
Neuroprotective Strategies after Neonatal Hypoxic Ischemic Encephalopathy

Abstract
:1. Introduction
1.1. Neonatal Hypoxic Ischemic Encephalopathy
1.2. Incidence and Prognosis of HIE
1.3. Clinical Presentation
1.4. Current Therapeutic Strategies
2. Potential Intervention Targets
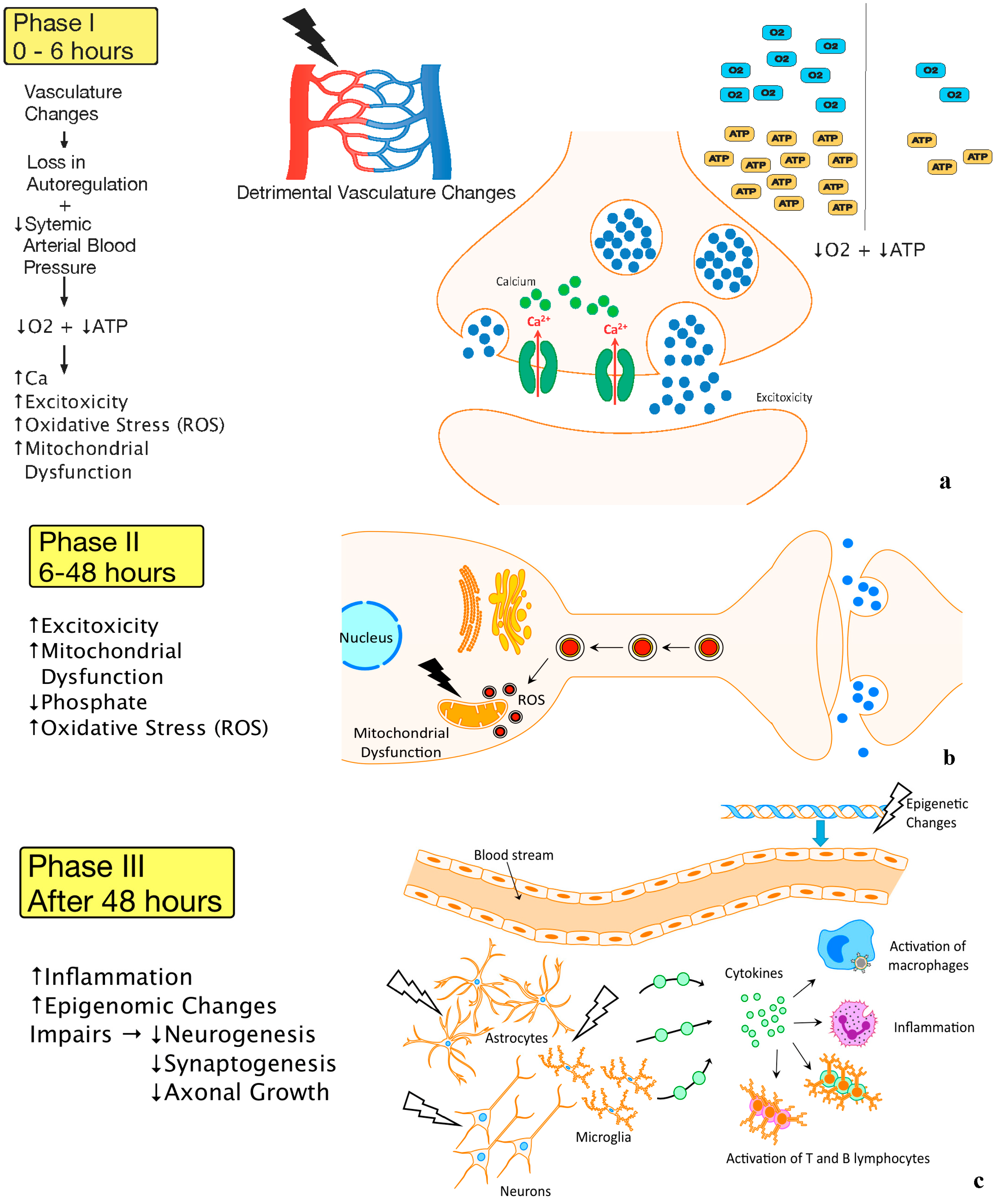
2.1. Pathophysiology


2.2. Blood-Brain Barrier
2.3. Angiogenesis
2.4. Neurogenesis
2.5. Autophagy
3. Limitations to Intervention Sites
3.1. Limitations to in Vitro and in Vivo
3.2. Limitations to Animal Models
3.3. Limitations to Central Nervous System Drug Delivery
3.4. Limitations to Neonatal Drugs and Dosage
4. Potential Novel Molecules and Strategies for Neuroprotection after HIE

