
Genetic and Transcriptomic Profiles of Inflammation in Neurodegenerative Diseases: Alzheimer, Parkinson, Creutzfeldt-Jakob and Tauopathies


Abstract
:1. Introduction
2. Inflammatory Genes as Risk Factors of Sporadic AD
3. Inflammatory Genes as Risk Factors of Sporadic PD
4. Inflammatory Genes as Risk Factors of Tauopathies
5. Systemic Infections as Risk Factors of AD, PD, and CJD
6. General Aspects of Inflammation in Sporadic AD
7. General Aspects of Inflammation in Sporadic PD
8. General Aspects of Inflammation in Sporadic Creutzfeldt-Jakob Disease (sCJD)
9. General Aspects of Inflammation in Tauopathies
10. A Comparative Inflammatory Transcriptome Study in Neurodegenerative Diseases with Abnormal Protein Aggregates
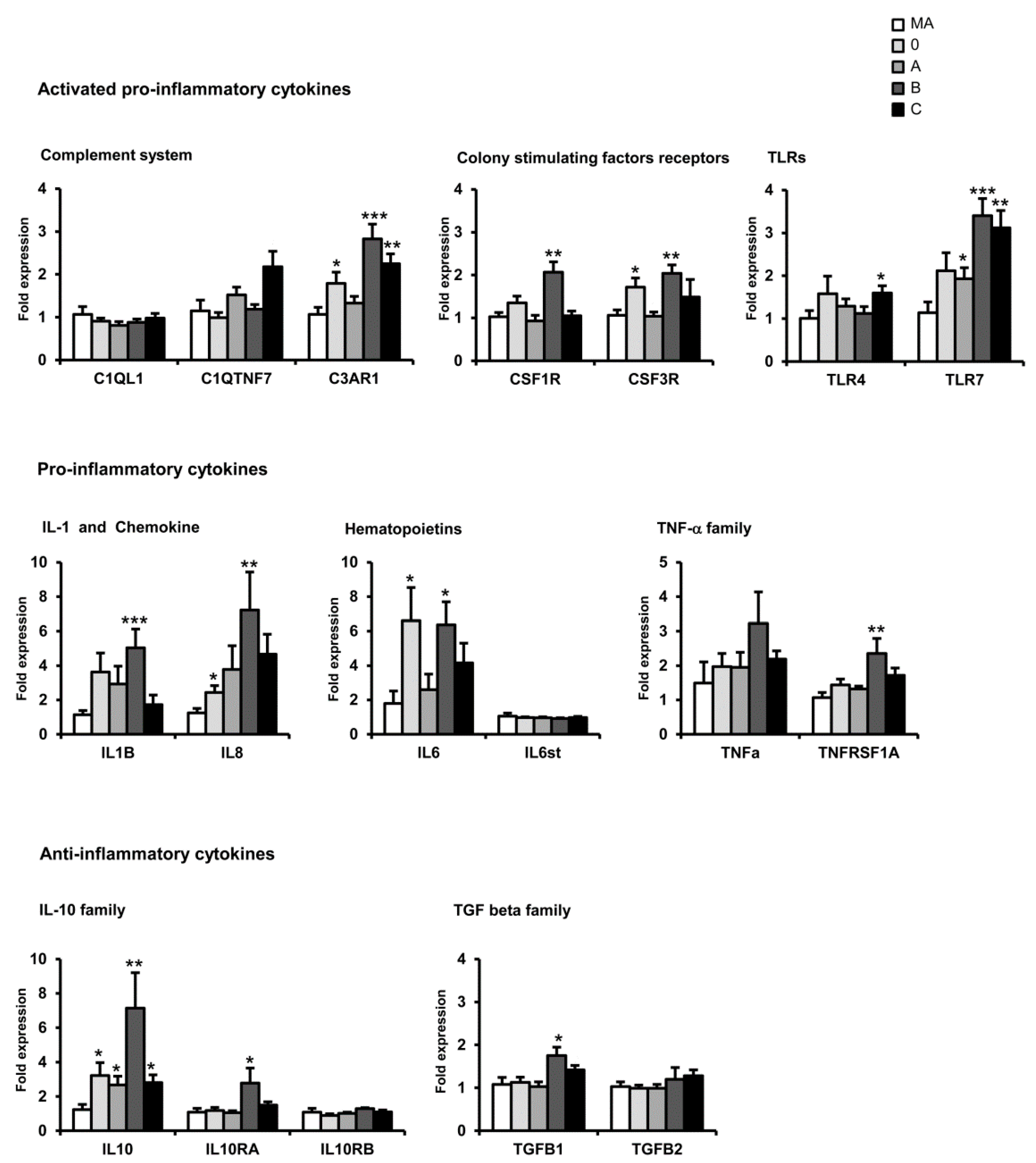
11. mRNA Expression of Selected Immune- and Inflammatory-Related Genes sAD: Regional Differences and Disease Progression



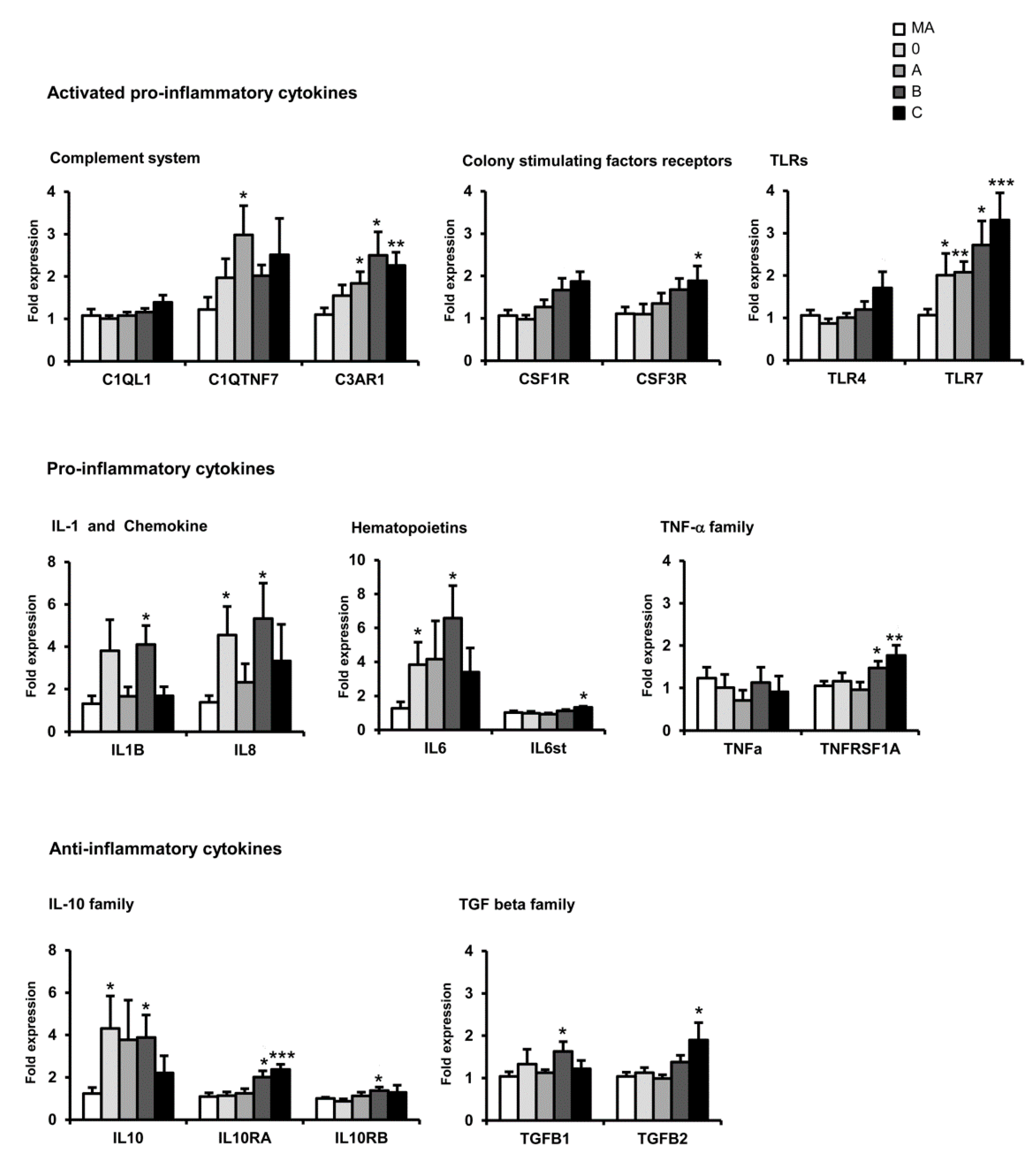
12. mRNA Expression of Selected Immune- and Inflammatory-Related Genes in sPD: Regional Differences
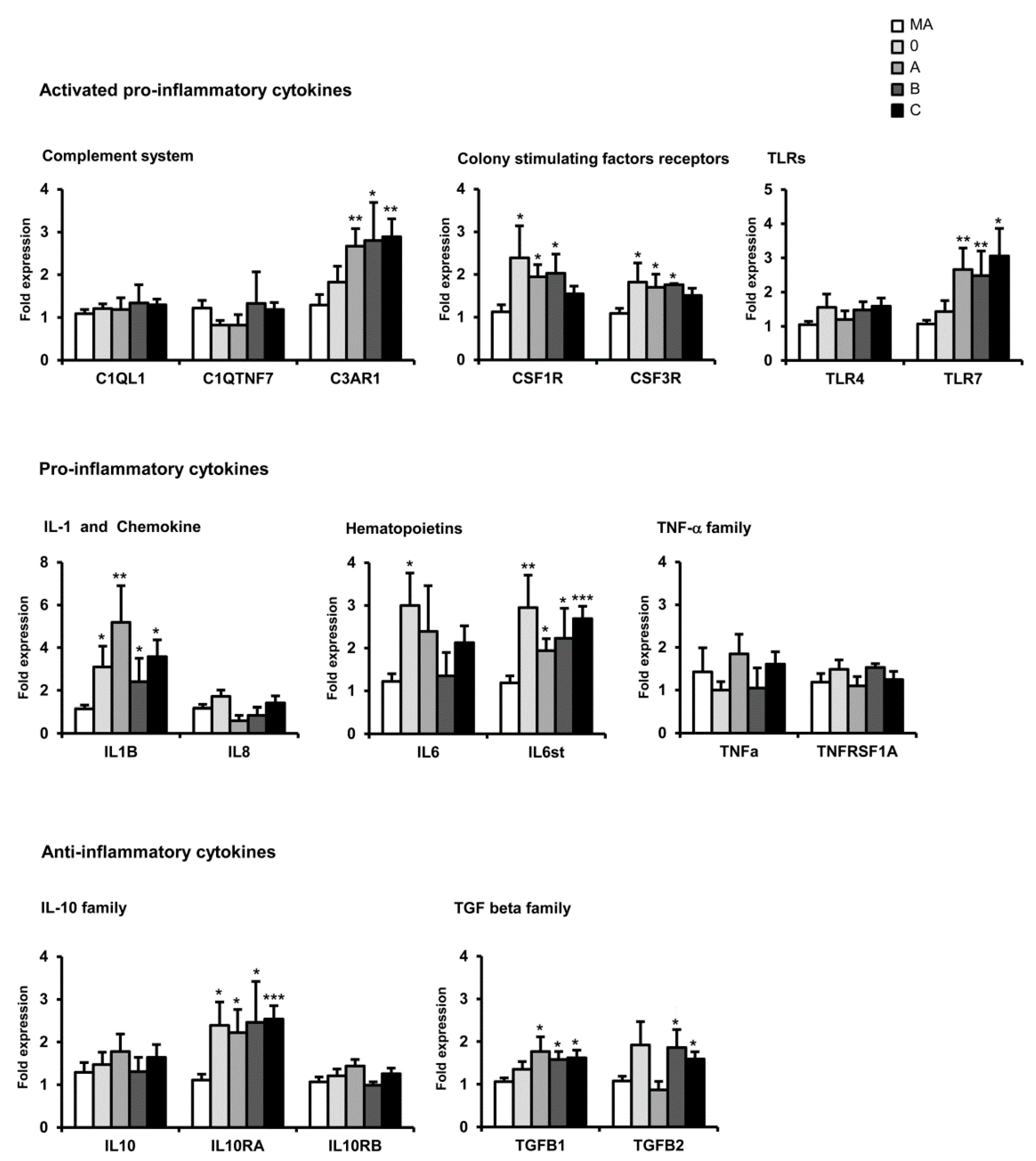
13. mRNA Expression of Selected Immune- and Inflammatory-Related Genes in CJD: Regional Differences Depending on the Genotype
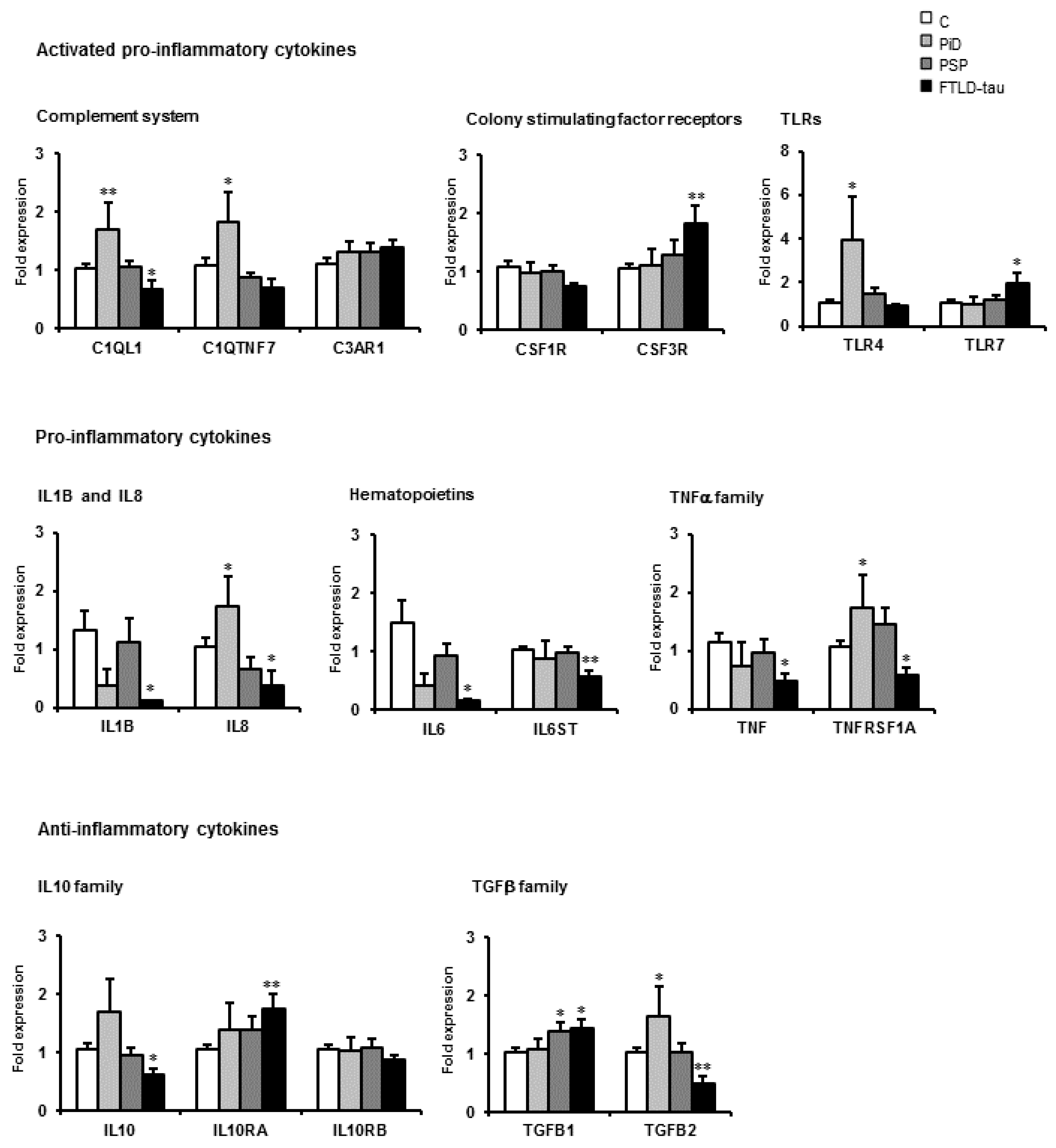
14. mRNA Expression of Selected Immune- and Inflammatory-Related Genes in Tauopathies: Disease Differences

15. Comparative Aspects of Inflammatory Gene Regulation in the Frontal Cortex in Neurodegenerative Diseases with Abnormal Protein Aggregates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Frontal Cortex Area 8 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AD A | AD B | AD C | PD | CJD MM1 | CJD VV2 | PiD | PSP | FTLD-tau | ||
| Complement system | C1QL1 | = | = | = | = | = | = | ↑ | = | ↓ |
| C1QTNF7 | = | = | = | = | = | = | = | = | = | |
| C3AR1 | ↑ | ↑ | ↑ | = | ↑ | ↑ | = | = | = | |
| Colony stimulating factors | CSF1R | ↑ | ↑ | = | = | ↑ | ↑ | = | = | = |
| CSF3R | ↑ | ↑ | = | ↓ | ↑ | ↑ | ↑ | = | ↑ | |
| Toll-like receptors | TLR4 | = | = | = | ↓ | ↑ | = | = | = | |
| TLR7 | ↑ | ↑ | ↑ | = | ↑ | = | = | = | ↑ | |
| Cytokines | IL8 | = | = | = | = | = | = | ↑ | = | ↓ |
| IL1B | ↑ | ↑ | ↑ | = | = | ↑ | = | = | ↓ | |
| IL6 | ↑ | = | = | = | ↑ | = | = | = | ↓ | |
| IL6ST | ↑ | ↑ | ↑ | = | ↑ | = | = | = | ↓ | |
| TNF family | TNFα | = | = | = | = | ↑ | ↑ | = | = | ↓ |
| TNFRS1A | = | = | = | = | ↑ | ↑ | ↑ | = | ↓ | |
| IL10 family | IL10 | = | = | = | = | ↑ | = | = | = | ↓ |
| L10RA | ↑ | ↑ | ↑ | ↑ | ↑ | = | = | = | ↑ | |
| L10RB | = | = | = | = | = | = | = | = | = | |
| TGF-β | - | ↑ | ↑ | ↑ | = | ↑ | = | = | ↑ | ↑ |
| - | = | ↑ | ↑ | = | ↑ | = | ↑ | = | ↓ | |
16. Increased Expression of Inflammatory Markers in Blood and Serum in AD and PD
17. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| C1QL1 | complement component 1, q subcomponent-like 1 |
| C1QTNF7 | C1q and tumor necrosis factor related protein 7 |
| C3AR1 | complement component 3a receptor 1 |
| CLEC7A | C-type lectin domain family 7, member A |
| CSF1R | colony stimulating factor 1 receptor |
| CSF3R | colony stimulating factor 3 receptor |
| IL1B | interleukin 1β |
| IL6 | interleukin 6 |
| IL6ST | interleukin 6 signal transducer |
| IL8 | interleukin 8 |
| IL10 | interleukin 10 |
| IL10RA | interleukin 10 receptor α |
| IL10RB | inerkeukin 10 receptor β |
| NFκB | nuclear factor of kappa light polypeptide gene enhancer in B-cells |
| TGFB1 | transforming growth factor β1 |
| TGFB2 | transforming growth factor β2 |
| TLR4 | toll-like receptor 4 |
| TLR7 | toll-like receptor 7 |
| TNF, TNFα | tumor necrosis factor α |
| TNFRSF1A | tumor necrosis factor receptor superfamily, member 1A |
References
- Love, S.; Budka, H.; Ironside, J.W.; Perry, A. Greenfield’s Neuropathology, 9th ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2015. [Google Scholar]
- Streit, W.J.; Greber, M.B.; Kreutzberg, G.W. Functional plasticity of microglia: A review. Glia 1988, 1, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Relat. Disord. 2004, 10 (Suppl. 1), S3–S7. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglia and neuroprotection: Implications for Alzheimer’s disease. Brain Res. Rev. 2005, 48, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B. Chanching face of microglia. Science 2010, 330, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Streit, W.J. Microglia: Biology and pathology. Acta Neuropathol. 2010, 119, 89–105. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. History of innate immunity in neurodegenerative disorders. Front. Pharmacol. 2011, 2, 77. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Li, W.; Michael, L.; Rodríguez, M.L. Role of microglia in CNS inflammation. FEBS Lett. 2011, 585, 3798–3805. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Muñoz, D.G. Non-steroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology 1998, 50, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, M.L.; Bliss, M.R.; Brain, A.T.; Scott, D.L. Rheumatoid arthritis and senile dementia of the Alzheimer’s type. Br. J. Rheumatol. 1989, 28, 86–88. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.; Rogers, J.; Sibley, J. Anti-inflammatory drugs and Alzheimer disease. Lancet 1990, 335, 1037. [Google Scholar] [CrossRef]
- Myllykangas-Luosujarvi, R.; Isomaki, H. Alzheimer’s disease and rheumatoid arthritis. Rheumatology 1994, 33, 501–502. [Google Scholar] [CrossRef]
