
Extracellular Vesicles in Chronic Obstructive Pulmonary Disease

Abstract
:1. Introduction
2. Pathogenesis of COPD
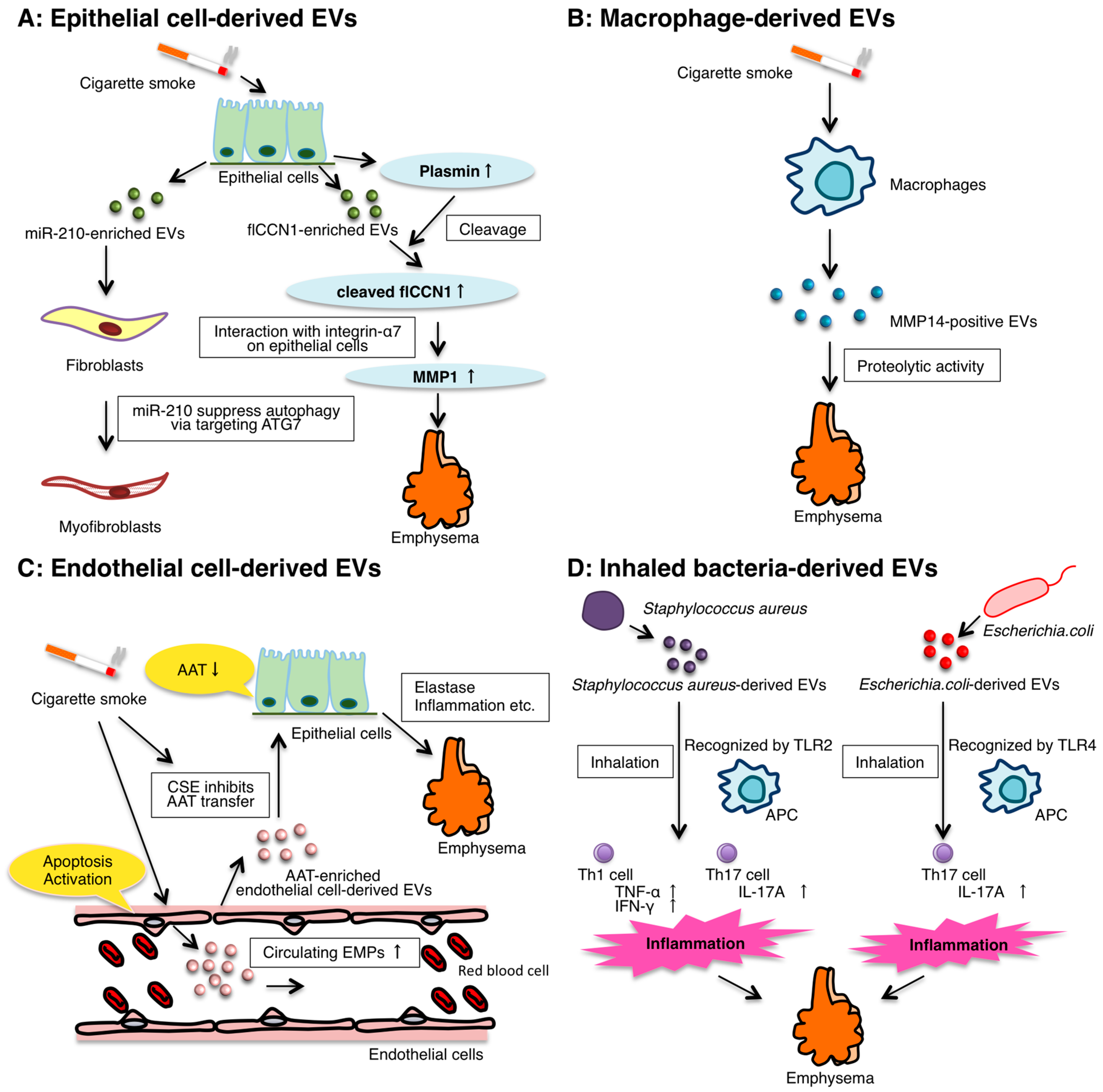
3. The Role of Epithelial EVs
4. The Role of Macrophage-Derived EVs
5. The Role of Endothelial Cell-Derived EVs
6. The Role of Inhaled Bacteria-Derived EVs
7. EVs in COPD Exacerbation
8. Circulating EVs and miRNAs as Potential Biomarkers for COPD
9. EVs as Potential Therapeutics for COPD
10. Conclusions
Acknowledgments
Conflicts of Interest
References
- Vestbo, J.; Hurd, S.S.; Agustí, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.; Ito, K.; Barnes, P.J. Accelerated ageing of the lung in COPD: New concepts. Thorax 2015, 70, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.; Flaxman, A.D.; Naghavi, M.; Lozano, R.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; Aboyans, V.; et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2163–2196. [Google Scholar] [CrossRef]
- López-Campos, J.L.; Tan, W.; Soriano, J.B. Global burden of COPD. Respirology 2016, 21, 14–23. [Google Scholar] [CrossRef] [PubMed]
- GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar]
- Hogg, J.C.; Timens, W. The pathology of chronic obstructive pulmonary disease. Annu. Rev. Pathol. 2009, 4, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.C. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J. Cell Biol. 1969, 41, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Crawford, N. The presence of contractile proteins in platelet microparticles isolated from human and animal platelet-free plasma. Br. J. Haematol. 1971, 21, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, Y.; Kosaka, N.; Araya, J.; Kuwano, K.; Ochiya, T. Extracellular vesicles in lung microenvironment and pathogenesis. Trends Mol. Med. 2015, 21, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Konishi, Y.; Kosaka, N.; Katsuda, T.; Kato, T.; Ochiya, T. Comparative marker analysis of extracellular vesicles in different human cancer types. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials—An ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, É.; Pap, E.; Kittel, Á.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Yoshioka, Y.; Ochiya, T. Extracellular vesicle transfer of cancer pathogenic components. Cancer Sci. 2016, 107, 385–390. [Google Scholar] [CrossRef] [PubMed]
- De Jong, O.G.; Verhaar, M.C.; Chen, Y.; Vader, P.; Gremmels, H.; Posthuma, G.; Schiffelers, R.M.; Gucek, M.; van Balkom, B.W.M. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J. Extracell. Vesicles 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Beninson, L.A.; Fleshner, M. Exosomes: An emerging factor in stress-induced immunomodulation. Semin. Immunol. 2014, 26, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Caby, M.P. Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 2005, 17, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Admyre, C.; Grunewald, J.; Thyberg, J.; Gripenbäck, S.; Tornling, G.; Eklund, A.; Scheynius, A.; Gabrielsson, S. Exosomes with major histocompatibility complex class II and co-stimulatory molecules are present in human BAL fluid. Eur. Respir. J. 2003, 22, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Porro, C.; Lepore, S.; Trotta, T.; Castellani, S.; Ratclif, L.; Battaglino, A.; di Gioia, S.; Martínez, M.C.; Conese, M.; Maffione, A.B. Isolation and characterization of microparticles in sputum from cystic fibrosis patients. Respir. Res. