition of influenza virus PA endonuclease. J. Biol. Inorg. Chem. 2015, 20, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Rogolino, D.; Carcelli, M.; Bacchi, A.; Compari, C.; Contardi, L.; Fisicaro, E.; Gatti, A.; Sechi, M.; Stevaert, A.; Naesens, L. A versatile salicyl hydrazonic ligand and its metal complexes as antiviral agents. J. Inorg. Biochem. 2015, 150, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Sechi, M.; Innocenti, A.; Pala, N.; Rogolino, D.; Carcelli, M.; Scozzafava, A.; Supuran, C.T. Inhibition of alpha-class cytosolic human carbonic anhydrases I, II, IX and XII, and β-class fungal enzymes by carboxylic acids and their derivatives: New isoform-I selective nanomolar inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5801–5806. [Google Scholar] [CrossRef] [PubMed]
- Pala, N.; Micheletto, L.; Sechi, M.; Aggarwal, M.; Carta, F.; McKenna, R.; Supuran, C.T. Carbonic anhydrase inhibition with benzenesulfonamides and tetrafluorobenzenesulfonamides obtained via click chemistry. ACS Med. Chem. Lett. 2014, 5, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Moses, J.E.; Moorhouse, A.D. The growing applications of click chemistry. Chem. Soc. Rev. 2007, 36, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Southan, C.; Stracz, A. Extracting and connecting chemical structures from text sources using chemicalize.org. J. Cheminform. 2013, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Gerondelis, P.; Johns, B.A. The development of novel pyrido-pyrimidinone antiretrovirals with selective activity against HIV ribonuclease H. In Procedeedings of the Conference on Retroviruses in Cold Spring Harbor Laboratories, New York, NY, USA, 21–26 May 2012; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 2012. [Google Scholar]