- Breitner, J.C.; Welsh, K.A.; Helms, M.J.; Gaskell, P.C.; Gau, B.A.; Roses, A.D.; Pericak-Vance, M.A.; Saunders, A.M. Delayed onset of Alzheimer’s disease with non-steroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol. Aging 1995, 16, 523–530. [Google Scholar] [CrossRef]
- Stewart, W.F.; Kawas, C.; Corrada, M.; Metter, E.J. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997, 48, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; McGeer, P.L. Inflammatory aspects of Alzheimer disease and other neurodegenerative disorders. J. Alzheimers Dis. 2008, 13, 359–369. [Google Scholar] [PubMed]
- Zandi, P.P.; Anthony, J.C.; Hayden, K.M.; Mehta, K.; Mayer, L.; Breitner, J.C. Cache County Study Investigator. Reduced incidence of AD with NSAID but not H2 receptor antagonists: The Cache County Study. Neurology 2002, 59, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.M.; Hernan, M.A.; Schwarzschild, M.A.; Willett, W.C.; Colditz, G.A.; Speizer, F.E.; Ascherio, A. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch. Neurol. 2003, 60, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Samii, A.; Etminan, M.; Wiens, M.O.; Jafari, S. NSAID use and the risk of Parkinson’s disease: Systematic review and meta-analysis of observational studies. Drugs Aging 2009, 26, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.S.; Cupples, A.; van Duijn, C.M.; Kurz, A.; Green, R.C.; Chui, H.; Duara, R.; Auerbach, S.A.; Volicer, L.; Wells, J.; et al. Evidence for major gene inheritance of Alzheimer disease in families of patients with and without apolipoprotein E epsilon 4. Am. J. Hum. Genet. 1996, 59, 664–675. [Google Scholar] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Jhoo, J.H.; Lee, K.U.; Lee, D.Y.; Lee, J.H.; Youn, J.Y.; Lee, B.J.; Han, S.H.; Woo, J.I. Association between apolipoprotein E polymorphism and Alzheimer’s disease in Koreans. Neurosci. Lett. 1999, 277, 145–148. [Google Scholar] [CrossRef]
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic meta-analyses of Alzheimer disease genetic association studies, the AlzGene database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Coon, K.D.; Myers, A.J.; Craig, D.W.; Webster, J.A.; Pearson, J.V.; Lince, D.H.; Zismann, V.L.; Beach, T.G.; Leung, D.; Bryden, L.; et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J. Clin. Psychiatry 2007, 68, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Chouraki, V.; Seshadri, S. Genetics of Alzheimer’s disease. Adv. Genet. 2014, 87, 245–294. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; Paul, S.M. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Maeda, N.; Montine, T.J.; Montine, K.S. Apolipoprotein E-specific innate immune response in astrocytes from targeted replacement mice. J. Neuroinflamm. 2006, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Nivison, M.; Montine, K.S.; Maeda, N.; Montine, T.J. Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J. 2006, 20, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Lee, C.Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lin, S.; Bales, K.R.; Gelfanova, V.; Koger, D.; Delong, C.; Hale, J.; Liu, F.; Hunter, J.M.; Paul, S.M. Macrophage-mediated degradation of beta-amyloid via an apolipoprotein E isoform-dependent mechanism. J. Neurosci. 2009, 29, 3603–3612. [Google Scholar] [CrossRef] [PubMed]
- Terwel, D.; Steffensen, K.R.; Verghese, P.B.; Kummer, M.P.; Gustafsson, J.Å.; Holtzman, D.M.; Heneka, M.T. Critical role of astroglial apolipoprotein E and liver X receptor-α expression for microglial Aβ phagocytosis. J. Neurosci. 2011, 31, 7049–7059. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; LaDu, M.J.; van Eldik, L.J. A dual role for apolipoprotein E in neuroinflammation: Anti- and pro-inflammatory activity. J. Mol. Neurosci. 2004, 23, 205–212. [Google Scholar] [CrossRef]
- Licastro, F.; Porcellini, E.; Caruso, C.; Lio, D.; Corder, E.H. Genetic risk profiles for Alzheimer’s disease: Integration of APOE genotype and variants that up-regulate inflammation. Neurobiol Aging 2007, 28, 1637–1643. [Google Scholar] [CrossRef] [PubMed]
- Dorey, E.; Chang, N.; Liu, Q.Y.; Yang, Z.; Zhang, W. Apolipoprotein E, amyloid-beta, and neuroinflammation in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.M.; Ghura, S.; Koster, K.P.; Liakaite, V.; Maienschein-Cline, M.; Kanabar, P.; Collins, N.; Ben-Aissa, M.; Lei, A.Z.; Bahroos, N.; et al. APOE-modulated Aβ-induced neuroinflammation in Alzheimer’s disease, current landscape, novel data, and future perspective. J. Neurochem. 2015, 133, 465–488. [Google Scholar] [CrossRef] [PubMed]
