
Insights on Molecular Mechanisms of Chondrocytes Death in Osteoarthritis

 ,
,
Abstract
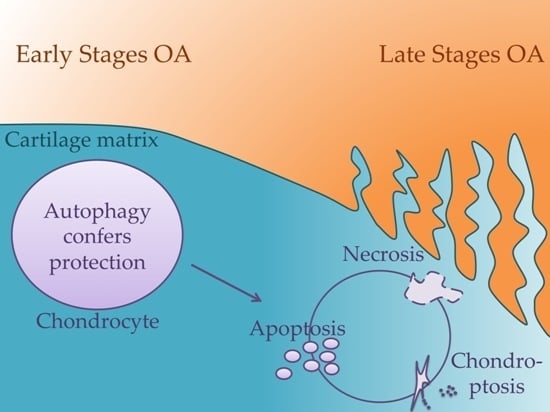
:
1. Osteoarthritis
2. Cell Death Mechanisms
2.1. Apoptosis
2.1.1. The Death Receptors Pathway (Extrinsic)
2.1.2. The Mitochondrial Pathway (Intrinsic)
2.1.3. Granzyme B Pathway
2.2. Necrosis
Discrimination between Apoptosis and Necrosis
2.3. Autophagic Cell Death
3. Apoptosis in Chondrocytes and Osteoarthritis (OA) Development
3.1. Apoptosis Incidence and Contribution to OA Development
3.1.1. Experiment Type
3.1.2. Kinetic Inconsistency
3.1.3. Spaciotemporality
3.1.4. Fate of Apoptotic Bodies
3.1.5. Chondroptosis
3.1.6. Cause or Consequence
3.2. Apoptosis as a Consequence of OA Progression
3.3. Apoptosis as a Cause of OA Progression
3.4. Chondrocytes Apoptosis in OA Compared with Rheumatoid Arthritis (RA)
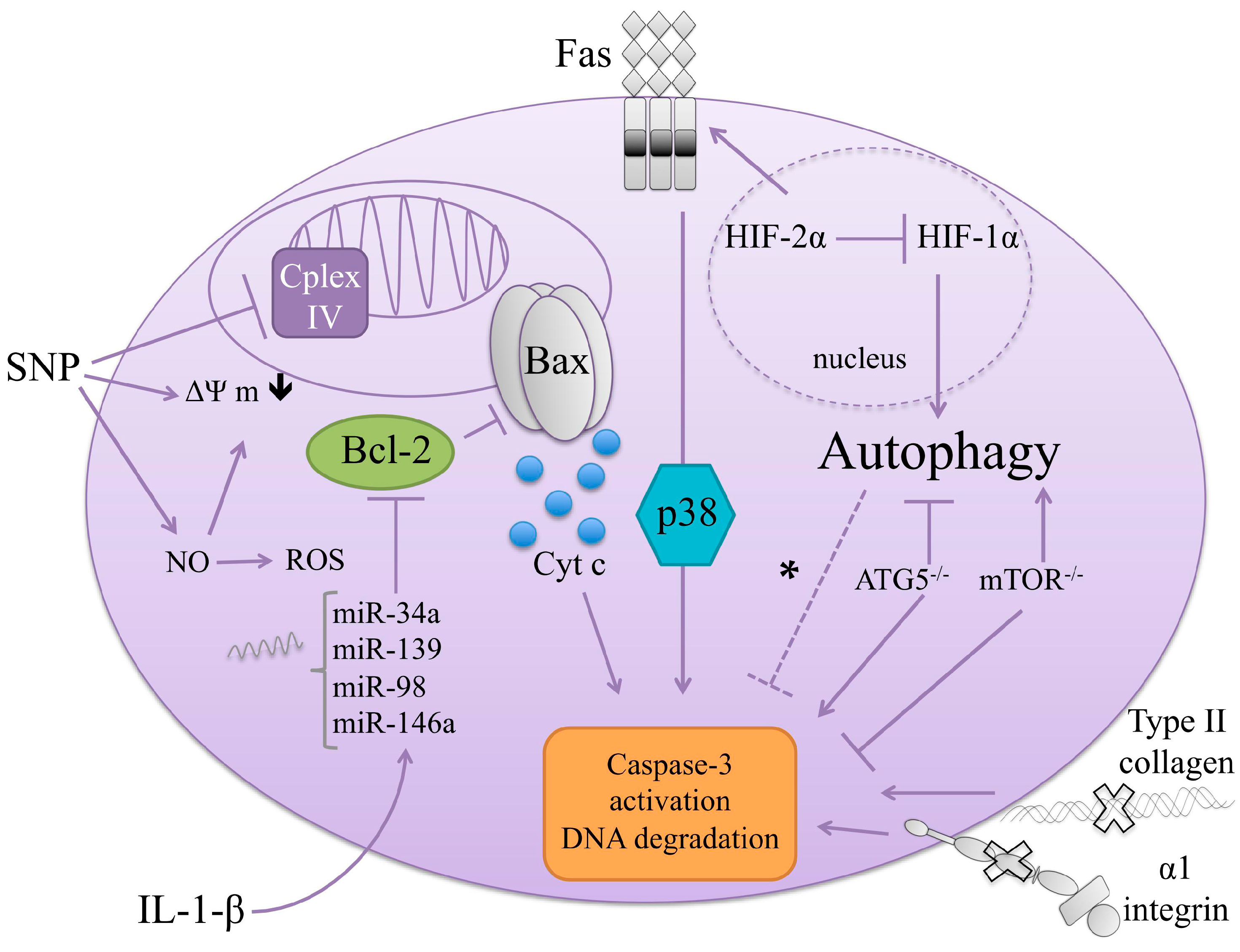
4. Autophagy in Chondrocytes and OA Development
Autophagy Regulators in Chondrocytes: The Hypoxia Inducible Factors (HIFs)
5. Cell Death Regulators in Chondrocytes
5.1. Inducers
5.1.1. Fas-FasL
5.1.2. Tumor Necrosis Factor-α (TNF-α) and Interleukin-1β (IL-1β)
5.1.3. Leptin
5.1.4. Nitric Oxide (NO) and Reactive Oxygen Species (ROS)
5.1.5. Mechanical Stresses
5.2. Inhibitory Pathways
5.2.1. Wnt Signaling
5.2.2. Bcl-2 Signaling
5.2.3. Insulin-Like Growth Factor-1 (IGF-1) Signaling
5.2.4. TGF-β Signaling
6. Targeting Chondrocyte Apoptosis for OA Treatment
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Glyn-Jones, S.; Palmer, A.J.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Michaelis, M.; Ladel, C.; Siebuhr, A.S.; Bihlet, A.R.; Andersen, J.R.; Guehring, H.; Christiansen, C.; Bay-Jensen, A.C.; Kraus, V.B. Disease-modifying treatments for osteoarthritis (DMOADs) of the knee and hip: Lessons learned from failures and opportunities for the future. Osteoarthr. Cartil. 2016, 24, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Bellamy, N.; Campbell, J.; Robinson, V.; Gee, T.; Bourne, R.; Wells, G. Intraarticular corticosteroid for treatment of osteoarthritis of the knee. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef]
- Maheu, E.; Rannou, F.; Reginster, J.Y. Efficacy and safety of hyaluronic acid in the management of osteoarthritis: Evidence from real-life setting trials and surveys. Semin. Arthritis Rheum. 2016, 45, S28–S33. [Google Scholar] [CrossRef] [PubMed]
- Portal-Nunez, S.; Esbrit, P.; Alcaraz, M.J.; Largo, R. Oxidative stress, autophagy, epigenetic changes and regulation by miRNAs as potential therapeutic targets in osteoarthritis. Biochem. Pharmacol. 2016, 108, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Doherty, M.; Leeb, B.F.; Alekseeva, L.; Arden, N.K.; Bijlsma, J.W.; Dincer, F.; Dziedzic, K.; Hauselmann, H.J.; Kaklamanis, P.; et al. EULAR evidence-based recommendations for the diagnosis of hand osteoarthritis: Report of a task force of ESCISIT. Ann. Rheum. Dis. 2009, 68, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Ort, T.; Ma, K.; Picha, K.; Carton, J.; Marsters, P.A.; Lohmander, L.S.; Baribaud, F.; Song, X.Y.; Blake, S. Resistin is elevated following traumatic joint injury and causes matrix degradation and release of inflammatory cytokines from articular cartilage in vitro. Osteoarthr. Cartil. 2009, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.Z.; Guan, J.; Wang, H.J.; Ma, W.; Li, F.; Xu, F.; Ding, L.B.; Xie, L.; Liu, B.; Liu, K.; et al. Possible involvement of serum and synovial fluid resistin in knee osteoarthritis: Cartilage damage, clinical, and radiological links. J. Clin. Lab. Anal. 2016, 30, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Ryu, J.H.; Oh, H.; Jeon, J.; Kwak, J.S.; Kim, J.H.; Kim, H.A.; Chun, C.H.; Chun, J.S. NAMPT (visfatin), a direct target of hypoxia-inducible factor-2α, is an essential catabolic regulator of osteoarthritis. Ann. Rheum. Dis. 2015, 74, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Raynauld, J.P.; Dorais, M.; Abram, F.; Pelletier, J.P. The levels of the adipokines adipsin and leptin are associated with knee osteoarthritis progression as assessed by MRI and incidence of total knee replacement in symptomatic osteoarthritis patients: A post hoc analysis. Rheumatology 2016, 55, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Dumond, H.; Presle, N.; Terlain, B.; Mainard, D.; Loeuille, D.; Netter, P.; Pottie, P. Evidence for a key role of leptin in osteoarthritis. Arthritis Rheum. 2003, 48, 3118–3129. [Google Scholar] [CrossRef] [PubMed]
- Zeddou, M.; Relic, B.; Malaise, O.; Charlier, E.; Desoroux, A.; Beguin, Y.; de Seny, D.; Malaise, M.G. Differential signalling through ALK-1 and ALK-5 regulates leptin expression in mesenchymal stem cells. Stem Cells Dev. 2012, 21, 1948–1955. [Google Scholar] [CrossRef] [PubMed]
- De Seny, D.; Cobraiville, G.; Charlier, E.; Neuville, S.; Esser, N.; Malaise, D.; Malaise, O.; Calvo, F.Q.; Relic, B.; Malaise, M.G. Acute-phase serum amyloid a in osteoarthritis: Regulatory mechanism and proinflammatory properties. PLoS ONE 2013, 8, e66769. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Goldring, S.R. Osteoarthritis. J. Cell. Physiol. 2007, 213, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Archer, C.W.; Francis-West, P. The chondrocyte. Int. J. Biochem. Cell Biol. 2003, 35, 401–404. [Google Scholar] [CrossRef]
- Eyre, D. Collagen of articular cartilage. Arthritis Res. 2002, 4, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roughley, P.J. Articular cartilage and changes in arthritis: Noncollagenous proteins and proteoglycans in the extracellular matrix of cartilage. Arthritis Res. 2001, 3, 342–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speziali, A.; Delcogliano, M.; Tei, M.; Placella, G.; Chillemi, M.; Tiribuzi, R.; Cerulli, G. Chondropenia: Current concept review. Musculoskelet. Surg. 2015, 99, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, H.; Richter, W. Osteoarthritis: Cellular and molecular changes in degenerating cartilage. Prog. Histochem. Cytochem. 2006, 40, 135–163. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M.; Plumb, D.A.; Dragomir, C.; Favero, M.; El Hachem, K.; Hashimoto, K.; Roach, H.I.; Olivotto, E.; Borzi, R.M.; et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: Signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur. Cells Mater. 2011, 21, 202–220. [Google Scholar] [CrossRef]
- Aigner, T.; Kim, H.A.; Roach, H.I. Apoptosis in osteoarthritis. Rheum. Dis. Clin. N. Am. 2004, 30, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.J.; Aigner, T. Articular cartilage and changes in arthritis. An introduction: Cell biology of osteoarthritis. Arthritis Res. 2001, 3, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.M.; Fuller, C.J.; Whittles, C.E.; Sharif, M. Chondrocyte death by apoptosis is associated with cartilage matrix degradation. Osteoarthr. Cartil. 2007, 15, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Lee, Y.J.; Seong, S.C.; Choe, K.W.; Song, Y.W. Apoptotic chondrocyte death in human osteoarthritis. J. Rheumatol. 2000, 27, 455–462. [Google Scholar] [PubMed]
- Roach, H.I.; Aigner, T.; Kouri, J.B. Chondroptosis: A variant of apoptotic cell death in chondrocytes? Apoptosis 2004, 9, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Burton-Wurster, N.; Borden, C.; Hueffer, K.; Bloom, S.E.; Lust, G. Chondrocyte necrosis and apoptosis in impact damaged articular cartilage. J. Orthop. Res. 2001, 19, 703–711. [Google Scholar] [CrossRef]
- Mitrovic, D.; Quintero, M.; Stankovic, A.; Ryckewaert, A. Cell density of adult human femoral condylar articular cartilage. Joints with normal and fibrillated surfaces. Lab. Investig. 1983, 49, 309–316. [Google Scholar] [PubMed]
- Chang, J.; Wang, W.; Zhang, H.; Hu, Y.; Wang, M.; Yin, Z. The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int. J. Mol. Med. 2013, 32, 1311–1318. [Google Scholar] [PubMed]