- Vernekar, S.K.; Liu, Z.; Nagy, E.; Miller, L.; Kirby, K.A.; Wilson, D.J.; Kankanala, J.; Sarafianos, S.G.; Parniak, M.A.; Wang, Z. Design, synthesis, biochemical, and antiviral evaluations of C6 benzyl and C6 biarylmethyl substituted 2-hydroxylisoquinoline-1,3-diones: Dual inhibition against HIV reverse transcriptase-associated RNase H and polymerase with antiviral activities. J. Med. Chem. 2015, 58, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E.; Grootenhuis, P.D. Predicting passive transport in silico—History, hype, hope. Curr. Top. Med. Chem. 2003, 3, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Palm, K.; Luthman, K.; Ungell, A.L.; Strandlund, G.; Artursson, P. Correlation of drug absorption with molecular surface properties. J. Pharm. Sci. 1996, 85, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Palm, K.; Stenberg, P.; Luthman, K.; Artursson, P. Polar molecular surface properties predict the intestinal absorption of drugs in humans. Pharm. Res. 1997, 14, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Palm, K.; Luthman, K.; Ungell, A.L.; Strandlund, G.; Beigi, F.; Lundahl, P.; Artursson, P. Evaluation of dynamic polar molecular surface area as predictor of drug absorption: Comparison with other computational and experimental predictors. J. Med. Chem. 1998, 41, 5382–5392. [Google Scholar] [CrossRef] [PubMed]
- Kharlamova, T.; Esposito, F.; Zinzula, L.; Floris, G.; Cheng, Y.-C.; Dutschman, G.E.; Tramontano, E. Inhibition of HIV-1 Ribonuclease H activity by novel frangula-emodine derivatives. Med. Chem. 2009, 5, 398–410. [Google Scholar]