- Rogaeva, E.; Meng, Y.; Lee, J.H.; Gu, Y.; Kawarai, T.; Zou, F.; Katayama, T.; Baldwin, C.T.; Cheng, R.; Hasegawa, H.; et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 2007, 39, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rowland, C.; Catanese, J.; Morris, J.; Lovestone, S.; O’Donovan, M.C.; Goate, A.; Owen, M.; Williams, J.; Grupe, A. SORL1 variants and risk of late-onset Alzheimer’s disease. Neurobiol. Dis. 2008, 29, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, G.; Naj, A.C.; Beecham, G.W.; Wang, L.S.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Ertekin-Taner, N.; Fallin, M.D.; Friedland, R.; et al. Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol. 2010, 67, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Cheng, R.; Rogaeva, E.; Lee, J.H.; Tokuhiro, S.; Zou, F.; Bettens, K.; Sleegers, K.; Tan, E.K.; Kimura, R.; et al. Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch. Neurol. 2011, 68, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, N.; van Cauwenberghe, C.; Engelborghs, S.; Lambert, J.C.; Bettens, K.; Le Bastard, N.; Pasquier, F.; Montoya, A.G.; Peeters, K.; Mattheijssens, M.; et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol. Psychiatry 2012, 17, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Liu, X.; Zhang, F.; Wu, Y.; Yuan, J.; Zhu, J.; Zhang, F.; Wang, G.; Cheng, Z. An updated meta-analysis of the association between SORL1 variants and the risk for sporadic Alzheimer’s disease. J. Alzheimers Dis. 2013, 37, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, P.; Hibar, D.P.; Thompson, P.M. TREM2 and neurodegenerative disease. N. Engl. J. Med. 2013, 369, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Simpson, J.F.; Parikh, I.; Wilfred, B.R.; Fardo, D.W.; Nelson, P.T.; Estus, S. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J. Neurosci. 2013, 33, 13320–13325. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, A.; Koike, A.; Jun, G.; Wang, L.S.; Takahashi, S.; Matsubara, E.; Kawarabayashi, T.; Shoj, M.; Tomita, N.; Arai, H.; et al. SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS ONE 2013, 8, e58618. [Google Scholar] [CrossRef]
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Dols-Icardo, O.; Bullido, M.J.; Pastor, P.; Rodríguez-Rodríguez, E.; López de Munain, A.; de Pancorbo, M.M.; Pérez-Tur, J.; Alvarez, V.; Antonell, A.; et al. Assessing the role of the TREM2 p.R47H variant as a risk factor for Alzheimer’s disease and frontotemporal dementia. Neurobiol. Aging 2014, 35, 444–e1. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Jun, G.; Reitz, C.; Kunkle, B.W.; Perry, W.; Park, Y.S.; Beecham, G.W.; Rajbhandary, R.A.; Hamilton-Nelson, K.L.; Wang, L.S.; et al. Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease: A genome-wide association study. JAMA Neurol. 2014, 71, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.T.; Hu, N.; Tan, M.S.; Zhu, X.C.; Tan, L. CD33 in Alzheimer’s disease. Mol. Neurobiol. 2014, 49, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Cruchaga, C.; Karch, C.M.; Jin, S.C.; Benitez, B.A.; Cai, Y.; Guerreiro, R.; Harari, O.; Norton, J.; Budde, J.; Bertelsen, S.; et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 2014, 505, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C. Genetic loci associated with Alzheimer’s disease. Future Neurol. 2014, 9, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Benitez, B.A.; Karch, C.M.; Cooper, B.; Skorupa, T.; Carrell, D.; Norton, J.B.; Hsu, S.; Harari, O.; Cai, Y.; et al. Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum. Mol. Genet. 2014, 23, 5838–5846. [Google Scholar] [CrossRef] [PubMed]
- International Genomics of Alzheimer’s Disease Consortium (IGAP). Convergent genetic and expression data implicate immunity in Alzheimer’s disease. Alzheimers Dement. 2015, 11, 658–671. [Google Scholar] [CrossRef]
- Zhu, X.C.; Yu, J.T.; Jiang, T.; Wang, P.; Cao, L.; Tan, L. CR1 in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, W.; Wang, X. TREM2 variants and risk of Alzheimer’s disease: A meta-analysis. Neurol. Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Gil, S.C.; Yan, P.; Wang, Y.; Han, S.; Gonzales, E.; Perez, R.; Cirrito, J.R.; Lee, J.M. Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J. Biol. Chem. 2012, 287, 21279–21289. [Google Scholar] [CrossRef] [PubMed]
- Lio, D.; Licastro, F.; Scola, L.; Chiappelli, M.; Grimaldi, L.M.; Crivello, A.; Colonna-Romano, G.; Candore, G.; Franceschi, C.; Caruso, C. Interleukin-10 promoter polymorphism in sporadic Alzheimer’s disease. Genes Immun. 2003, 4, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.L.; Tang, N.L.; Lam, L.C.; Chiu, H.F. The association between promoter polymorphism of the interleukin-10 gene and Alzheimer’s disease. Neurobiol. Aging 2005, 26, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Sardi, F.; Fassina, L.; Venturini, L.; Inguscio, M.; Guerriero, F.; Rolfo, E.; Ricevuti, G. Alzheimer’s disease, autoimmunity and inflammation. The good, the bad and the ugly. Autoimmun. Rev. 2011, 11, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Flex, A.; Giovannini, S.; Biscetti, F.; Liperoti, R.; Spalletta, G.; Straface, G.; Landi, F.; Angelini, F.; Caltagirone, C.; Ghirlanda, G.; et al. Effect of proinflammatory gene polymorphisms on the risk of Alzheimer’s disease. Neurodegener. Dis. 2014, 13, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Hamza, T.H.; Zabetian, C.P.; Tenesa, A.; Laederach, A.; Montimurro, J.; Yearout, D.; Kay, D.M.; Doheny, K.F.; Paschall, J.; Pugh, E.; et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 2010, 42, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Saiki, M.; Baker, A.; Williams-Gray, C.H.; Foltynie, T.; Goodman, R.S.; Taylor, C.J.; Compston, D.A.; Barker, R.A.; Sawcer, S.J.; Goris, A. Association of the human leucocyte antigen region with susceptibility to Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Hill-Burns, E.M.; Factor, S.A.; Zabetian, C.P.; Thomson, G.; Payami, H. Evidence for more than one Parkinson’s disease-associated variant within the HLA region. PLoS ONE 2011, 6, e27109. [Google Scholar] [CrossRef] [PubMed]