- Almonte-Becerril, M.; Navarro-Garcia, F.; Gonzalez-Robles, A.; Vega-Lopez, M.A.; Lavalle, C.; Kouri, J.B. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis 2010, 15, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Kaiser, W.J.; Bertrand, M.J.; Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell. Oncol. 2015, 2, e975093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Perez, H.E.; Luna, M.J.; Rojas, M.L.; Kouri, J.B. Chondroptosis: An immunohistochemical study of apoptosis and Golgi complex in chondrocytes from human osteoarthritic cartilage. Apoptosis 2005, 10, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Kurosaka, K.; Takahashi, M.; Watanabe, N.; Kobayashi, Y. Silent cleanup of very early apoptotic cells by macrophages. J. Immunol. 2003, 171, 4672–4679. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G. Active role of chondrocyte apoptosis in endochondral ossification. Microsc. Res. Tech. 1998, 43, 191–204. [Google Scholar] [CrossRef]
- Norbury, C.J.; Hickson, I.D. Cellular responses to DNA damage. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 367–401. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Declercq, W.; Kalai, M.; Saelens, X.; Vandenabeele, P. Alice in caspase land. A phylogenetic analysis of caspases from worm to man. Cell Death Differ. 2002, 9, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [PubMed]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [PubMed]
- Porter, A.G.; Janicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Kroemer, G. Mechanisms of apoptotic phosphatidylserine exposure. Cell Res. 2013, 23, 1247–1248. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Moitra, P.K.; Saha, S.; Basu, J. Caspase 3 regulates phosphatidylserine externalization and phagocytosis of oxidatively stressed erythrocytes. FEBS Lett. 2002, 513, 184–188. [Google Scholar] [CrossRef]
- Gaur, U.; Aggarwal, B.B. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem. Pharmacol. 2003, 66, 1403–1408. [Google Scholar] [CrossRef]
- Lavrik, I.; Golks, A.; Krammer, P.H. Death receptor signaling. J. Cell Sci. 2005, 118 Pt 2, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P.; Scaffidi, C.; Kischkel, F.C.; Shevchenko, A.; Mann, M.; Krammer, P.H.; Peter, M.E. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J. 1997, 16, 2794–2804. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Prior, P.; Salvesen, G.S. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 2004, 384 Pt 2, 201–232. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998, 17, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, G.; van Gurp, M.; Depuydt, B.; Srinivasula, S.M.; Rodriguez, I.; Alnemri, E.S.; Gevaert, K.; Vandekerckhove, J.; Declercq, W.; Vandenabeele, P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ. 2002, 9, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, G.; Demol, H.; van Gurp, M.; Hoorelbeke, B.; Schotte, P.; Beyaert, R.; Zhivotovsky, B.; Gevaert, K.; Declercq, W.; Vandekerckhove, J.; et al. A matrix-assisted laser desorption ionization post-source decay (MALDI-PSD) analysis of proteins released from isolated liver mitochondria treated with recombinant truncated Bid. Cell Death Differ. 2002, 9, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. Overview: Apoptotic signaling pathways in the immune system. Immunol. Rev. 2003, 193, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.; Shi, Y. Apoptosome: A platform for the activation of initiator caspases. Cell Death Differ. 2007, 14, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J. The ABCs of granule-mediated cytotoxicity: New weapons in the arsenal. Nat. Rev. Immunol. 2003, 3, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Boivin, W.A.; Cooper, D.M.; Hiebert, P.R.; Granville, D.J. Intracellular versus extracellular granzyme B in immunity and disease: Challenging the dogma. Lab. Investig. 2009, 89, 1195–1220. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Saito, S.; Sasaki, R.; Tomatsu, T.; Toyama, Y. Expression of granzyme B in human articular chondrocytes. J. Rheumatol. 2003, 30, 1799–1810. [Google Scholar] [PubMed]
- Saito, S.; Murakoshi, K.; Kotake, S.; Kamatani, N.; Tomatsu, T. Granzyme B induces apoptosis of chondrocytes with natural killer cell-like cytotoxicity in rheumatoid arthritis. J. Rheumatol. 2008, 35, 1932–1943. [Google Scholar] [PubMed]
- Kummer, J.A.; Tak, P.P.; Brinkman, B.M.; van Tilborg, A.A.; Kamp, A.M.; Verweij, C.L.; Daha, M.R.; Meinders, A.E.; Hack, C.E.; Breedveld, F.C. Expression of granzymes A and B in synovial tissue from patients with rheumatoid arthritis and osteoarthritis. Clin. Immunol. Immunopathol. 1994, 73, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Festjens, N.; Vanden Berghe, T.; Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 2006, 1757, 1371–1387. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Laster, S.M.; Wood, J.G.; Gooding, L.R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 1988, 141, 2629–2634. [Google Scholar] [PubMed]
- Roach, H.I.; Clarke, N.M. Physiological cell death of chondrocytes in vivo is not confined to apoptosis. New observations on the mammalian growth plate. J. Bone Jt. Surg. Br. 2000, 82, 601–613. [Google Scholar] [CrossRef]
- Coustry, F.; Posey, K.L.; Liu, P.; Alcorn, J.L.; Hecht, J.T. D469del-COMP retention in chondrocytes stimulates caspase-independent necroptosis. Am. J. Pathol. 2012, 180, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Rho, J.H.; Lee, S.Y.; Kim, J.H.; Cheong, J.H.; Kim, H.Y.; Jeong, N.Y.; Chung, W.T.; Yoo, Y.H. Leptin protects rat articular chondrocytes from cytotoxicity induced by TNF-α in the presence of cyclohexamide. Osteoarthr. Cartil. 2015, 23, 2269–2278. [Google Scholar] [CrossRef] [PubMed]
- Zeiss, C.J. The apoptosis-necrosis continuum: Insights from genetically altered mice. Vet. Pathol. 2003, 40, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Single, B.; Castoldi, A.F.; Kuhnle, S.; Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: A switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Denecker, G.; Vercammen, D.; Declercq, W.; Vandenabeele, P. Apoptotic and necrotic cell death induced by death domain receptors. Cell. Mol. Life Sci. 2001, 58, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Maneiro, E.; Lopez-Armada, M.J.; de Andres, M.C.; Carames, B.; Martin, M.A.; Bonilla, A.; del Hoyo, P.; Galdo, F.; Arenas, J.; Blanco, F.J. Effect of nitric oxide on mitochondrial respiratory activity of human articular chondrocytes. Ann. Rheum. Dis. 2005, 64, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Vanden Berghe, T.; D’Herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Grogan, S.P.; Aklin, B.; Frenz, M.; Brunner, T.; Schaffner, T.; Mainil-Varlet, P. In vitro model for the study of necrosis and apoptosis in native cartilage. J. Pathol. 2002, 198, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T. Apoptosis, necrosis, or whatever: How to find out what really happens? J. Pathol. 2002, 198, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Denecker, G.; Dooms, H.; van Loo, G.; Vercammen, D.; Grooten, J.; Fiers, W.; Declercq, W.; Vandenabeele, P. Phosphatidyl serine exposure during apoptosis precedes release of cytochrome c and decrease in mitochondrial transmembrane potential. FEBS Lett. 2000, 465, 47–52. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Layfield, R.; Lotz, M.; Settembre, C.; Whitehouse, C. Boning up on autophagy: The role of autophagy in skeletal biology. Autophagy 2014, 10, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Physiological functions of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 71–84. [Google Scholar] [PubMed]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [PubMed]
- Hatori, M.; Klatte, K.J.; Teixeira, C.C.; Shapiro, I.M. End labeling studies of fragmented DNA in the avian growth plate: Evidence of apoptosis in terminally differentiated chondrocytes. J. Bone Miner. Res. 1995, 10, 1960–1968. [Google Scholar] [CrossRef] [PubMed]