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.K.; Agusti, A.; Calverley, P.M.; Celli, B.R.; Criner, G.; Curtis, J.L.; Fabbri, L.M.; Goldin, J.G.; Jones, P.W.; MacNee, W.; et al. Chronic obstructive pulmonary disease phenotypes: The future of COPD. Am. J. Respir. Crit. Care Med. 2010, 182, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Miravitlles, M.; Soler-Cataluña, J.J.; Calle, M.; Molina, J.; Almagro, P.; Quintano, J.A.; Riesco, J.A.; Trigueros, J.A.; Piñera, P.; Simón, A.; et al. Spanish guideline for COPD (GesEPOC). Update 2014. Arch. Bronconeumol. 2014, 50, 1–16. [Google Scholar] [CrossRef]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Brusselle, G.G.; Joos, G.F.; Bracke, K.R. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 2011, 378, 1015–1026. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Liang, J.; Li, Y.; Noble, P.W. The role of Toll-like receptors in non-infectious lung injury. Cell Res. 2006, 16, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Cosio, M.G.; Saetta, M.; Agusti, A. Immunologic aspects of chronic obstructive pulmonary disease. N. Engl. J. Med. 2009, 360, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Caramori, G.; Oates, T.; Capelli, A.; Lusuardi, M.; Gnemmi, I.; Ioli, F.; Chung, K.F.; Donner, C.F.; Barnes, P.J.; et al. Increased expression of nuclear factor-κB in bronchial biopsies from smokers and patients with COPD. Eur. Respir. J. 2002, 20, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Hara, H.; Araya, J.; Takasaka, N.; Kojima, J.; Ito, S.; Minagawa, S.; Yumino, Y.; Ishikawa, T.; Numata, T.; et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. OncoImmunology 2014, 1, 630–641. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, K.; Araya, J.; Hara, H.; Minagawa, S.; Takasaka, N.; Ito, S.; Nakayama, K. Japan Autophagy and Cellular Senescence in Lung Diseases. J. Biochem. Mol. Biol. Res. 2015, 1, 54–66. [Google Scholar] [CrossRef]
- Aoshiba, K.; Nagai, A. Senescence hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2009, 6, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.C.; Welker, L.; Paasch, K.; Feindt, B.; Erpenbeck, V.J.; Hohlfeld, J.M.; Krug, N.; Nakashima, M.; Branscheid, D.; Magnussen, H.; et al. Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respir. Res. 2006, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Alveolar cell senescence in patients with pulmonary emphysema. Am. J. Respir. Crit. Care Med. 2006, 174, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 177, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Yoshii, Y.; Yumino, Y.; Fujii, S.; et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Araya, J.; Ito, S.; Kobayashi, K.; Takasaka, N.; Yoshii, Y.; Wakui, H.; Kojima, J.; Shimizu, K.; Numata, T.; et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol. 2013, 305, L737–L746. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S.; et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Jobling, W.A.; Chen, B.P.C.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21 (CIP1), but not p16 (INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Seeger, W.; Voswinckel, R. Senescence-associated secretory phenotype and its possible role in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2014, 51, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Cambier, S.; Markovics, J.A.; Wolters, P.; Jablons, D.; Hill, A.; Finkbeiner, W.; Jones, K.; Broaddus, V.C.; Sheppard, D.; et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J. Clin. Investig. 2007, 117, 3551–3562. [Google Scholar] [CrossRef] [PubMed]