- Wissemann, W.T.; Hill-Burns, E.M.; Zabetian, C.P.; Factor, S.A.; Patsopoulos, N.; Hoglund, B.; Holcomb, C.; Donahue, R.J.; Thomson, G.; Erlich, H.; et al. Association of Parkinson disease with structural and regulatory variants in the HLA region. Am. J. Hum. Genet. 2013, 93, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Krügler, R.; Hardt, C.; Tschentscher, F.; Jäckel, S.; Kuhn, W.; Müller, T.; Werner, J.; Woitalla, D.; Berg, D.; Kühnl, N.; et al. Genetic analysis of immunomodulating factors in sporadic Parkinson’s disease. J. Neural Transm. 2000, 107, 553–562. [Google Scholar]
- McGeer, P.L.; Yasojima, K.; McGeer, E.G. Association of interleukin-1 beta polymorphisms with idiopathic Parkinson’s disease. Neurosci. Lett. 2002, 326, 67–69. [Google Scholar] [CrossRef]
- Mattila, K.M.; Rinne, J.O.; Lehtimäki, T.; Röyttä, M.; Ahonen, J.P.; Hurme, M. Association of an interleukin 1B gene polymorphism (-511) with Parkinson’s disease in Finnish patients. J. Med. Genet. 2002, 39, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Hakansson, A.; Westberg, L.; Nilsson, S.; Buervenich, S.; Carmine, A.; Holmberg, B.; Sydow, O.; Olson, L.; Johnels, B.; Eriksson, E.; et al. Interaction of polymorphisms in the genes encoding interleukin-6 and estrogen receptor beta on the susceptibility to Parkinson’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 133B, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Wahner, A.D.; Sinsheimer, J.S.; Bronstein, J.M.; Ritz, B. Inflammatory cytokine gene polymorphisms and increased risk of Parkinson disease. Arch. Neurol. 2007, 64, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Bialecka, M.; Klodowska-Duda, G.; Kurzawski, M.; Slawek, J.; Gorzkowska, A.; Opala, G.; Bialecki, P.; Sagan, L.; Droździk, M. Interleukin-10 (IL10) and tumor necrosis factor alpha (TNF) gene polymorphisms in Parkinson’s disease patients. Parkinsonism Relat. Disord. 2008, 14, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, D.; He, Q.; Gao, J.; Chen, B.; Xie, A. Interleukin-18 promoter polymorphisms and risk of Parkinson’s disease in a Han Chinese population. Brain Res. 2011, 1381, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Holmans, P.; Moskvina, V.; Jones, L.; Sharma, M.; International Parkinson’s Disease Genomics Consortium.; Vedernikov, A.; Buchel, F.; Saad, M.; Bras, J.M.; Bettella, F.; et al. A pathway-based analysis provides additional support for an immune-related genetic susceptibility to Parkinson’s disease. Hum. Mol. Genet. 2013, 22, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Nie, K.; Zhang, Y.; Gan, R.; Wang, L.; Zhao, J.; Huang, Z.; Tang, H.; Wang, L. Polymorphisms in immune/inflammatory cytokine genes are related to Parkinson’s disease with cognitive impairment in the Han Chinese population. Neurosci. Lett. 2013, 541, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.; García-Gorostiaga, I.; Sánchez-Juan, P.; Sánchez-Quintana, C.; Gurpegui, J.L.; Rodríguez-Rodríguez, E.; Mateo, I.; Berciano, J.; Combarros, O. Inflammation-related genes and the risk of Parkinson’s disease: A multilocus approach. Eur. J. Neurol. 2008, 15, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Im, S.Y.; Kim, Y.E.; Kim, Y.J. Genetics of progressive supranuclear palsy. J. Mov. Disord. 2015, 8, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Davidson, Y.; Gibbons, L.; Payton, A.; Richardson, A.M.; Varma, A.; Julien, C.; Stopford, C.; Thompson, J.; Horan, M.A.; et al. The apolipoprotein E epsilon4 allele selectively increases the risk of frontotemporal lobar degeneration in males. J. Neurol. Neurosurg. Psychiatry 2006, 77, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, L.; Maletta, R.G.; Tomaino, C.; Gallo, M.; Geracitano, S.; Costanzo, A.; Colao, R.; Puccio, G.; Frangipane, F.; Curcio, S.A.; et al. The effects of APOE and tau gene variability on risk of frontotemporal dementia. Neurobiol. Aging 2006, 27, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Verpillat, P.; Camuzat, A.; Hannequin, D.; ThomasAnterion, C.; Puel, M.; Belliard, S.; Dubois, B.; Didic, M.; Lacomblez, L.; Moreaud, O.; et al. Apolipoprotein E gene in frontotemporal dementia: An association study and meta-analysis. Eur. J. Hum. Genet. 2002, 10, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Cunningham, C.; Holmes, C. Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 2007, 7, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J. Emerging roles of pathogens in Alzheimer disease. Expert Rev. Mol. Med. 2011, 13, e30. [Google Scholar] [CrossRef] [PubMed]
- Widman, C.N.; Heneka, M.T. Long-term cerebral consequences of sepsis. Lancet Neurol. 2014, 13, 630–636. [Google Scholar] [CrossRef]
- Sparks Stein, P.; Steffen, M.J.; Smith, C.; Jicha, G.; Ebersole, J.L.; Abner, E.; Dawson, D. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimers Dement. 2012, 8, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Pirraglia, E.; Tsui, W.; Rusinek, H.; Vallabhajosula, S.; Mosconi, L.; Yi, L.; McHugh, P.; Craig, R.G.; Svetcov, S.; et al. Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol. Aging 2015, 36, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F.; Wozniak, M.A. Herpes simplex virus type 1 in Alzheimer’s disease: The enemy within. J. Alzheimers Dis. 2008, 13, 393–340. [Google Scholar] [CrossRef] [PubMed]