- Zenmyo, M.; Komiya, S.; Kawabata, R.; Sasaguri, Y.; Inoue, A.; Morimatsu, M. Morphological and biochemical evidence for apoptosis in the terminal hypertrophic chondrocytes of the growth plate. J. Pathol. 1996, 180, 430–433. [Google Scholar] [CrossRef]
- Aizawa, T.; Kokubun, S.; Tanaka, Y. Apoptosis and proliferation of growth plate chondrocytes in rabbits. J. Bone Jt. Sur. Br. 1997, 79, 483–486. [Google Scholar] [CrossRef]
- Adams, C.S.; Horton, W.E., Jr. Chondrocyte apoptosis increases with age in the articular cartilage of adult animals. Anat. Rec. 1998, 250, 418–425. [Google Scholar] [CrossRef]
- Heraud, F.; Heraud, A.; Harmand, M.F. Apoptosis in normal and osteoarthritic human articular cartilage. Ann. Rheum. Dis. 2000, 59, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Guitian, R.; Vazquez-Martul, E.; de Toro, F.J.; Galdo, F. Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis Rheum. 1998, 41, 284–289. [Google Scholar] [CrossRef]
- Hashimoto, S.; Ochs, R.L.; Komiya, S.; Lotz, M. Linkage of chondrocyte apoptosis and cartilage degradation in human osteoarthritis. Arthritis Rheum. 1998, 41, 1632–1638. [Google Scholar] [CrossRef]
- Millucci, L.; Giorgetti, G.; Viti, C.; Ghezzi, L.; Gambassi, S.; Braconi, D.; Marzocchi, B.; Paffetti, A.; Lupetti, P.; Bernardini, G.; et al. Chondroptosis in alkaptonuric cartilage. J. Cell. Physiol. 2015, 230, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Sharif, M.; Whitehouse, A.; Sharman, P.; Perry, M.; Adams, M. Increased apoptosis in human osteoarthritic cartilage corresponds to reduced cell density and expression of caspase-3. Arthritis Rheum. 2004, 50, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Kouri, J.B.; Aguilera, J.M.; Reyes, J.; Lozoya, K.A.; Gonzalez, S. Apoptotic chondrocytes from osteoarthrotic human articular cartilage and abnormal calcification of subchondral bone. J. Rheumatol. 2000, 27, 1005–1019. [Google Scholar] [PubMed]
- Aigner, T.; Hemmel, M.; Neureiter, D.; Gebhard, P.M.; Zeiler, G.; Kirchner, T.; McKenna, L. Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritis human articular knee cartilage: A study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage. Arthritis Rheum. 2001, 44, 1304–1312. [Google Scholar] [PubMed]
- Grasl-Kraupp, B.; Ruttkay-Nedecky, B.; Koudelka, H.; Bukowska, K.; Bursch, W.; Schulte-Hermann, R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: A cautionary note. Hepatology 1995, 21, 1465–1468. [Google Scholar] [PubMed]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Blanco, F.J. Cell death and apoptosis in osteoarthritic cartilage. Curr. Drug Targets 2007, 8, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Kim, H.A. Apoptosis and cellular vitality: Issues in osteoarthritic cartilage degeneration. Arthritis Rheum. 2002, 46, 1986–1996. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Gannon, F.H.; Sokoloff, L. Histomorphometry of the aging human patella: Histologic criteria and controls. Osteoarthr. Cartil. 1999, 7, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Stolberg-Stolberg, J.A.; Furman, B.D.; Garrigues, N.W.; Lee, J.; Pisetsky, D.S.; Stearns, N.A.; DeFrate, L.E.; Guilak, F.; Olson, S.A. Effects of cartilage impact with and without fracture on chondrocyte viability and the release of inflammatory markers. J. Orthop. Res. 2013, 31, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.; Zhang, J.; Zhang, M.; Wei, Y.; Wu, Y.; Qiu, Z.Y.; He, J.; Cao, Y.; Hu, J.; Zhu, H.; et al. The identification of CD163 expressing phagocytic chondrocytes in joint cartilage and its novel scavenger role in cartilage degradation. PLoS ONE 2013, 8, e53312. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Ochs, R.L.; Rosen, F.; Quach, J.; McCabe, G.; Solan, J.; Seegmiller, J.E.; Terkeltaub, R.; Lotz, M. Chondrocyte-derived apoptotic bodies and calcification of articular cartilage. Proc. Natl. Acad. Sci. USA 1998, 95, 3094–3099. [Google Scholar] [CrossRef] [PubMed]
- Wilsman, N.J.; Farnum, C.E.; Hilley, H.D.; Carlson, C.S. Ultrastructural evidence of a functional heterogeneity among physeal chondrocytes in growing swine. Am. J. Vet. Res. 1981, 42, 1547–1553. [Google Scholar] [PubMed]
- Farnum, C.E.; Wilsman, N.J. Condensation of hypertrophic chondrocytes at the chondro-osseous junction of growth plate cartilage in Yucatan swine: Relationship to long bone growth. Am. J. Anat. 1989, 186, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Kouri-Flores, J.B.; Abbud-Lozoya, K.A.; Roja-Morales, L. Kinetics of the ultrastructural changes in apoptotic chondrocytes from an osteoarthrosis rat model: A window of comparison to the cellular mechanism of apoptosis in human chondrocytes. Ultrastruct. Pathol. 2002, 26, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Nishida, K.; Yoshida, A.; Murakami, T.; Inoue, H. Expression of caspase-3 and -9 relevant to cartilage destruction and chondrocyte apoptosis in human osteoarthritic cartilage. Acta Medica Okayama 2001, 55, 333–340. [Google Scholar] [PubMed]
- Sitte, I.; Kathrein, A.; Pfaller, K.; Pedross, F.; Roberts, S. Intervertebral disc cell death in the porcine and human injured cervical spine after trauma: A histological and ultrastructural study. Spine 2009, 34, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, P.A.; Glasson, S.S.; Trubetskoy, O.V.; Haimes, H.B. Spontaneous osteoarthritis in Dunkin Hartley guinea pigs: Histologic, radiologic, and biochemical changes. Lab. Anim. Sci. 1997, 47, 598–601. [Google Scholar] [PubMed]
- Mobasheri, A. Role of chondrocyte death and hypocellularity in ageing human articular cartilage and the pathogenesis of osteoarthritis. Med. Hypotheses 2002, 58, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Takahashi, K.; Amiel, D.; Coutts, R.D.; Lotz, M. Chondrocyte apoptosis and nitric oxide production during experimentally induced osteoarthritis. Arthritis Rheum. 1998, 41, 1266–1274. [Google Scholar] [CrossRef]
- Yatsugi, N.; Tsukazaki, T.; Osaki, M.; Koji, T.; Yamashita, S.; Shindo, H. Apoptosis of articular chondrocytes in rheumatoid arthritis and osteoarthritis: Correlation of apoptosis with degree of cartilage destruction and expression of apoptosis-related proteins of p53 and c-myc. J. Orthop. Sci. 2000, 5, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Zamli, Z.; Sharif, M. Chondrocyte apoptosis: A cause or consequence of osteoarthritis? Int. J. Rheum. Dis. 2011, 14, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Zemmyo, M.; Meharra, E.J.; Kuhn, K.; Creighton-Achermann, L.; Lotz, M. Accelerated, aging-dependent development of osteoarthritis in α1 integrin-deficient mice. Arthritis Rheum. 2003, 48, 2873–2880. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, S.W.; Helminen, H.J.; Khillan, J.S.; Bao, Y.; Prockop, D.J. Apoptosis of chondrocytes in transgenic mice lacking collagen II. Exp. Cell Res. 1997, 235, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Withofs, N.; Charlier, E.; Simoni, P.; Alvarez-Miezentseva, V.; Mievis, F.; Giacomelli, F.; Mella, C.; Gambhir, S.S.; Malaise, O.; de Seny, D.; et al. 18F-FPRGD2 PET/CT imaging of musculoskeletal disorders. Ann. Nucl. Med. 2015, 29, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bryan, J.; Franz, C.; Havlioglu, N.; Sandell, L.J. Type IIB procollagen NH2-propeptide induces death of tumor cells via interaction with integrins αVβ3 and αVβ5. J. Biol. Chem. 2010, 285, 20806–20817. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Olmer, M.; Kiosses, W.B.; Lotz, M.K. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. 2015, 67, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Lopez de Figueroa, P.; Blanco, F.J.; Mendes, A.F.; Carames, B. Insulin decreases autophagy and leads to cartilage degradation. Osteoarthr. Cartil. 2016, 24, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Zamli, Z.; Adams, M.A.; Tarlton, J.F.; Sharif, M. Increased chondrocyte apoptosis is associated with progression of osteoarthritis in spontaneous Guinea pig models of the disease. Int. J. Mol. Sci. 2013, 14, 17729–17743. [Google Scholar] [CrossRef] [PubMed]