- Shaykhiev, R.; Otaki, F.; Bonsu, P.; Dang, D.T.; Teater, M.; Strulovici-Barel, Y.; Salit, J.; Harvey, B.G.; Crystal, R.G. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell. Mol. Life Sci. 2011, 68, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Leopold, P.L.; O’Mahony, M.J.; Lian, X.J.; Tilley, A.E.; Harvey, B.G.; Crystal, R.G. Smoking is associated with shortened airway cilia. PLoS ONE 2009, 4, e8157. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, A.; Ahmad, T.; Agrawal, A.; Ghosh, B. Proinflammatory role of epithelial cell-derived exosomes in allergic airway inflammation. J. Allergy Clin. Immunol. 2013, 131, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Kesimer, M.; Gupta, R. Physical characterization and profiling of airway epithelial derived exosomes using light scattering. Methods 2015, 87, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Kesimer, M.; Scull, M.; Brighton, B.; DeMaria, G.; Burns, K.; O’Neal, W.; Pickles, R.J.; Sheehan, J.K. Characterization of exosome-like vesicles released from human tracheobronchial ciliated epithelium: A possible role in innate defense. FASEB J. 2009, 23, 1858–1868. [Google Scholar] [CrossRef] [PubMed]
- Hattrup, C.L.; Gendler, S.J. Structure and function of the cell surface (tethered) mucins. Annu. Rev. Physiol. 2008, 70, 431–457. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.G.; Kim, S.H.; Gao, J.; Quan, T.; Qin, Z.; Osorio, J.C.; Rosas, I.O.; Wu, M.; Tesfaigzi, Y.; Jin, Y. CCN1 secretion and cleavage regulate the lung epithelial cell functions after cigarette smoke. Am. J. Physiol. 2014, 307, L326–L337. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Araya, J.; Ito, S.; Kobayashi, K.; Kosaka, N.; Yoshioka, Y.; Kadota, T.; Hara, H.; Kuwano, K.; Ochiya, T. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. J. Extracell. Vesicles 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Araya, J.; Minagawa, S.; Hara, H.; Saito, N.; Kadota, T.; Sato, N.; Yoshida, M.; Tsubouchi, K.; Kurita, Y.; et al. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J. Immunol. 2016, 197, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.F. CCN1/CYR61: The very model of a modern matricellular protein. Cell. Mol. Life Sci. 2011, 68, 3149–3163. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Tuder, R.M.; Taraseviciene-Stewart, L.; Le Cras, T.D.; Abman, S.; Hirth, P.K.; Waltenberger, J.; Voelkel, N.F. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Investig. 2000, 106, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H.; Yoshikawa, J. Elevated oxidative stress and reciprocal reduction of vascular endothelial growth factor levels with severity of COPD. Chest 2005, 128, 3191–3197. [Google Scholar] [CrossRef] [PubMed]
- D’Armiento, J.; Dalal, S.S.; Okada, Y.; Berg, R.A.; Chada, K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell 1992, 71, 955–961. [Google Scholar] [CrossRef]