- Honjo, K.; van Reekum, R.; Verhoeff, N.P. Alzheimer’s disease and infection: Do infectious agents contribute to progression of Alzheimer’s disease? Alzheimers Dement. 2009, 5, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.L.; Yao, X.Q.; Jiao, S.S.; Zeng, F.; Liu, Y.H.; Xiang, Y.; Liang, C.R.; Wang, Q.H.; Wang, X.; Cao, H.Y.; et al. A study on the association between infectious burden and Alzheimer’s disease. Eur. J. Neurol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Carbone, I.; Lazzarotto, T.; Ianni, M.; Porcellini, E.; Forti, P.; Masliah, E.; Gabrielli, L.; Licastro, F. Herpes virus in Alzheimer’s disease: Relation to progression of the disease. Neurobiol. Aging 2014, 35, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Lövheim, H.; Gilthorpe, J.; Johansson, A.; Eriksson, S.; Hallmans, G.; Elgh, F. Herpes simplex infection and the risk of Alzheimer’s disease: A nested case-control study. Alzheimers Dement. 2015, 11, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Barnes, L.L.; Capuano, A.W.; Aiello, A.E.; Turner, A.D.; Yolken, R.H.; Torrey, E.F.; Bennett, D.A. Cytomegalovirus infection and risk of Alzheimer disease in older black and white individuals. J. Infect. Dis. 2015, 211, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Boltz, D.A.; Webster, R.G.; Smeyne, R.J. Viral parkinsonism. Biochim. Biophys. Acta 2009, 1792, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Vlajinac, H.; Dzoljic, E.; Maksimovic, J.; Marinkovic, J.; Sipetic, S.; Kostic, V. Infections as a risk factor for Parkinson’s disease: A case-control study. Int. J. Neurosci. 2013, 123, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Rugbjerg, K.; Friis, S.; Ritz, B.; Schernhammer, E.S.; Korbo, L.; Olsen, J.H. Autoimmune disease and risk for Parkinson disease: A population-based case-control study. Neurology 2009, 73, 1462–1468. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sundquist, J.; Sundquist, K. Subsequent risks of Parkinson disease in patients with autoimmune and related disorders: A nationwide epidemiological study from Sweden. Neurodgener. Dis. 2012, 10, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Ravenholt, R.T.; Foege, W.H. 1918 influenza, encephalitis lethargica, parkinsonism. Lancet 1982, 2, 860–864. [Google Scholar] [CrossRef]
- Miman, O.; Kusbeci, O.Y.; Aktepe, O.C.; Cetinkaya, Z. The probable relation between Toxoplasma gondii and Parkinson’s disease. Neurosci. Lett. 2010, 475, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Mosher, K.I.; Wyss-Coray, T. Go with your gut: Microbiota meet microglia. Nat. Neurosci. 2015, 18, 930–931. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Stappenbeck, T.S.; Hsieh, C.S. Peripheral education of the immune system by colonic commensal microbiota. Nature 2011, 478, 250–254. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.; Sheng, J.G.; Royston, M.C.; Gentleman, S.M.; McKenzie, J.E.; Graham, D.I.; Roberts, G.W.; Mrak, R.E. Glial-neuronal interactions in Alzheimer’s disease: The potential role of a “cytokine cycle” in disease progression. Brain Pathol. 1998, 8, 65–72. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Inflammation: Autotoxicity and Alzheimer disease. Neurobiol. Aging 2001, 22, 799–809. [Google Scholar] [CrossRef]
- Streit, W.J.; Conde, J.R.; Harrison, J.K. Chemokines and Alzheimer’s disease. Neurobiol. Aging 2001, 22, 909–913. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Lee, Y.J.; Han, S.B.; Nam, S.Y.; Oh, K.W.; Hong, J.T. Inflammation and Alzheimer’s disease. Arch. Pharm. Res. 2010, 33, 1539–1556. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.; Guo, L.; Bu, J.; Maloof, M.; El Khoury, J.; Geula, C. Microglia activation mediates fibrillar amyloid-beta toxicity in the aged primate cortex. Neurobiol. Aging 2011, 32, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Horio, J.; Satoh, H.; Sue, L.; Beach, T.; Arita, S.; Tooyama, I.; Konishi, Y. Expression profiles of cytokines in the brains of Alzheimer’s disease (AD) patients, compared to the brains of non-demented patients with and without increasing AD pathology. J. Alzheimer Dis. 2011, 25, 59–76. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol. 2013, 126, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Prokop, S.; Miller, K.R.; Heppner, F.L. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nature 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, P.; van Exel, E.; Hoozemans, J.J.; Veerhuis, R.; Rozemuller, A.J.; van Gool, W.A. Neuroinflammation—An early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener. Dis. 2010, 7, 38–44. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s disease. J. Neural Transm. Suppl. 2000, 60, 277–290. [Google Scholar] [PubMed]
- Hirsch, E.C.; Hunot, S.; Damier, P.; Faucheux, B. Glial cells and inflammation in Parkinson’s disease: A role in neurodegeneration? Ann. Neurol. 1998, 44, S115–S120. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S.; Damier, P.; Brugg, B.; Faucheux, B.A.; Michel, P.P.; Ruberg, M.; Muriel, M.P.; Mouatt-Prigent, A.; Agid, Y. Glial cell participation in the degeneration of dopaminergic neurons in Parkinson’s disease. Adv. Neurol. 1999, 80, 9–18. [Google Scholar] [PubMed]