- Zamli, Z.; Robson Brown, K.; Tarlton, J.F.; Adams, M.A.; Torlot, G.E.; Cartwright, C.; Cook, W.A.; Vassilevskaja, K.; Sharif, M. Subchondral bone plate thickening precedes chondrocyte apoptosis and cartilage degradation in spontaneous animal models of osteoarthritis. BioMed Res. Int. 2014, 2014, 606870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mani, S.B.; He, Y.; Hall, A.M.; Xu, L.; Li, Y.; Zurakowski, D.; Jay, G.D.; Warman, M.L. Induced superficial chondrocyte death reduces catabolic cartilage damage in murine posttraumatic osteoarthritis. J. Clin. Investig. 2016, 126, 2893–2902. [Google Scholar] [CrossRef] [PubMed]
- Bobinac, D.; Spanjol, J.; Zoricic, S.; Maric, I. Changes in articular cartilage and subchondral bone histomorphometry in osteoarthritic knee joints in humans. Bone 2003, 32, 284–290. [Google Scholar] [CrossRef]
- Aigner, T.; Fundel, K.; Saas, J.; Gebhard, P.M.; Haag, J.; Weiss, T.; Zien, A.; Obermayr, F.; Zimmer, R.; Bartnik, E. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006, 54, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Bouderlique, T.; Vuppalapati, K.K.; Newton, P.T.; Li, L.; Barenius, B.; Chagin, A.S. Targeted deletion of Atg5 in chondrocytes promotes age-related osteoarthritis. Ann. Rheum. Dis. 2016, 75, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Loening, A.M.; James, I.E.; Levenston, M.E.; Badger, A.M.; Frank, E.H.; Kurz, B.; Nuttall, M.E.; Hung, H.H.; Blake, S.M.; Grodzinsky, A.J.; et al. Injurious mechanical compression of bovine articular cartilage induces chondrocyte apoptosis. Arch. Biochem. Biophys. 2000, 381, 205–212. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. Chondrocyte apoptosis in rheumatoid arthritis: Is preventive therapy possible? Immunotherapy 2015, 1, 102. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Song, Y.W. Apoptotic chondrocyte death in rheumatoid arthritis. Arthritis Rheum. 1999, 42, 1528–1537. [Google Scholar] [CrossRef]
- Kuhn, K.; Shikhman, A.R.; Lotz, M. Role of nitric oxide, reactive oxygen species, and p38 MAP kinase in the regulation of human chondrocyte apoptosis. J. Cell. Physiol. 2003, 197, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Schuerwegh, A.J.; Dombrecht, E.J.; Stevens, W.J.; van Offel, J.F.; Bridts, C.H.; de Clerck, L.S. Influence of pro-inflammatory (IL-1α, IL-6, TNF-α, IFN-γ) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthr. Cartil. 2003, 11, 681–687. [Google Scholar] [CrossRef]
- Wei, L.; Sun, X.; Kanbe, K.; Wang, Z.; Sun, C.; Terek, R.; Chen, Q. Chondrocyte death induced by pathological concentration of chemokine stromal cell-derived factor-1. J. Rheumatol. 2006, 33, 1818–1826. [Google Scholar] [PubMed]
- Schuerwegh, A.J.; Dombrecht, E.J.; Stevens, W.J.; van Offel, J.F.; Kockx, M.M.; Bridts, C.H.; de Clerck, L.S. Synovial fluid and peripheral blood immune complexes of patients with rheumatoid arthritis induce apoptosis in cytokine-activated chondrocytes. Rheumatol. Int. 2007, 27, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Zhu, S.; Liao, C.; Xiao, J.; Wu, Q.; Lin, Z.; Chen, J. Advanced oxidation protein products induce apoptosis of human chondrocyte through reactive oxygen species-mediated mitochondrial dysfunction and endoplasmic reticulum stress pathways. Fundam. Clin. Pharmacol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhong, Z.M.; Zhu, S.Y.; Liao, C.R.; Pan, Y.; Zeng, J.H.; Zheng, S.; Ding, R.T.; Lin, Q.S.; Ye, Q.; et al. Advanced oxidation protein products induce chondrocyte apoptosis via receptor for advanced glycation end products-mediated, redox-dependent intrinsic apoptosis pathway. Apoptosis 2016, 21, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Zhong, Z.; Zhu, S.; Zheng, S.; Xiao, J.; Song, S.; Yu, H.; Wu, Q.; Lin, Z.; Chen, J. Advanced oxidation protein products induce chondrocyte death through a redox-dependent, poly (ADP-ribose) polymerase-1-mediated pathway. Apoptosis 2016. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Zhong, Z.; Zhu, S.; Zheng, S.; Xiao, J.; Song, S.; Yu, H.; Wu, Q.; Lin, Z.; Chen, J. Advanced oxidation protein products induce catabolic effect through oxidant-dependent activation of NF-κB pathway in human chondrocyte. Int. Immunopharmacol. 2016, 39, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, K.; Ishijima, M.; Rittling, S.R.; Tsuji, K.; Tsuchiya, Y.; Kon, S.; Nifuji, A.; Uede, T.; Denhardt, D.T.; Noda, M. Osteopontin deficiency protects joints against destruction in anti-type II collagen antibody-induced arthritis in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 4556–4561. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.Y.; He, Z.N.; Zhang, B.; Li, Y.Q.; Guo, J.; Xu, Y.L.; Han, M.Y.; Chen, Z. Adenovirus-mediated osteoprotegerin ameliorates cartilage destruction by inhibiting proteoglycan loss and chondrocyte apoptosis in rats with collagen-induced arthritis. Cell Tissue Res. 2015, 362, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Rong, C.; Chen, F.H.; Jiang, S.; Hu, W.; Wu, F.R.; Chen, T.Y.; Yuan, F.L. Inhibition of acid-sensing ion channels by amiloride protects rat articular chondrocytes from acid-induced apoptosis via a mitochondrial-mediated pathway. Cell Biol. Int. 2012, 36, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Adan, N.; Guzman-Morales, J.; Ledesma-Colunga, M.G.; Perales-Canales, S.I.; Quintanar-Stephano, A.; Lopez-Barrera, F.; Mendez, I.; Moreno-Carranza, B.; Triebel, J.; Binart, N.; et al. Prolactin promotes cartilage survival and attenuates inflammation in inflammatory arthritis. J. Clin. Investig. 2013, 123, 3902–3913. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Hu, S. Recent insights into the role of autophagy in the pathogenesis of rheumatoid arthritis. Rheumatology 2016, 55, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.Y.; Beyer, C.; Giessl, A.; Kireva, T.; Scholtysek, C.; Uderhardt, S.; Munoz, L.E.; Dees, C.; Distler, A.; Wirtz, S.; et al. Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann. Rheum. Dis. 2013, 72, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, W.B. Lessons from animal models of osteoarthritis. Curr. Rheumatol. Rep. 2008, 10, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Guan, Z.P.; Zhang, S.L.; Pei, Z.; Chen, Y.Y.; Pan, H. Programmed cell death 5 correlates with disease activity and interleukin-17 in serum and synovial fluid of rheumatoid arthritis patients. Chin. Med. J. 2013, 126, 296–299. [Google Scholar] [PubMed]
- Malemud, C.J. The PI3K/Akt/PTEN/mTOR pathway: A fruitful target for inducing cell death in rheumatoid arthritis? Future Med. Chem. 2015, 7, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Lopez de Figueroa, P.; Nogueira-Recalde, U.; Centeno, A.; Mendes, A.F.; Blanco, F.J.; Carames, B. Diabetes-accelerated experimental osteoarthritis is prevented by autophagy activation. Osteoarthr. Cartil. 2016, 24, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Lopez de Figueroa, P.; Lotz, M.K.; Blanco, F.J.; Carames, B. Autophagy activation and protection from mitochondrial dysfunction in human chondrocytes. Arthritis Rheumatol. 2015, 67, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Takayama, K.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Fujita, N.; Oka, S.; Kurosaka, M.; Kuroda, R. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012, 64, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Bohensky, J.; Shapiro, I.M.; Leshinsky, S.; Terkhorn, S.P.; Adams, C.S.; Srinivas, V. HIF-1 regulation of chondrocyte apoptosis: Induction of the autophagic pathway. Autophagy 2007, 3, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Vasheghani, F.; Li, Y.H.; Blati, M.; Simeone, K.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Pelletier, J.P.; et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Vasheghani, F.; Zhang, Y.; Li, Y.H.; Blati, M.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Endisha, H.; Saffar, B.; et al. PPARγ deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann. Rheum. Dis. 2015, 74, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, I.B.; Jimenez, S.A.; Haw kins, D.F.; Piera-Velazquez, S.; Stokes, D.G. Hypoxia inducible factor-1α expression in human normal and osteoarthritic chondrocytes. Osteoarthr. Cartil. 2004, 12, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Kiaer, T.; Gronlund, J.; Sorensen, K.H. Subchondral pO2, pCO2, pressure, pH, and lactate in human osteoarthritis of the hip. Clin. Orthop. Relat. Res. 1988, 229, 149–155. [Google Scholar]