- Esposito, F.; Corona, A.; Zinzula, L.; Kharlamova, T.; Tramontano, E. New anthraquinone derivatives as inhibitors of the HIV-1 reverse transcriptase-associated Ribonuclease H function. Chemotherapy 2012, 58, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Sanna, C.; Del Vecchio, C.; Cannas, V.; Venditti, A.; Corona, A.; Bianco, A.; Serrilli, A.M.; Guarcini, L.; Parolin, C.; et al. Hypericum hircinum L. components as new single molecule inhibitors of both HIV-1 reverse transcriptase-associated DNA polymerase and ribonuclease H activities. Pathog. Dis. 2013, 68, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Tintori, C.; Ferrarese, R.; Cabiddu, G.; Corona, A.; Ceresola, E.R.; Calcaterra, A.; Iovine, V.; Botta, B.; Clementi, M.; et al. Kuwanon-L as a new allosteric HIV-1 integrase inhibitor: Molecular modeling and biological evaluation. ChemBioChem 2015, 16, 2507–2512. [Google Scholar] [CrossRef] [PubMed]
- Tintori, C.; Esposito, F.; Morreale, F.; Martini, R.; Tramontano, E.; Botta, M. Investigation on the sucrose binding pocket of HIV-1 Integrase by molecular dynamics and synergy experiments. Bioorg. Med. Chem. Lett. 2015, 25, 3013–3016. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group. Molecular Operating Environment, MOE 2009.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2009. [Google Scholar]
- Meleddu, R.; Distinto, S.; Corona, A.; Bianco, G.; Cannas, V.; Esposito, F.; Artese, A.; Alcaro, S.; Matyus, P.; Bogdan, D.; et al. (3Z)-3-(2-[4-(aryl)-1,3-thiazol-2-yl]hydrazin-1-ylidene)-2,3-dihydro-1H-indol-2-one derivatives as dual inhibitors of HIV-1 reverse transcriptase. Eur. J. Med. Chem. 2015, 93, 452–460. [Google Scholar] [CrossRef] [PubMed]






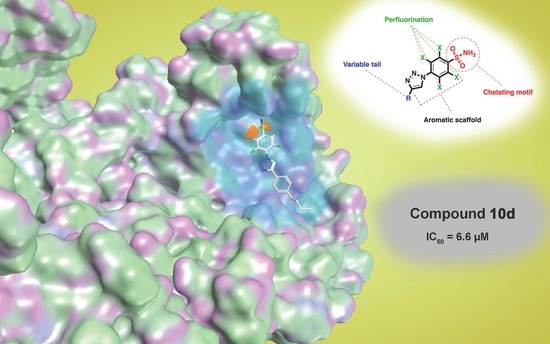
{kind=link}
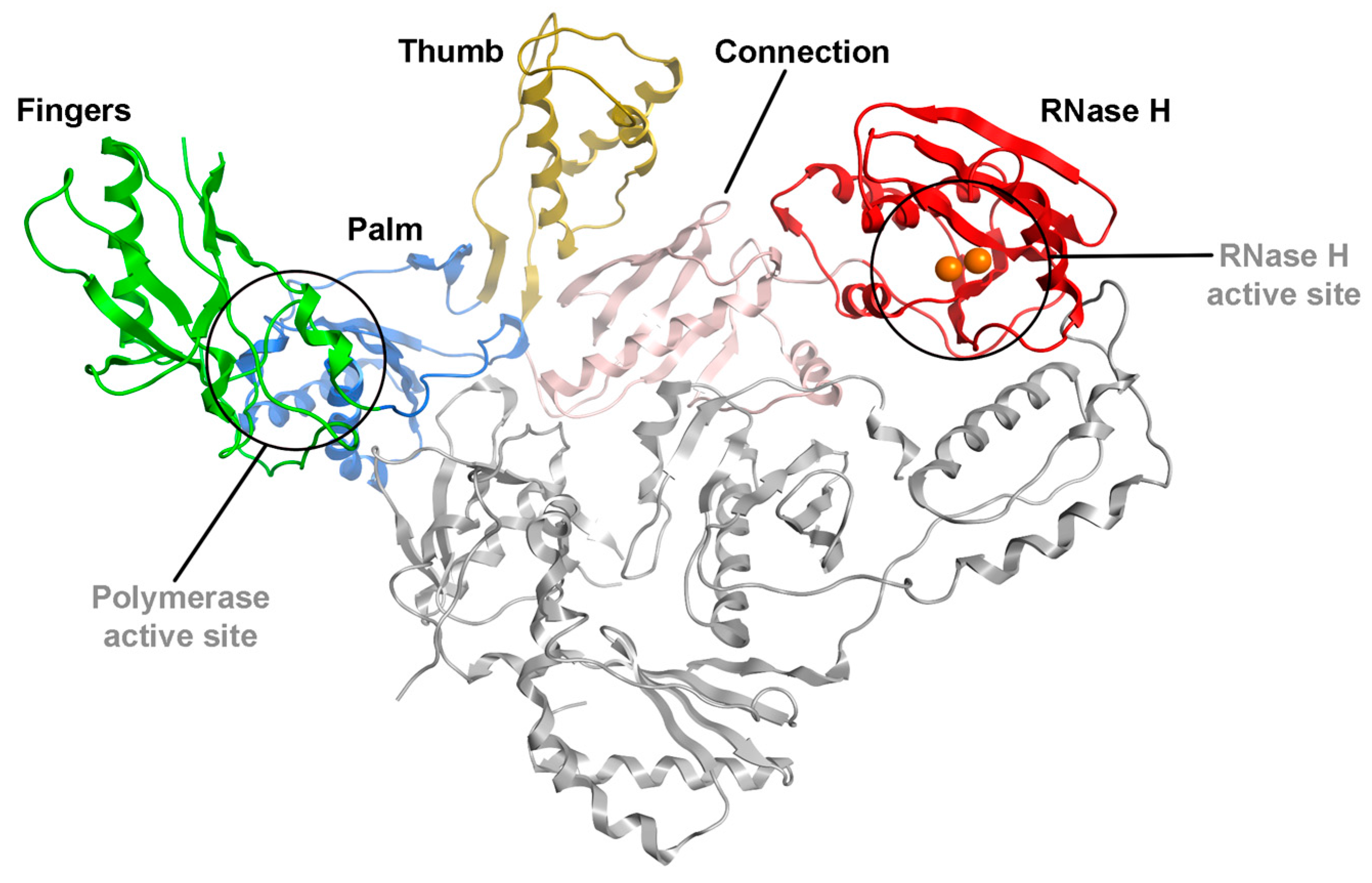{kind=link}
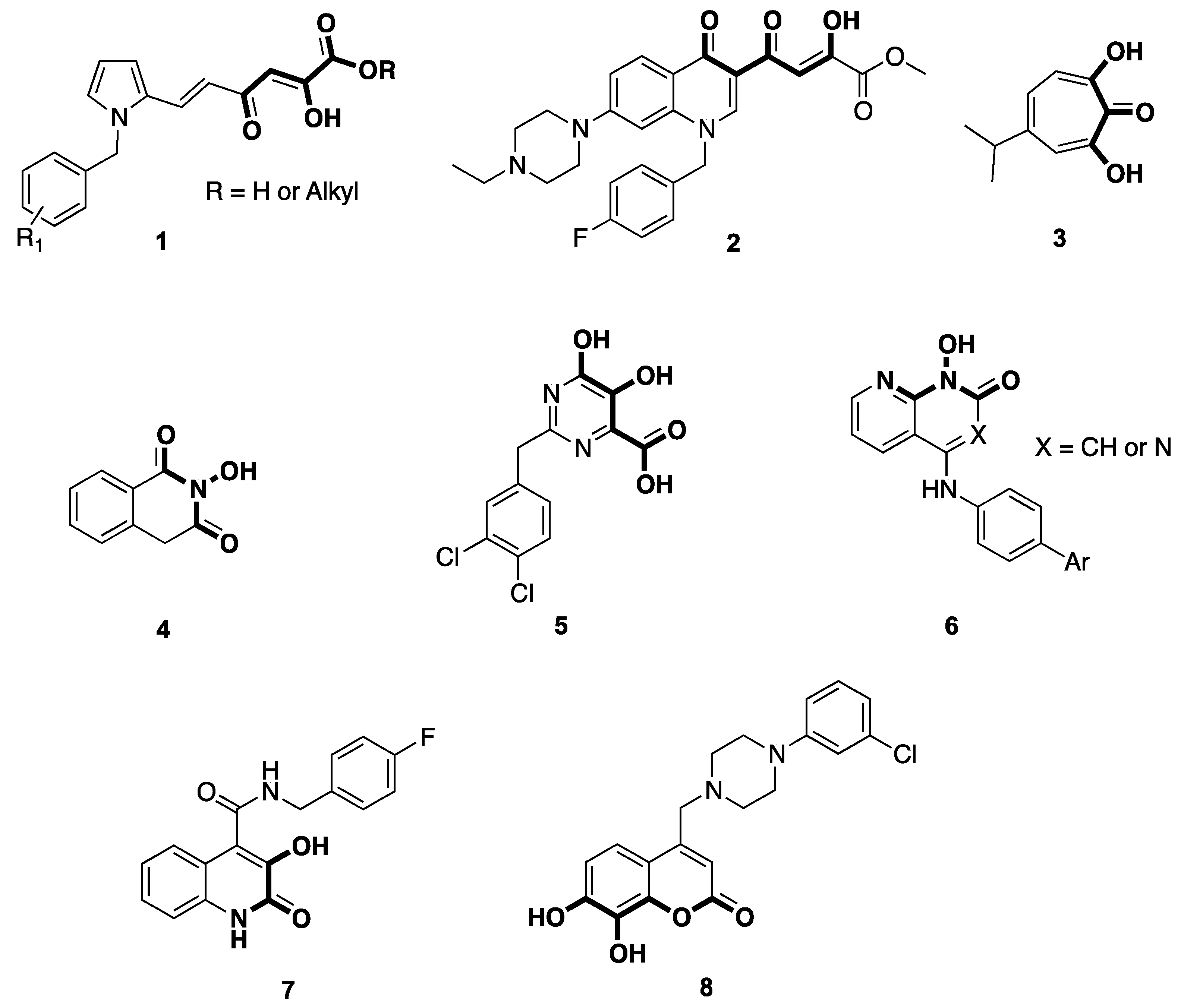{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | RNase H IC50 (µM) a | DP IC50 (µM) b | IN-LEDGF IC50 (µM) c |
|---|---|---|---|
| 9a | >100 | ND | ND |
| 9b | >100 | ND | ND |