- Hirsch, E.C.; Breidert, T.; Rousselet, E.; Hunot, S.; Hartmann, A.; Michel, P.P. The role of glial reaction and inflammation in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Imamura, K.; Nagatsu, T. Role of cytokines in inflammatory process in Parkinson’s disease. J. Neural Transm. Suppl. 2006, 70, 373–381. [Google Scholar] [PubMed]
- Whitton, P.S. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br. J. Pharmacol. 2007, 150, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Long-Smith, C.M.; Sullivan, A.M.; Nolan, Y.M. The influence of microglia on the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2009, 89, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.U.; Meuwissen, R.; Genc, S.; Genc, K. Inflammation in Parkinson’s disease. Adv. Protein Chem. Struct. Biol. 2012, 88, 69–132. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. 1), S210–S212. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of MHC class II-positive microglia and cytokine profile of Parkinson’s disease v brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Hishikawa, N.; Ono, K.; Suzuki, H.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol. 2005, 109, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C] (R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Yagi, S.; Yokokura, M.; Sakamoto, M. Neuroinflammation in the living brain of Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15 (Suppl. 3), S200–S204. [Google Scholar] [CrossRef]
- Brooks, D.J. Imaging approaches to Parkinson disease. J. Nucl. Med. 2010, 51, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Blum-Degen, D.; Muller, T.; Kuhn, W.; Gerlach, M.; Przuntek, H.; Riederer, P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 1995, 202, 17–20. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riedered, P.; Nagatsu, T. Interleukin-2 but not basic fibroblast growth factor is elevated in parkinsonian brain. J. Neural Transm. 1996, 103, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Sawada, M. Inflammatory process in Parkinson’s disease: Role for cytokines. Curr. Pharm. Des. 2005, 11, 999–1016. [Google Scholar] [CrossRef] [PubMed]
- Duke, D.C.; Moran, L.B.; Pearce, R.K.; Graeber, M.B. The medial and lateral substantia nigra in Parkinson’s disease: mRNA profiles associated with higher brain tissue vulnerability. Neurogenetics 2007, 8, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Sawada, M. Biochemistry of post-mortem brains in Parkinson’s disease: Historical overview and future prospects. J. Neural Transm. Suppl. 2007, 72, 113–120. [Google Scholar] [PubMed]
- Miklossy, J.; Doudet, D.D.; Schwab, C.; Yu, S.; McGeer, E.G.; McGeer, P.L. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp. Neurol. 2006, 197, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Phani, S.; Loike, J.D.; Przedborski, S. Neurodegeneration and inflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, S207–S209. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Guiroy, DC.; Wakayama, I.; Liberski, P.P.; Gajdusek, D.C. Relationship of microglia and scrapie amyloid-immunoreactive plaques in kuru, Creutzfeldt-Jakob disease and Gerstmann-Straussler syndrome. Acta Neuropathol. 1994, 87, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.; Brown, D.R.; Groschup, M.H.; Feldmann, C.; Haist, I.; Kretzschmar, H.A. Role of microglia in neuronal cell death in prion disease. Brain Pathol. 1998, 8, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, saboteur, or something else? Science 2013, 339, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Nuvolone, M.; Zhu, C. The immunobiology of prion diseases. Nat. Rev. Immunol. 2013, 13, 888–902. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Kobayashi, K.; Isaki, K. Microglial and astrocytic change in brains of Creutzfeldt-Jakob disease: An immunocytochemical and quantitative study. Clin. Neuropathol. 1999, 18, 51–60. [Google Scholar] [PubMed]
- Muhleisen, H.; Gehrmann, J.; Meyermann, R. Reactive microglia in Creutzfeldt-Jakob disease. Neuropathol. Appl. Neurobiol. 1995, 21, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Schmidt, B.; Kretzschmar, H.A. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature 1996, 380, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Eitzen, U.; Egensperger, R.; Kosel, S.; Grasbon-Frodl, E.M.; Imai, Y.; Bise, K.; Kohsaka, S.; Mehraein, P.; Graeber, M.B. Microglia and the development of spongiform change in Creutzfeldt-Jakob disease. J. Neuropathol. Exp. Neurol. 1998, 57, 246–256. [Google Scholar] [CrossRef]
- Baker, C.A.; Lu, Z.Y.; Zaitsev, I.; Manuelidis, L. Microglial activation varies in different models of Creutzfeldt-Jakob disease. J. Virol. 1999, 73, 5089–5097. [Google Scholar] [PubMed]
- Rezaie, P.; Lantos, P.L. Microglia and the pathogenesis of spongiform encephalopathies. Brain Res. Brain Res. Rev. 2001, 35, 55–72. [Google Scholar] [CrossRef]
- Van Everbroeck, B.; Dewulf, E.; Pals, P.; Lübke, U.; Martin, J.J.; Cras, P. The role of cytokines, astrocytes, microglia and apoptosis in Creutzfeldt-Jakob disease. Neurobiol. Aging 2002, 23, 59–64. [Google Scholar] [CrossRef]