- Wang, G.L.; Semenza, G.L. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J. Biol. Chem. 1993, 268, 21513–21518. [Google Scholar] [PubMed]
- Schipani, E.; Ryan, H.E.; Didrickson, S.; Kobayashi, T.; Knight, M.; Johnson, R.S. Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genes Dev. 2001, 15, 2865–2876. [Google Scholar] [PubMed]
- Bohensky, J.; Leshinsky, S.; Srinivas, V.; Shapiro, I.M. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr. Nephrol. 2010, 25, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, J.; Chu, J.; Yang, C.; Xiao, H.; Zhao, C.; Sun, Z.; Gao, X.; Chen, G.; Han, Z.; et al. MicroRNA-146a Induced by Hypoxia Promotes Chondrocyte Autophagy through Bcl-2. Cell. Physiol. Biochem. 2015, 37, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Bohensky, J.; Terkhorn, S.P.; Freeman, T.A.; Adams, C.S.; Garcia, J.A.; Shapiro, I.M.; Srinivas, V. Regulation of autophagy in human and murine cartilage: Hypoxia-inducible factor 2 suppresses chondrocyte autophagy. Arthritis Rheum. 2009, 60, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.J.; Luo, W.; Lei, G.H. Role of HIF-1α and HIF-2α in osteoarthritis. J. Bone Spine 2015, 82, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Hirata, M.; Kugimiya, F.; Fukai, A.; Saito, T.; Yano, F.; Ikeda, T.; Mabuchi, A.; Sapkota, B.R.; Akune, T.; Nishida, N.; et al. C/EBPβ and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2α as the inducer in chondrocytes. Hum. Mol. Genet. 2012, 21, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Yang, S.; Shin, Y.; Rhee, J.; Chun, C.H.; Chun, J.S. Interleukin-6 plays an essential role in hypoxia-inducible factor 2α-induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum. 2011, 63, 2732–2743. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kim, J.; Ryu, J.H.; Oh, H.; Chun, C.H.; Kim, B.J.; Min, B.H.; Chun, J.S. Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 2010, 16, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Shin, Y.; Huh, Y.H.; Yang, S.; Chun, C.H.; Chun, J.S. Hypoxia-inducible factor-2α regulates Fas-mediated chondrocyte apoptosis during osteoarthritic cartilage destruction. Cell Death Differ. 2012, 19, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Todd Allen, R.; Robertson, C.M.; Harwood, F.L.; Sasho, T.; Williams, S.K.; Pomerleau, A.C.; Amiel, D. Characterization of mature vs aged rabbit articular cartilage: Analysis of cell density, apoptosis-related gene expression and mechanisms controlling chondrocyte apoptosis. Osteoarthr. Cartil. 2004, 12, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Sun, X.J.; Wang, Z.; Chen, Q. CD95-induced osteoarthritic chondrocyte apoptosis and necrosis: dependency on p38 mitogen-activated protein kinase. Arthritis Res. Ther. 2006, 8, R37. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Setareh, M.; Ochs, R.L.; Lotz, M. Fas/Fas ligand expression and induction of apoptosis in chondrocytes. Arthritis Rheum. 1997, 40, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, K.; Lotz, M. Regulation of CD95 (Fas/APO-1)-induced apoptosis in human chondrocytes. Arthritis Rheum. 2001, 44, 1644–1653. [Google Scholar] [CrossRef]
- Kuhn, K.; Hashimoto, S.; Lotz, M. Cell density modulates apoptosis in human articular chondrocytes. J. Cell. Physiol. 1999, 180, 439–447. [Google Scholar] [CrossRef]
- Charlier, E.; Conde, C.; Zhang, J.; Deneubourg, L.; di Valentin, E.; Rahmouni, S.; Chariot, A.; Agostinis, P.; Pang, P.C.; Haslam, S.M.; et al. SHIP-1 inhibits CD95/APO-1/Fas-induced apoptosis in primary T lymphocytes and T leukemic cells by promoting CD95 glycosylation independently of its phosphatase activity. Leukemia 2010, 24, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Tanaka, M.; Suda, T.; Tomita, T.; Hayashida, K.; Takeuchi, E.; Kaneko, M.; Takano, H.; Nagata, S.; Ochi, T. Soluble Fas ligand in the joints of patients with rheumatoid arthritis and osteoarthritis. Arthritis Rheum. 1998, 41, 657–662. [Google Scholar] [CrossRef]
- Vaillancourt, F.; Fahmi, H.; Shi, Q.; Lavigne, P.; Ranger, P.; Fernandes, J.C.; Benderdour, M. 4-Hydroxynonenal induces apoptosis in human osteoarthritic chondrocytes: The protective role of glutathione-S-transferase. Arthritis Res. Ther. 2008, 10, R107. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Kim, Y.H.; Song, Y.W. Facilitation of Fas mediated apoptosis of human chondrocytes by the proteasome inhibitor and actinomycin D. J. Rheumatol. 2003, 30, 550–558. [Google Scholar] [PubMed]
- Morquette, B.; Shi, Q.; Lavigne, P.; Ranger, P.; Fernandes, J.C.; Benderdour, M. Production of lipid peroxidation products in osteoarthritic tissues: New evidence linking 4-hydroxynonenal to cartilage degradation. Arthritis Rheum. 2006, 54, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733. [Google Scholar] [CrossRef] [PubMed]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Peluso, D.; Kanki, K.; Yang, Z.; et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Servais, J.; Polur, I.; Kim, D.; Lee, P.L.; Chung, K.; Li, Y. Attenuation of osteoarthritis progression by reduction of discoidin domain receptor 2 in mice. Arthritis Rheum. 2010, 62, 2736–2744. [Google Scholar] [CrossRef] [PubMed]
- Van Lent, P.L.; Grevers, L.; Blom, A.B.; Sloetjes, A.; Mort, J.S.; Vogl, T.; Nacken, W.; van den Berg, W.B.; Roth, J. Myeloid-related proteins S100A8/S100A9 regulate joint inflammation and cartilage destruction during antigen-induced arthritis. Ann. Rheum. Dis. 2008, 67, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Echtermeyer, F.; Bertrand, J.; Dreier, R.; Meinecke, I.; Neugebauer, K.; Fuerst, M.; Lee, Y.J.; Song, Y.W.; Herzog, C.; Theilmeier, G.; et al. Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat. Med. 2009, 15, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, W.B. Osteoarthritis year 2010 in review: Pathomechanisms. Osteoarthr. Cartil. 2011, 19, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Birkhead, J.; Sandell, L.J.; Kimura, T.; Krane, S.M. Interleukin 1 suppresses expression of cartilage-specific types II and IX collagens and increases types I and III collagens in human chondrocytes. J. Clin. Investig. 1988, 82, 2026–2037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mao, Z.; Yu, C. Suppression of early experimental osteoarthritis by gene transfer of interleukin-1 receptor antagonist and interleukin-10. J. Orthop. Res. 2004, 22, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B. Anticytokine therapy for osteoarthritis. Expert Opin. Biol. Ther. 2001, 1, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.; Hashimoto, S.; Kuhn, K. Mechanisms of chondrocyte apoptosis. Osteoarthr. Cartil. 1999, 7, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Lopez-Armada, M.J.; Cillero-Pastor, B.; Lires-Dean, M.; Vaamonde, C.; Galdo, F.; Blanco, F.J. Differential effects of tumor necrosis factor-α and interleukin-1β on cell death in human articular chondrocytes. Osteoarthr. Cartil. 2008, 16, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, K.; Hashimoto, S.; Lotz, M. IL-1β protects human chondrocytes from CD95-induced apoptosis. J. Immunol. 2000, 164, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Armada, M.J.; Carames, B.; Lires-Dean, M.; Cillero-Pastor, B.; Ruiz-Romero, C.; Galdo, F.; Blanco, F.J. Cytokines, tumor necrosis factor-α and interleukin-1β, differentially regulate apoptosis in osteoarthritis cultured human chondrocytes. Osteoarthr. Cartil. 2006, 14, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, F.; Kanno, H.; Uzuki, M.; Tajima, K.; Shimamura, T.; Sawai, T. Downregulation of inhibitor of apoptosis proteins in apoptotic human chondrocytes treated with tumor necrosis factor-α and actinomycin D. Osteoarthr. Cartil. 2006, 14, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Ochs, R.L.; Schwarz, H.; Lotz, M. Chondrocyte apoptosis induced by nitric oxide. Am. J. Pathol. 1995, 146, 75–85. [Google Scholar] [PubMed]