- Finkelstein, R.; Fraser, R.S.; Ghezzo, H.; Cosio, M.G. Alveolar inflammation and its relation to emphysema in smokers. Am. J. Respir. Crit. Care Med. 1995, 152, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Capelli, A.; Lusuardi, M.; Balbo, P.; Vecchio, C.; Maestrelli, P.; Mapp, C.E.; Fabbri, L.M.; Donner, C.F.; Saetta, M. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am. J. Respir. Crit. Care Med. 1998, 158, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.; Wang, Y.; Dakhlallah, D.; Moldovan, L.; Agarwal, K.; Batte, K.; Shah, P.; Wisler, J.; Eubank, T.D.; Tridandapani, S.; et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood 2013, 121, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Ramachandra, L.; Mohr, S.; Franchi, L.; Harding, C.V.; Nunez, G.; Dubyak, G.R. P2X7 receptor-stimulated secretion of MHC class II-containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase-1. J. Immunol. 2009, 182, 5052–5062. [Google Scholar] [CrossRef] [PubMed]
- Zeitvogel, J.; Dalpke, A.; Eiz-Vesper, B.; Kracht, M.; Dittrich-Breiholz, O.; Werfel, T.; Wittmann, M. Human primary keratinocytes show restricted ability to up-regulate suppressor of cytokine signaling (SOCS)3 protein compared with autologous macrophages. J. Biol. Chem. 2012, 287, 9923–9930. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, D.; Williams, K.J.; Liu, M.L. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Cordazzo, C.; Petrini, S.; Neri, T.; Lombardi, S.; Carmazzi, Y.; Pedrinelli, R.; Paggiaro, P.; Celi, A. Rapid shedding of proinflammatory microparticles by human mononuclear cells exposed to cigarette smoke is dependent on Ca2+ mobilization. Inflamm. Res. 2014, 63, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Liu, Y.; Chen, Y.; Yu, D.; Williams, K.J.; Liu, M.L. Novel proteolytic microvesicles released from human macrophages after exposure to tobacco smoke. Am. J. Pathol. 2013, 182, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Loh, E.; Roddy, M.A.; Currie, K.E.; Creager, M.A. Nitric oxide regulates basal systemic and pulmonary vascular resistance in healthy humans. Circulation 1994, 89, 2035–2040. [Google Scholar] [CrossRef] [PubMed]
- Cremona, G.; Wood, A.M.; Hall, L.W.; Bower, E.A.; Higenbottam, T. Effect of inhibitors of nitric oxide release and action on vascular tone in isolated lungs of pig, sheep, dog and man. J. Physiol. 1994, 481, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Peinado, V.I.; Pizarro, S.; Barberà, J.A. Pulmonary vascular involvement in COPD. Chest 2008, 134, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Peinado, V.I.; Barberá, J.A.; Abate, P.; Ramírez, J.; Roca, J.; Santos, S.; Rodriguez-Roisin, R. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999, 159, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Tuder, R.M.; Cool, C.D.; Lynch, D.A.; Flores, S.C.; Voelkel, N.F. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am. J. Respir. Crit. Care Med. 2001, 163, 737–744. [Google Scholar] [CrossRef] [PubMed]
- McAllister, D.A.; Maclay, J.D.; Mills, N.L.; Mair, G.; Miller, J.; Anderson, D.; Newby, D.E.; Murchison, J.T.; MacNee, W. Arterial stiffness is independently associated with emphysema severity in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2007, 176, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Sabit, R.; Bolton, C.E.; Edwards, P.H.; Pettit, R.J.; Evans, W.D.; McEniery, C.M.; Wilkinson, I.B.; Cockcroft, J.R.; Shale, D.J. Arterial stiffness and osteoporosis in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2007, 175, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Gupta, A.; Strollo, P.J., Jr.; Fuhrman, C.R.; Leader, J.K.; Bon, J.; Slivka, W.A.; Shoushtari, A.H.; Avolio, J.; Kip, K.E.; et al. Airflow limitation and endothelial dysfunction. unrelated and independent predictors of atherosclerosis. Am. J. Respir. Crit. Care Med. 2016, 194, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Heiss, C.; Amabile, N.; Lee, A.C.; Real, W.M.; Schick, S.F.; Lao, D.; Wong, M.L.; Jahn, S.; Angeli, F.S.; Minasi, P.; et al. Brief secondhand smoke exposure depresses endothelial progenitor cells activity and endothelial function. J. Am. Coll. Cardiol. 