- Baker, C.A.; Manuelidis, L. Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc. Natl. Acad. Sci. USA 2003, 100, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Puoti, G.; Giaccone, G.; Mangieri, M.; Limido, L.; Fociani, P.; Zerbi, P.; Suardi, S.; Rossi, G.; Iussich, S.; Capobianco, R.; et al. Sporadic Creutzfeldt-Jakob disease: The extent of microglia activation is dependent on the biochemical type of PrPSc. J. Neuropathol. Exp. Neurol. 2005, 64, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Szpak, G.M.; Lewandowska, E.; Lechowicz, W.; Wierzba-Bobrowicz, T.; Kulczycki, J.; Bertrand, E.; Pasennik, E.; Dymecki, J. The brain immune response in human prion diseases. Microglial activation and microglial disease. I. Sporadic Creutzfeldt-Jakob disease. Folia Neuropathol. 2006, 44, 202–213. [Google Scholar] [PubMed]
- Wojtera, M.; Sobów, T.; Kłoszewska, I.; Liberski, P.P.; Brown, D.R.; Sikorska, B. Expression of immunohistochemical markers on microglia in Creutzfeldt-Jakob disease and Alzheimer’s disease: Morphometric study and review of the literature. Folia Neuropathol. 2012, 50, 74–84. [Google Scholar] [PubMed]
- Shi, Q.; Xie, W.L.; Zhang, B.; Chen, L.N.; Xu, Y.; Wang, K.; Ren, K.; Zhang, X.M.; Chen, C.; Zhang, J.; et al. Brain microglia were activated in sporadic CJD but almost unchanged in fatal familial insomnia and G114V genetic CJD. Virol. J. 2013, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Sharief, M.K.; Green, A.; Dick, J.P.; Gawler, J.; Thompson, E.J. Heightened intrathecal release of proinflammatory cytokines in Creutzfeldt-Jakob disease. Neurology 1999, 52, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.A.; Martin, D.; Manuelidis, L. Microglia from Creutzfeldt-Jakob disease-infected brains are infectious and show specific mRNA activation profiles. J. Virol. 2002, 76, 10905–10913. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Dua, P.; Pogue, A.I.; Eicken, C.; Hill, J.M. Upregulation of micro RNA-146a (miRNA-146a), a marker for inflammatory neurodegeneration, in sporadic Creutzfeldt-Jakob disease (sCJD) and Gerstmann-Straussler-Scheinker (GSS) syndrome. J. Toxicol. Environ. Health A 2011, 74, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Stoeck, K.; Bodemer, M.; Zerr, I. Pro- and anti-inflammatory cytokines in the CSF of patients with Creutzfeldt-Jakob disease. J. Neuroimmunol. 2006, 172, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Stoec, K.; Schmitz, M.; Ebert, E.; Schmidt, C.; Zerr, I. Immune responses in rapidly progressive dementia: A comparative study of neuroinflammatory markers in Creutzfeldt-Jakob disease, Alzheimer’s disease and multiple sclerosis. J. Neuroinflamm. 2014, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.; Bancher, C.; Jellinger, K. Microglial reaction in Pick’s disease. Neurosci. Lett. 1993, 161, 89–92. [Google Scholar] [CrossRef]
- Schofield, E.; Kersaitis, C.; Shepherd, C.E.; Kril, J.J.; Halliday, G.M. Severity of gliosis in Pick’s disease and frontotemporal lobar degeneration: Tau-positive glia differentiate these disorders. Brain 2003, 126, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Botrán, R.; Ahmed, Z.; Crespo, F.A.; Gatenbee, C.; Gonzalez, J.; Dickson, D.W.; Litvan, I. Cytokine expression and microglial activation in progressive supranuclear palsy. Parkinsonism Relat. Disord. 2011, 17, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Lant, S.B.; Robinson, A.C.; Thompson, J.C.; Rollinson, S.; Pickering-Brown, S.; Snowden, J.S.; Davidson, Y.S.; Gerhard, A.; Mann, D.M. Patterns of microglial cell activation in frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 2014, 40, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Bugiani, O.; Ghetti, B.; Spillantini, M.G. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener. Dis. 2011, 8, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Esparcia, P.; Llorens, F.; Carmona, M.; Ferrer, I. Complex deregulation and expression of cytokines and mediators of the immune response in Parkinson’s disease brain is region dependent. Brain Pathol. 2014, 24, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; López-González, I.; Thüne, K.; Carmona, M.; Zafar, S.; Andréoletti, O.; Zerr, I.; Ferrer, I. Subtype and regional-specific neuroinflammation in sporadic Creutzfeldt-Jakob disease. Front. Aging Neurosci. 2014, 6, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-González, I.; Schlüter, A.; Aso, E.; Garcia-Esparcia, P.; Ansoleaga, B.; LLorens, F.; Carmona, M.; Moreno, J.; Fuso, A.; Portero-Otin, M.; et al. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: Correlations with plaques, tangles, and oligomeric species. J. Neuropathol. Exp. Neurol. 2015, 74, 319–344. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Samkons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Braak, H.; Xue, Q.S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Schuitemaker, A.; Dik, M.G.; Veerhuis, R.; Scheltens, P.; Schoonenboom, N.S.; Hack, C.E.; Blankenstein, M.A.; Jonker, C. Inflammatory markers in AD and MCI patients with different biomarker profiles. Neurobiol. Aging 2009, 30, 1885–1889. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Chung, J.H.; Choi, T.K.; Suh, S.Y.; Oh, B.H.; Hong, C.H. Peripheral cytokines and chemokines in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2009, 28, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, O.V.; Diniz, B.S.; Talib, L.L.; Mendonça, V.A.; Ojopi, E.B.; Gattaz, W.F.; Teixeira, A.L. Increased serum IL-1beta level in Alzheimer’s disease and mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2009, 28, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Fiala, M.; Veerhuis, R. Biomarkers of inflammation and amyloid-beta phagocytosis in patients at risk of Alzheimer disease. Exp. Gerontol. 