- Qin, J.; Shang, L.; Ping, A.S.; Li, J.; Li, X.J.; Yu, H.; Magdalou, J.; Chen, L.B.; Wang, H. TNF/TNFR signal transduction pathway-mediated anti-apoptosis and anti-inflammatory effects of sodium ferulate on IL-1β-induced rat osteoarthritis chondrocytes in vitro. Arthritis Res. Ther. 2012, 14, R242. [Google Scholar] [CrossRef] [PubMed]
- Swingler, T.E.; Wheeler, G.; Carmont, V.; Elliott, H.R.; Barter, M.J.; Abu-Elmagd, M.; Donell, S.T.; Boot-Handford, R.P.; Hajihosseini, M.K.; Munsterberg, A.; et al. The expression and function of microRNAs in chondrogenesis and osteoarthritis. Arthritis Rheum. 2012, 64, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Watkins, G.; Le Good, N.; Roberts, S.; Murphy, C.L.; Brockbank, S.M.; Needham, M.R.; Read, S.J.; Newham, P. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF-α and MMP13. Osteoarthr. Cartil. 2009, 17, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Wang, M.; Zhao, J.; Zhang, H.; Zhou, C.; Jin, L.; Zhang, Y.; Qiu, X.; Ma, B.; Fan, Q. MicroRNA-34a affects chondrocyte apoptosis and proliferation by targeting the SIRT1/p53 signaling pathway during the pathogenesis of osteoarthritis. Int. J. Mol. Med. 2016, 38, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, J.; Dai, L.; Yu, D.; Chen, Q.; Zhang, X.; Dai, K. miR-146a, an IL-1β responsive miRNA, induces vascular endothelial growth factor and chondrocyte apoptosis by targeting Smad4. Arthritis Res. Ther. 2012, 14, R75. [Google Scholar] [CrossRef] [PubMed]
- Makki, M.S.; Haqqi, T.M. miR-139 modulates MCPIP1/IL-6 expression and induces apoptosis in human OA chondrocytes. Exp. Mol. Med. 2015, 47, e189. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, L.; Jin, S.; Lin, J.; Zheng, H.; Zhang, H.; Fan, H.; He, F.; Ma, S.; Li, Q. MiR-98 promotes chondrocyte apoptosis by decreasing Bcl-2 expression in a rat model of osteoarthritis. Acta Biochim. Biophys. Sin. 2016, 48, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhao, J.; Jing, W.; Yan, S.; Wang, X.; Xiao, C.; Ma, B. Role of miR-146a in human chondrocyte apoptosis in response to mechanical pressure injury in vitro. Int. J. Mol. Med. 2014, 34, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, T.; Kon, T.; Einhorn, T.A.; Gerstenfeld, L.C. Induction of apoptosis in chondrocytes by tumor necrosis factor-α. J. Orthop. Res. 2001, 19, 785–796. [Google Scholar] [CrossRef]
- Fischer, B.A.; Mundle, S.; Cole, A.A. Tumor necrosis factor-α induced DNA cleavage in human articular chondrocytes may involve multiple endonucleolytic activities during apoptosis. Microsc. Res. Tech. 2000, 50, 236–242. [Google Scholar] [CrossRef]
- Relic, B.; Bentires-Alj, M.; Ribbens, C.; Franchimont, N.; Guerne, P.A.; Benoit, V.; Merville, M.P.; Bours, V.; Malaise, M.G. TNF-α protects human primary articular chondrocytes from nitric oxide-induced apoptosis via nuclear factor-κB. Lab. Investig. 2002, 82, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Song, Y.S.; Lee, S.Y.; Yoon, Y.G.; Lee, S.H.; Park, B.S.; Yun, I.; Choi, H.; Kim, K.; Chung, W.T.; et al. Downregulation of protein kinase CK2 activity facilitates tumor necrosis factor-α-mediated chondrocyte death through apoptosis and autophagy. PLoS ONE 2011, 6, e19163. [Google Scholar] [CrossRef] [PubMed]
- Relic, B.; Zeddou, M.; Desoroux, A.; Beguin, Y.; de Seny, D.; Malaise, M.G. Genistein induces adipogenesis but inhibits leptin induction in human synovial fibroblasts. Lab. Investig. 2009, 89, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Charlier, E.; Malaise, O.; Zeddou, M.; Neuville, S.; Cobraiville, G.; Deroyer, C.; Sanchez, C.; Gillet, P.; Kurth, W.; de Seny, D.; et al. Restriction of spontaneous and prednisolone-induced leptin production to dedifferentiated state in human hip OA chondrocytes: Role of Smad1 and β-catenin activation. Osteoarthr. Cartil. 2016, 24, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Malaise, O.; Relic, B.; Charlier, E.; Zeddou, M.; Neuville, S.; Deroyer, C.; Gillet, P.; Louis, E.; Malaise, M.G.; de Seny, D. Glucocorticoid-induced leucine zipper (GILZ) is involved in glucocorticoid-induced and mineralocorticoid-induced leptin production by osteoarthritis synovial fibroblasts. Arthritis Res. Ther. 2016, 18, 219. [Google Scholar] [CrossRef] [PubMed]
- Scotece, M.; Mobasheri, A. Leptin in osteoarthritis: Focus on articular cartilage and chondrocytes. Life Sci. 2015, 140, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.M.; Du, S.H.; Huang, L.G.; Li, J.H.; Xiao, L.; Tong, P. Leptin promotes apoptosis and inhibits autophagy of chondrocytes through upregulating lysyl oxidase-like 3 during osteoarthritis pathogenesis. Osteoarthr. Cartil. 2016, 24, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Z.; Wang, J.; Xu, S. DUSP19, a downstream effector of leptin, inhibits chondrocyte apoptosis via dephosphorylating JNK during osteoarthritis pathogenesis. Mol. Biosyst. 2016, 12, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Li, J.; Du, S.; Chen, G.; Qi, Y.; Huang, L.; Xiao, L.; Tong, P. Effects of UCP4 on the Proliferation and Apoptosis of Chondrocytes: Its Possible Involvement and Regulation in Osteoarthritis. PLoS ONE 2016, 11, e0150684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.M.; Shen, C.; Li, H.; Fan, Q.; Ding, J.; Jin, F.C.; Sha, L. Leptin induces the apoptosis of chondrocytes in an in vitro model of osteoarthritis via the JAK2STAT3 signaling pathway. Mol. Med. Rep. 2016, 13, 3684–3690. [Google Scholar] [PubMed]
- Roszer, T.; Jozsa, T.; Kiss-Toth, E.D.; de Clerck, N.; Balogh, L. Leptin receptor deficient diabetic (db/db) mice are compromised in postnatal bone regeneration. Cell Tissue Res. 2014, 356, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Del Carlo, M., Jr.; Loeser, R.F. Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 2002, 46, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M. The role of nitric oxide in articular cartilage damage. Rheum. Dis. Clin. N. Am. 1999, 25, 269–282. [Google Scholar] [CrossRef]
- Murrell, G.A.; Jang, D.; Williams, R.J. Nitric oxide activates metalloprotease enzymes in articular cartilage. Biochem. Biophys. Res. Commun. 1995, 206, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Taskiran, D.; Stefanovic-Racic, M.; Georgescu, H.; Evans, C. Nitric oxide mediates suppression of cartilage proteoglycan synthesis by interleukin-1. Biochem. Biophys. Res. Commun. 1994, 200, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Farrell, A.J.; Blake, D.R.; Palmer, R.M.; Moncada, S. Increased concentrations of nitrite in synovial fluid and serum samples suggest increased nitric oxide synthesis in rheumatic diseases. Ann. Rheum. Dis. 1992, 51, 1219–1222. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, K.; Lotz, M. Mechanisms of sodium nitroprusside-induced death in human chondrocytes. Rheumatol. Int. 2003, 23, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Lotz, M. IL-1-induced nitric oxide inhibits chondrocyte proliferation via PGE2. Exp. Cell Res. 1995, 218, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.; Stefanovic-Racic, M.; Billiar, T.R.; Curran, R.D.; McIntyre, L.A.; Georgescu, H.I.; Simmons, R.L.; Evans, C.H. Articular chondrocytes synthesize nitric oxide in response to cytokines and lipopolysaccharide. J. Immunol. 1991, 147, 3915–3920. [Google Scholar] [PubMed]