2008, 51, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Chironi, G.N.; Boulanger, C.M.; Simon, A.; Dignat-George, F.; Freyssinet, J.M.; Tedgui, A. Endothelial microparticles in diseases. Cell Tissue Res. 2009, 335, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kobayashi, S.; Fujino, N.; Suzuki, T.; Ota, C.; He, M.; Yamada, M.; Suzuki, S.; Yanai, M.; Kurosawa, S.; et al. Increased circulating endothelial microparticles in COPD patients: A potential biomarker for COPD exacerbation susceptibility. Thorax 2012, 67, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.; Gudi, K.; Krause, A.; Sackrowitz, R.; Harvey, B.-G.; Strulovici-Barel, Y.; Mezey, J.G.; Crystal, R.G. Circulating endothelial microparticles as a measure of early lung destruction in cigarette smokers. Am. J. Respir. Crit. Care Med. 2011, 184, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Lockett, A.D.; Brown, M.B.; Santos-Falcon, N.; Rush, N.I.; Oueini, H.; Oberle, A.J.; Bolanis, E.; Fragoso, M.A.; Petrusca, D.N.; Serban, K.A.; et al. Active trafficking of α 1 antitrypsin across the lung endothelium. PLoS ONE 2014, 9, e93979. [Google Scholar] [CrossRef] [PubMed]
- Strulovici-Barel, Y.; Staudt, M.R.; Krause, A.; Gordon, C.; Tilley, A.E.; Harvey, B.G.; Kaner, R.J.; Hollmann, C.; Mezey, J.G.; Bitter, H.; et al. Persistence of circulating endothelial microparticles in COPD despite smoking cessation. Thorax 2016. [Google Scholar] [CrossRef] [PubMed]
- American Thoracic Society; European Respiratory Society American Thoracic Society. European Respiratory Society statement: Standards for the diagnosis and management of individuals with α-1 antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 2003, 168, 818–900. [Google Scholar]
- Kim, J.H.; Lee, J.; Park, J.; Gho, Y.S. Gram-negative and Gram-positive bacterial extracellular vesicles. Semin. Cell Dev. Biol. 2015, 40, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Wolf, J.M.; Prados-Rosales, R.; Casadevall, A. Through the wall: Extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Publ. Group 2015, 13, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C.; Kuehn, M.J. Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat. Publ. Group 2015, 13, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Amano, A.; Takeuchi, H.; Furuta, N. Outer membrane vesicles function as offensive weapons in host-parasite interactions. Microbes Infect. 2010, 12, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Schooling, S.R.; Beveridge, T.J. Membrane vesicles: An overlooked component of the matrices of biofilms. J. Bacteriol. 2006, 188, 5945–5957. [Google Scholar] [CrossRef] [PubMed]
- Namork, E.; Brandtzaeg, P. Fatal meningococcal septicaemia with “blebbing” meningococcus. Lancet 2002, 360, 1741. [Google Scholar] [CrossRef]