2010, 45, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yin, Y.; Zhao, Z.; Huang, L.; Huang, S.; Zhuang, J.; Wu, H.; Peng, H.; Li, P. Elevation of serum TNF-α levels in mild and moderate Alzheimer patients with daytime sleepiness. J. Neuroimmunol. 2012, 244, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Sudduth, T.L.; Schmitt, F.A.; Nelson, P.T.; Wilcock, D.M. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Dursun, E.; Gezen-Ak, D.; Hanağası, H.; Bilgiç, B.; Lohmann, E.; Ertan, S.; Atasoy, İ.L.; Alaylıoğlu, M.; Araz, Ö.S.; Önal, B.; et al. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J. Neuroimmunol. 2015, 283, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Diniz, B.S.; Teixeira, A.L.; Ojopi, E.B.; Talib, L.L.; Mendonça, V.A.; Gattaz, W.F.; Forlenza, O.V. Higher serum sTNFR1 level predicts conversion from mild cognitive impairment to Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Cunningham, C.; Zotova, E.; Culliford, D.; Perry, V.H. Pro-inflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology 2011, 77, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Kannarkat, G.T.; Boss, J.M.; Tansey, M.G. The role of innate and adaptive immunity in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 493–514. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Pefetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.H.; Rowe, D.; Morel-Kopp, M.C.; Orr, C.; Russell, T.; Ranola, M.; Ward, C.; Halliday, G.M. Reduced T helper and B lymphocytes in Parkinson’s disease. J. Neuroimmunol. 2012, 252, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Saunders, J.A.; Estes, K.A.; Kosloski, L.M.; Allen, H.E.; Dempsey, K.M.; Torres-Russotto, D.R.; Meza, J.L.; Santamaria, P.M.; Bertoni, J.M.; Murman, D.L.; et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J. Neuroimmune Pharmacol. 2012, 7, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Bessler, H.; Djaldetti, R.; Salman, H.; Bergman, M.; Djaldetti, M. IL-1 beta, IL-2, IL-6 and TNF-alpha production by peripheral blood mononuclear cells from patients with Parkinson’s disease. Biomed. Pharmacother. 1999, 53, 141–145. [Google Scholar] [CrossRef]
- Chen, H.; O’Reilly, E.J.; Schwarzchild, M.A.; Adscherio, A. Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am. J. Epidemiol. 2008, 167, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Dufek, M.; Hamanová, M.; Lokaj, J.; Goldemund, D.; Rektorová, I.; Michálková, Z.; Sheardová, K.; Rektor, I. Serum inflammatory biomarkers in Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Rentzos, M.; Nikolaou, C.; Andreadou, E.; Paraskevas, G.P.; Rombos, A.; Zoga, M.; Tsoutsou, A.; Boufidou, F.; Kapaki, E.; Vassilopoulos, D. Circulating interleukin-15 and RANTES chemokine in Parkinson’s disease. Acta Neurol. Scand. 2007, 116, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Rentzos, M.; Nikolaou, C.; Andreadou, E.; Paraskevas, G.P.; Rombos, A.; Zoga, M.; Tsoutsou, A.; Boufidou, F.; Kapaki, E.; Vassilopoulos, D. Circulating interleukin-10 and interleukin-12 in Parkinson’s disease. Acta Neurol. Scand. 2009, 119, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Brodacki, B.; Staszewski, J.; Toczyłowska, B.; Kozłowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, P.; Kümmer, A.; Cardoso, F.; Teixeira, A.L. Increased serum levels of soluble tumor necrosis factor-alpha receptor-1 in patients with Parkinson’s disease. J. Neuroimmunol. 2009, 216, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Koziorowski, D.; Tomasiuk, R.; Szlufik, S.; Friedman, A. Inflammatory cytokines and NT-proCNP in Parkinson’s disease patients. Cytokine 2012, 60, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Menza, M.; Dobkin, R.D.; Marin, H.; Mark, M.H.; Gara, M.; Bienfait, K.; Dicke, A.; Kusnekov, A. The role of inflammatory cytokines in cognition and other non-motor symptoms of Parkinson’s disease. Psychosomatics 2010, 51, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, P.; Stemberger, S.; Sprenger, F.; Rainer, J.; Hametner, E.; Kirchmair, R.; Grabmer, C.; Scherfler, C.; Wenning, G.K.; Seppi, K.; et al. An antibody microarray analysis of serum cytokines in neurodegenerative Parkinsonian syndromes. Proteome Sci. 2012, 10, 71. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López González, I.; Garcia-Esparcia, P.; Llorens, F.; Ferrer, I. Genetic and Transcriptomic Profiles of Inflammation in Neurodegenerative Diseases: Alzheimer, Parkinson, Creutzfeldt-Jakob and Tauopathies. Int. J. Mol. Sci. 2016, 17, 206. https://doi.org/10.3390/ijms17020206
López González I, Garcia-Esparcia P, Llorens F, Ferrer I. Genetic and Transcriptomic Profiles of Inflammation in Neurodegenerative Diseases: Alzheimer, Parkinson, Creutzfeldt-Jakob and Tauopathies. International Journal of Molecular Sciences. 2016; 17(2):206. https://doi.org/10.3390/ijms17020206
Chicago/Turabian StyleLópez González, Irene, Paula Garcia-Esparcia, Franc Llorens, and Isidre Ferrer. 2016. "Genetic and Transcriptomic Profiles of Inflammation in Neurodegenerative Diseases: Alzheimer, Parkinson, Creutzfeldt-Jakob and Tauopathies" International Journal of Molecular Sciences 17, no. 2: 206. https://doi.org/10.3390/ijms17020206