- Kim, J.; Xu, M.; Xo, R.; Mates, A.; Wilson, G.L.; Pearsall, A.W.T.; Grishko, V. Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthr. Cartil. 2010, 18, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.J.; Chen, T.G.; Chang, H.C.; Chiu, W.T.; Chang, C.C.; Chen, R.M. Nitric oxide from both exogenous and endogenous sources activates mitochondria-dependent events and induces insults to human chondrocytes. J. Cell. Biochem. 2007, 101, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.P.; Jovanovic, D.; Fernandes, J.C.; Manning, P.; Connor, J.R.; Currie, M.G.; di Battista, J.A.; Martel-Pelletier, J. Reduced progression of experimental osteoarthritis in vivo by selective inhibition of inducible nitric oxide synthase. Arthritis Rheum. 1998, 41, 1275–1286. [Google Scholar] [CrossRef]
- Notoya, K.; Jovanovic, D.V.; Reboul, P.; Martel-Pelletier, J.; Mineau, F.; Pelletier, J.P. The induction of cell death in human osteoarthritis chondrocytes by nitric oxide is related to the production of prostaglandin E2 via the induction of cyclooxygenase-2. J. Immunol. 2000, 165, 3402–3410. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Wang, X.P.; Chen, T.S. Resveratrol protects rabbit articular chondrocyte against sodium nitroprusside-induced apoptosis via scavenging ROS. Apoptosis 2014, 19, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- De Andres, M.C.; Maneiro, E.; Martin, M.A.; Arenas, J.; Blanco, F.J. Nitric oxide compounds have different effects profiles on human articular chondrocyte metabolism. Arthritis Res. Ther. 2013, 15, R115. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Arai, Y.; Mazda, O.; Kishida, T.; Takahashi, K.A.; Sakao, K.; Saito, M.; Honjo, K.; Imanishi, J.; Kubo, T. N-acetylcysteine prevents nitric oxide-induced chondrocyte apoptosis and cartilage degeneration in an experimental model of osteoarthritis. J. Orthop. Res. 2010, 28, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Sakata, S.; Hayashi, S.; Fujishiro, T.; Kawakita, K.; Kanzaki, N.; Hashimoto, S.; Iwasa, K.; Chinzei, N.; Kihara, S.; Haneda, M.; et al. Oxidative stress-induced apoptosis and matrix loss of chondrocytes is inhibited by eicosapentaenoic acid. J. Orthop. Res. 2015, 33, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Studer, R.K.; Levicoff, E.; Georgescu, H.; Miller, L.; Jaffurs, D.; Evans, C.H. Nitric oxide inhibits chondrocyte response to IGF-I: Inhibition of IGF-IRβ tyrosine phosphorylation. Am. J. Physiol. Cell Physiol. 2000, 279, C961–C969. [Google Scholar] [PubMed]
- Lee, S.W.; Song, Y.S.; Shin, S.H.; Kim, K.T.; Park, Y.C.; Park, B.S.; Yun, I.; Kim, K.; Lee, S.Y.; Chung, W.T.; et al. Cilostazol protects rat chondrocytes against nitric oxide-induced apoptosis in vitro and prevents cartilage destruction in a rat model of osteoarthritis. Arthritis Rheum. 2008, 58, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Jung, A.; Murphy, A.; Andreyev, A.; Dykens, J.; Terkeltaub, R. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum. 2000, 43, 1560–1570. [Google Scholar] [CrossRef]
- Tomita, M.; Sato, E.F.; Nishikawa, M.; Yamano, Y.; Inoue, M. Nitric oxide regulates mitochondrial respiration and functions of articular chondrocytes. Arthritis Rheum. 2001, 44, 96–104. [Google Scholar] [CrossRef]
- Kim, S.J.; Ju, J.W.; Oh, C.D.; Yoon, Y.M.; Song, W.K.; Kim, J.H.; Yoo, Y.J.; Bang, O.S.; Kang, S.S.; Chun, J.S. ERK-1/2 and p38 kinase oppositely regulate nitric oxide-induced apoptosis of chondrocytes in association with p53, caspase-3, and differentiation status. J. Biol. Chem. 2002, 277, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Relic, B.; Benoit, V.; Franchimont, N.; Ribbens, C.; Kaiser, M.J.; Gillet, P.; Merville, M.P.; Bours, V.; Malaise, M.G. 15-deoxy-Δ12,14-prostaglandin J2 inhibits Bay 11-7085-induced sustained extracellular signal-regulated kinase phosphorylation and apoptosis in human articular chondrocytes and synovial fibroblasts. J. Biol. Chem. 2004, 279, 22399–22403. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, S.Q.; Yu, L.; He, B.; Wu, S.H.; Zhao, Q.; Xia, S.Q.; Mei, H.J. Berberine prevents nitric oxide-induced rat chondrocyte apoptosis and cartilage degeneration in a rat osteoarthritis model via AMPK and p38 MAPK signaling. Apoptosis 2015, 20, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Armstrong, J.S.; Cheung, N.S.; Siau, J.L.; Rose, P.; Schantz, J.T.; Jones, D.P.; Halliwell, B. Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. FASEB J. 2004, 18, 1395–1397. [Google Scholar] [CrossRef] [PubMed]
- Gelber, A.C.; Hochberg, M.C.; Mead, L.A.; Wang, N.Y.; Wigley, F.M.; Klag, M.J. Joint injury in young adults and risk for subsequent knee and hip osteoarthritis. Ann. Intern. Med. 2000, 133, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, J.E.; Aspden, R.M. Cyclooxygenase inhibition lowers prostaglandin E2 release from articular cartilage and reduces apoptosis but not proteoglycan degradation following an impact load in vitro. Arthritis Res. Ther. 2007, 9, R129. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Sun, M.T.; Pang, S.T.; Ueng, S.W.; Chen, S.C.; Hwang, T.L.; Wang, T.H. Evaluation of differentially expressed genes by shear stress in human osteoarthritic chondrocytes in vitro. Chang. Gung Med. J. 2009, 32, 42–50. [Google Scholar] [PubMed]
- D’Lima, D.D.; Hashimoto, S.; Chen, P.C.; Colwell, C.W., Jr.; Lotz, M.K. Human chondrocyte apoptosis in response to mechanical injury. Osteoarthr. Cartil. 2001, 9, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Zheng, T.; Zhang, M.; Wang, D.; Du, S.; Li, X.; Fang, J.; Cao, X. Static mechanical stress induces apoptosis in rat endplate chondrocytes through MAPK and mitochondria-dependent caspase activation signaling pathways. PLoS ONE 2013, 8, e69403. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Liu, F.; Wang, J.; Guo, Z.; Ji, Z.; Yao, M. The effect of mechanical stretch stress on the differentiation and apoptosis of human growth plate chondrocytes. In Vitro Cell. Dev. Biol. Anim. 2016. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.L.; Carter, D.R.; Schurman, D.J. Pressure and shear differentially alter human articular chondrocyte metabolism: A review. Clin. Orthop. Relat. Res. 2004, 427, S89–S95. [Google Scholar]
- Zhang, M.; Zhang, J.; Lu, L.; Qiu, Z.Y.; Zhang, X.; Yu, S.B.; Wu, Y.P.; Wang, M.Q. Enhancement of chondrocyte autophagy is an early response in the degenerative cartilage of the temporomandibular joint to biomechanical dental stimulation. Apoptosis 2013, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Taniguchi, N.; Seino, D.; Blanco, F.J.; D’Lima, D.; Lotz, M. Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheum. 2012, 64, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Takebe, K.; Nishiyama, T.; Hayashi, S.; Hashimoto, S.; Fujishiro, T.; Kanzaki, N.; Kawakita, K.; Iwasa, K.; Kuroda, R.; Kurosaka, M. Regulation of p38 MAPK phosphorylation inhibits chondrocyte apoptosis in response to heat stress or mechanical stress. Int. J. Mol. Med. 2011, 27, 329–335. [Google Scholar] [PubMed]
- Martin, J.A.; Buckwalter, J.A. Post-traumatic osteoarthritis: The role of stress induced chondrocyte damage. Biorheology 2006, 43, 517–521. [Google Scholar] [PubMed]
- Beecher, B.R.; Martin, J.A.; Pedersen, D.R.; Heiner, A.D.; Buckwalter, J.A. Antioxidants block cyclic loading induced chondrocyte death. Iowa Orthop. J. 2007, 27, 1–8. [Google Scholar] [PubMed]