- Kim, Y.S.; Choi, E.J.; Lee, W.H.; Choi, S.J.; Roh, T.Y.; Park, J.; Jee, Y.K.; Zhu, Z.; Koh, Y.Y.; Gho, Y.S.; et al. Extracellular vesicles, especially derived from Gram-negative bacteria, in indoor dust induce neutrophilic pulmonary inflammation associated with both Th1 and Th17 cell responses. Clin. Exp. Allergy 2013, 43, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Choi, K.H.; Kim, Y.S.; Hong, B.S.; Kim, O.Y.; Kim, J.H.; Yoon, C.M.; Koh, G.Y.; Kim, Y.K.; Gho, Y.S. Outer membrane vesicles derived from Escherichia coli induce systemic inflammatory response syndrome. PLoS ONE 2010, 5, e11334. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Lee, W.H.; Choi, E.J.; Choi, J.P.; Heo, Y.J.; Gho, Y.S.; Jee, Y.K.; Oh, Y.M.; Kim, Y.K. Extracellular vesicles derived from Gram-negative bacteria, such as Escherichia coli, induce emphysema mainly via IL-17A-Mediated neutrophilic inflammation. J. Immunol. 2015, 194, 3361–3368. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Yoshii, Y.; Morozumi, M.; Chiba, N.; Ubukata, K.; Uruga, H.; Hanada, S.; Saito, N.; Kadota, T.; Wakui, H.; et al. Pathogens in COPD exacerbations identified by comprehensive real-time PCR plus older methods. COPD 2015, 10, 2009. [Google Scholar] [CrossRef] [PubMed]
- Eltom, S.; Dale, N.; Raemdonck, K.R.G.; Stevenson, C.S.; Snelgrove, R.J.; Sacitharan, P.K.; Recchi, C.; Wavre-Shapton, S.; McAuley, D.F.; O’Kane, C.; et al. Respiratory infections cause the release of extracellular vesicles: Implications in exacerbation of asthma/COPD. PLoS ONE 2014, 9, e101087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerveri, I.; Cosrsico, A.G.; Accordini, S.; Niniano, R.; Ansaldo, E.; Anto, J.M.; Kunzli, N.; Janson, C.; Sunyer, J.; Jarvis, D.; et al. Underestimation of airflow obstruction among young adults using FEV1/FVC. Thorax 2008, 63, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Iraci, N.; Leonardi, T.; Gessler, F.; Vega, B.; Pluchino, S. Focus on extracellular vesicles: Physiological role and signalling properties of extracellular membrane vesicles. Int. J. Mol. Sci. 2016, 17, 171. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Yoshioka, Y.; Fujita, Y.; Ochiya, T. Versatile roles of extracellular vesicles in cancer. J. Clin. Investig. 2016, 126, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Makiguchi, T.; Yamada, M.; Yoshioka, Y.; Sugiura, H.; Koarai, A.; Chiba, S.; Fujino, N.; Tojo, Y.; Ota, C.; Kubo, H.; et al. Serum extracellular vesicular miR-21-51-5p is a predictor of the prognosis in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 110. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Kosaka, N.; Konishi, Y.; Ohta, H.; Okamoto, H.; Sonoda, H.; Nonaka, R.; Yamamoto, H.; Ishii, H.; Mori, M.; et al. Ultra-sensitive liquid biopsy of circulating extracellular vesicles using ExoScreen. Nat. Commun. 2014, 5, 3591. [Google Scholar] [CrossRef] [PubMed]
- Thomashow, M.A.; Shimbo, D.; Parikh, M.A.; Hoffman, E.A.; Vogel-Claussen, J.; Hueper, K.; Fu, J.; Liu, C.Y.; Bluemke, D.A.; Ventetuolo, C.E.; et al. Endothelial microparticles in mild chronic obstructive pulmonary disease and emphysema. The multi-ethnic study of atherosclerosis chronic obstructive pulmonary disease study. Am. J. Respir. Crit. Care Med. 2013, 188, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Lacedonia, D.; Carpagnano, G.E.; Trotta, T.; Palladino, G.P.; Panaro, M.A.; Zoppo, L.D.; Foschino Barbaro, M.P.; Porro, C. Microparticles in sputum of COPD patients: A potential biomarker of the disease? COPD 2016, 11, 527–533. [Google Scholar] [CrossRef]