| 9c | >100 | ND | ND |
| 9d | 63 ± 7 | >100 | >100 |
| 10a | >100 | ND | ND |
| 10b | >100 | ND | ND |
| 10c | 26 ± 3 | 90 ± 5 | >100 |
| 10d | 6.6 ± 0.5 | 33.4 ± 5.8 | >100 |
| RDS1759 | 7.0 ± 1.5 | ND | ND |
| Compound | MW | H-acc | H-don | Rbond | log P (o/w) | log S | TPSA |
|---|---|---|---|---|---|---|---|
| 9d | 370.5 | 4 | 1 | 7 | 3.328 | −6.216 | 91 |
| 10c | 372.3 | 2 | 1 | 3 | 1.829 | −4.885 | 65 |
| 10d | 442.4 | 2 | 1 | 7 | 3.928 | −7.396 | 65 |
| RDS1759 | 359.8 | 3 | 2 | 8 | 4.466 | −4.103 | 69 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pala, N.; Esposito, F.; Rogolino, D.; Carcelli, M.; Sanna, V.; Palomba, M.; Naesens, L.; Corona, A.; Grandi, N.; Tramontano, E.; et al. Inhibitory Effect of 2,3,5,6-Tetrafluoro-4-[4-(aryl)-1H-1,2,3-triazol-1-yl]benzenesulfonamide Derivatives on HIV Reverse Transcriptase Associated RNase H Activities. Int. J. Mol. Sci. 2016, 17, 1371. https://doi.org/10.3390/ijms17081371
Pala N, Esposito F, Rogolino D, Carcelli M, Sanna V, Palomba M, Naesens L, Corona A, Grandi N, Tramontano E, et al. Inhibitory Effect of 2,3,5,6-Tetrafluoro-4-[4-(aryl)-1H-1,2,3-triazol-1-yl]benzenesulfonamide Derivatives on HIV Reverse Transcriptase Associated RNase H Activities. International Journal of Molecular Sciences. 2016; 17(8):1371. https://doi.org/10.3390/ijms17081371
Chicago/Turabian StylePala, Nicolino, Francesca Esposito, Dominga Rogolino, Mauro Carcelli, Vanna Sanna, Michele Palomba, Lieve Naesens, Angela Corona, Nicole Grandi, Enzo Tramontano, and et al. 2016. "Inhibitory Effect of 2,3,5,6-Tetrafluoro-4-[4-(aryl)-1H-1,2,3-triazol-1-yl]benzenesulfonamide Derivatives on HIV Reverse Transcriptase Associated RNase H Activities" International Journal of Molecular Sciences 17, no. 8: 1371. https://doi.org/10.3390/ijms17081371