- Healy, Z.R.; Lee, N.H.; Gao, X.; Goldring, M.B.; Talalay, P.; Kensler, T.W.; Konstantopoulos, K. Divergent responses of chondrocytes and endothelial cells to shear stress: Cross-talk among COX-2, the phase 2 response, and apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 14010–14015. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, P.; Hecht, B.A.; Pedersen, D.R.; Lavery, M.R.; Maynard, J.; Buckwalter, J.A.; Martin, J.A. Oxidant conditioning protects cartilage from mechanically induced damage. J. Orthop. Res. 2010, 28, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Trindade, M.C.; Ikenoue, T.; Goodman, S.B.; Schurman, D.J.; Smith, R.L. Regulation of nitric oxide and Bcl-2 expression by shear stress in human osteoarthritic chondrocytes in vitro. J. Cell. Biochem. 2003, 90, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Chen, M.; Zuscik, M.; Wu, Q.; Wang, Y.J.; Rosier, R.N.; O’Keefe, R.J.; Chen, D. Inhibition of β-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum. 2008, 58, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.H.; Wang, C.J.; Ko, J.Y.; Sun, Y.C.; Su, Y.S.; Wang, F.S. Inflammation induction of Dickkopf-1 mediates chondrocyte apoptosis in osteoarthritic joint. Osteoarthr. Cartil. 2009, 17, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.H.; Wang, C.J.; Ko, J.Y.; Sun, Y.C.; Wang, F.S. Control of Dkk-1 ameliorates chondrocyte apoptosis, cartilage destruction, and subchondral bone deterioration in osteoarthritic knees. Arthritis Rheum. 2010, 62, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Zhong, L.; van Blitterswijk, C.A.; Post, J.N.; Karperien, M. T cell factor 4 is a pro-catabolic and apoptotic factor in human articular chondrocytes by potentiating nuclear factor κB signaling. J. Biol. Chem. 2013, 288, 17552–17558. [Google Scholar] [CrossRef] [PubMed]
- Karaliotas, G.I.; Mavridis, K.; Scorilas, A.; Babis, G.C. Quantitative analysis of the mRNA expression levels of BCL2 and BAX genes in human osteoarthritis and normal articular cartilage: An investigation into their differential expression. Mol. Med. Rep. 2015, 12, 4514–4521. [Google Scholar] [CrossRef] [PubMed]
- Surendran, S.; Kim, S.H.; Jee, B.K.; Ahn, S.H.; Gopinathan, P.; Han, C.W. Anti-apoptotic Bcl-2 gene transfection of human articular chondrocytes protects against nitric oxide-induced apoptosis. J. Bone Jt. Surg. Br. 2006, 88, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Precht, P.; Balakir, R.; Horton, W.E., Jr. Evidence of a direct role for Bcl-2 in the regulation of articular chondrocyte apoptosis under the conditions of serum withdrawal and retinoic acid treatment. J. Cell. Biochem. 1998, 71, 302–309. [Google Scholar] [CrossRef]
- Iannone, F.; de Bari, C.; Scioscia, C.; Patella, V.; Lapadula, G. Increased Bcl-2/p53 ratio in human osteoarthritic cartilage: A possible role in regulation of chondrocyte metabolism. Ann. Rheum. Dis. 2005, 64, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Balakir, R.; Precht, P.; Horton, W.E., Jr. Bcl-2 regulates chondrocyte morphology and aggrecan gene expression independent of caspase activation and full apoptosis. J. Cell. Biochem. 1999, 74, 576–586. [Google Scholar] [CrossRef]
- Evans, C.H.; Ghivizzani, S.C.; Robbins, P.D. Getting arthritis gene therapy into the clinic. Nat. Rev. Rheumatol. 2011, 7, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.Y.; Kim, H.T. Chondrocyte apoptosis induced by collagen degradation: Inhibition by caspase inhibitors and IGF-1. J. Orthop. Res. 2004, 22, 140–144. [Google Scholar] [CrossRef]
- Kooijman, R. Regulation of apoptosis by insulin-like growth factor (IGF)-I. Cytokine Growth Factor Rev. 2006, 17, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Shakibaei, M.; Schulze-Tanzil, G.; de Souza, P.; John, T.; Rahmanzadeh, M.; Rahmanzadeh, R.; Merker, H.J. Inhibition of mitogen-activated protein kinase kinase induces apoptosis of human chondrocytes. J. Biol. Chem. 2001, 276, 13289–13294. [Google Scholar] [CrossRef] [PubMed]
- Hemphill, D.D.; McIlwraith, C.W.; Slayden, R.A.; Samulski, R.J.; Goodrich, L.R. Adeno-associated virus gene therapy vector scAAVIGF-I for transduction of equine articular chondrocytes and RNA-seq analysis. Osteoarthr. Cartil. 2016, 24, 902–911. [Google Scholar] [CrossRef] [PubMed]
- van der Kraan, P.M.; Blaney Davidson, E.N.; Blom, A.; van den Berg, W.B. TGF-β signaling in chondrocyte terminal differentiation and osteoarthritis: Modulation and integration of signaling pathways through receptor-Smads. Osteoarthr. Cartil. 2009, 17, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Remst, D.F.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, J.K.; Rey-Rico, A.; Schmitt, G.; Wezel, A.; Madry, H.; Cucchiarini, M. rAAV-mediated overexpression of TGF-β stably restructures human osteoarthritic articular cartilage in situ. J. Transl. Med. 2013, 11, 211. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Nakasa, T.; Miyaki, S.; Ishikawa, M.; Deie, M.; Adachi, N.; Yasunaga, Y.; Asahara, H.; Ochi, M. Expression of MicroRNA-146a in osteoarthritis cartilage. Arthritis Rheum. 2009, 60, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- D’Lima, D.; Hermida, J.; Hashimoto, S.; Colwell, C.; Lotz, M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006, 54, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.K.; Kraus, V.B. New developments in osteoarthritis. Posttraumatic osteoarthritis: Pathogenesis and pharmacological treatment options. Arthritis Res. Ther. 2010, 12, 211. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Kiosses, W.B.; Akasaki, Y.; Brinson, D.C.; Eap, W.; Koziol, J.; Lotz, M.K. Glucosamine activates autophagy in vitro and in vivo. Arthritis Rheum. 2013, 65, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Garcia, O.; Olmer, M.; Akagi, R.; Akasaki, Y.; Fisch, K.M.; Shen, T.; Su, A.I.; Lotz, M.K. Suppression of REDD1 in osteoarthritis cartilage, a novel mechanism for dysregulated mTOR signaling and defective autophagy. Osteoarthr. Cartil. 2016, 24, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guo, Y.; Wang, C.; Yu, H.; Yu, X.; Yu, H. MicroRNA-142-3p inhibits chondrocyte apoptosis and inflammation in osteoarthritis by targeting HMGB1. Inflammation 2016, 39, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, S.; Alvarez-Garcia, O.; Muramatsu, Y.; Flamigni, F.; Lotz, M.K. MicroRNA-155 suppresses autophagy in chondrocytes by modulating expression of autophagy proteins. Osteoarthr. Cartil. 2016, 24, 1082–1091. [Google Scholar]


{kind=link}
{kind=link}
| Cell Components/Events | Cell Death Types | |||
|---|---|---|---|---|
| Apoptosis | Necrosis | Autophagic Cell Death | Chondroptosis | |
| Chromatine | Marginal condensation | Fragmented | Absence of condensation | Patchy condensation |
| Nucleus | Fragmentation into apoptotic bodies | Nuclear condensation (pyknosis) | Intact until late stages | Convoluted nucleus Nuclear condensation |
| Apoptotic bodies | Yes | Cell explosion | No | Cellular remnants and vesicules |
| Inflammation | No | Yes | No | Not precised |
| ER | No enlargement | No enlargement | Enlargement | Increase in amount and expansion of lumen (*) |
| Golgi apparatus | No increase | No increase | Enlargement | Increase in early stages |
| Autophagic vacuoles | No | No | Abundant | Frequent |
| DNA | Intranucleosomal cleavage-DNA laddering | Random cleavage DNA Smear | DNA fragmentation occurs very late | Cleaved |
| Plasma membrane | Blebbing but intact | Loss of integrity | Participate to autophagosome formation | Vesicle blebs |
| cell | Shrinkage | Swelling | Shrinkage | Not precised |
| Lysosomial enzyme | Inside apoptotic bodies | Leakage | Inside autophagic vacuoles | Inside cytoplasmic ‘islands’(*) or autophagic Vacuoles |
| Elimination (cell fate) | Phagocytosis of apoptotic bodies | General lysis | Auto-elimination | Auto-elimination of chondrocytes in absence of phagocytes |
| Caspases involvement | Yes | No | No | Yes |
| ATP requirement | Yes | No | Yes | Not precised |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charlier, E.; Relic, B.; Deroyer, C.; Malaise, O.; Neuville, S.; Collée, J.; Malaise, M.G.; De Seny, D. Insights on Molecular Mechanisms of Chondrocytes Death in Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 2146. https://doi.org/10.3390/ijms17122146
Charlier E, Relic B, Deroyer C, Malaise O, Neuville S, Collée J, Malaise MG, De Seny D. Insights on Molecular Mechanisms of Chondrocytes Death in Osteoarthritis. International Journal of Molecular Sciences. 2016; 17(12):2146. https://doi.org/10.3390/ijms17122146
Chicago/Turabian StyleCharlier, Edith, Biserka Relic, Céline Deroyer, Olivier Malaise, Sophie Neuville, Julie Collée, Michel G. Malaise, and Dominique De Seny. 2016. "Insights on Molecular Mechanisms of Chondrocytes Death in Osteoarthritis" International Journal of Molecular Sciences 17, no. 12: 2146. https://doi.org/10.3390/ijms17122146