- Gibbings, D.J.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 2009, 11, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- De Smet, E.G.; Mestdagh, P.; Vandesompele, J.; Brusselle, G.G.; Bracke, K.R. Non-coding RNAs in the pathogenesis of COPD. Thorax 2015, 70, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Wu, M.; Lin, H.; Liu, C.; Yang, H.; Zhan, J.; Sun, S. An increased ratio of serum miR-21 to miR-181a levels is associated with the early pathogenic process of chronic obstructive pulmonary disease in asymptomatic heavy smokers. Mol. Biosyst. 2014, 10, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Akbas, F.; Coskunpinar, E.; Aynaci, E.; Oltulu, Y.M.; Yildiz, P. Analysis of serum micro-RNAs as potential biomarker in chronic obstructive pulmonary disease. Exp. Lung Res. 2012, 38, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Pottelberge, G.R.V.; Mestdagh, P.; Bracke, K.R.; Thas, O.; Durme, Y.M.T.A.V.; Joos, G.F.; Vandesompele, J.; Brusselle, G.G. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, M.; Chinchilli, V.; Banta, E.; Craig, T.; August, A.; Bascom, R.; Cantorna, M.; Harvill, E.; Ishmael, F.T. Differential expression of microRNAs in exhaled breath condensates of patients with asthma, patients with chronic obstructive pulmonary disease, and healthy adults. J. Allergy Clin. Immunol. 2013, 132, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Yoshioka, Y.; Tominaga, N.; Hagiwara, K.; Katsuda, T.; Ochiya, T. Dark side of the exosome: The role of the exosome in cancer metastasis and targeting the exosome as a strategy for cancer therapy. Future Oncol. 2014, 10, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. OncoImmunology 2016, 5, e1071008. [Google Scholar] [CrossRef] [PubMed]
- Kordelas, L.; Rebmann, V.; Ludwig, A.K.; Radtke, S.; Ruesing, J.; Doeppner, T.R.; Epple, M.; Horn, P.A.; Beelen, D.W.; Giebel, B. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 2014, 28, 970–973. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Body Fluids | Potential Biomarkers | Detection Methods | References |
|---|---|---|---|
| miRNAs | |||
| serum | miR-21/miR-181a ratio | realtime PCR | [112] |
| serum | upregulated: miR-7 downregulated: miR-20a, miR-28-3p, miR-34c-5p, miR-100 | realtime PCR | [113] |
| sputum | downregulated: let-7c, miR-125b | realtime PCR | [114] |
| exhaled breath condensates | dounregulated: let-7a, miR-21, miR-328 | realtime PCR | [115] |
| EVs | |||
| plasma | CD31+ EMPs, CD62E+/CD31+ EMPs ratio | flow cytometry | [86,88] |
| plasma | CD31+ EMPs, CD62E+ EMPs | flow cytometry | [106] |
| plasma | CD144+ EMPs, CD31+ EMPs, CD62E+ EMPs | flow cytometry | [85] |
| sputum | CD31+ EMPs, CD66+ EMPs, CD235ab+ EMPs | flow cytometry | [107] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadota, T.; Fujita, Y.; Yoshioka, Y.; Araya, J.; Kuwano, K.; Ochiya, T. Extracellular Vesicles in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2016, 17, 1801. https://doi.org/10.3390/ijms17111801
Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, Ochiya T. Extracellular Vesicles in Chronic Obstructive Pulmonary Disease. International Journal of Molecular Sciences. 2016; 17(11):1801. https://doi.org/10.3390/ijms17111801
Chicago/Turabian StyleKadota, Tsukasa, Yu Fujita, Yusuke Yoshioka, Jun Araya, Kazuyoshi Kuwano, and Takahiro Ochiya. 2016. "Extracellular Vesicles in Chronic Obstructive Pulmonary Disease" International Journal of Molecular Sciences 17, no. 11: 1801. https://doi.org/10.3390/ijms17111801