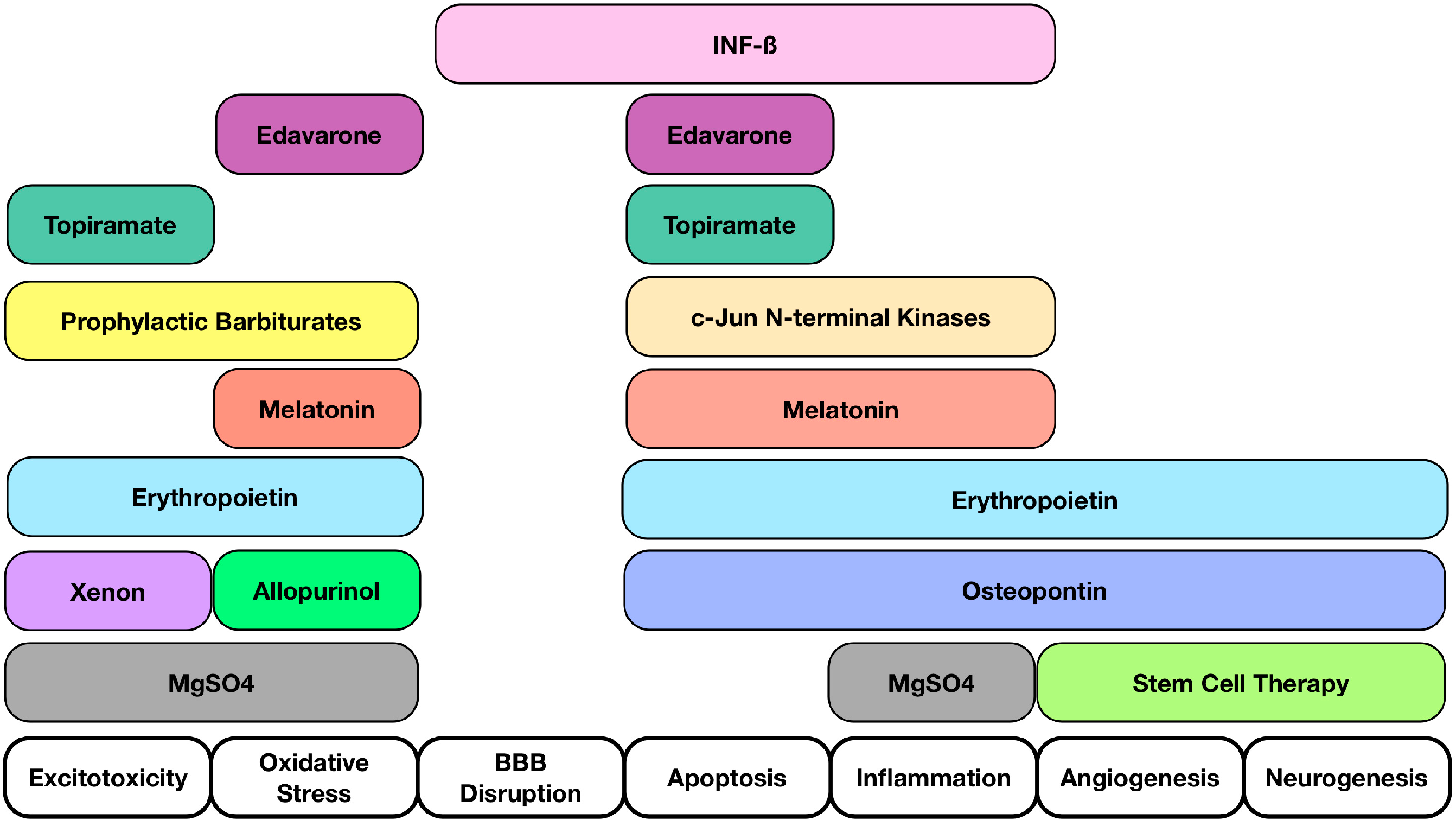{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules Studied | Possible Effects Related to Neuroprotection |
|---|---|
| Osteopontin (OPN) | OPN repairs brain injury after neonatal HIE by mediating regulation of cerebral cell proliferation, cell survival, and oligodendrocyte differentiation after injury [95] |
| Interferon Beta (INFβ) | Reduce TNF-α levels, proliferation and activation of T-cell lymphocytes, and pro-inflammatory cytokines produced by T-cells; Blood Brain Barrier integrity [96] |
| c-Jun N-terminal kinases (JNKs) | JNKs play a role in regulation of apoptosis [97]; Reductions in early neuronal damage [98]; Reduced inflammation and inhibition of apoptotic neuronal loss [99] |
| Prophylactic barbiturates | Diminishes moderate to severe neurodevelopmental impairment or death (HIE undergoing whole-body cooling) [100]; Multivariate analysis suggested its use to be associated with better outcomes [100] |
| Melatonin | Antioxidant, anti-inflammatory, and anti-apoptotic properties [101,102]; Protect the brain independently or in concert with therapeutic hypothermia [103]; Reducing oxidative stress and improved survival with favorable neurodevelopmental outcome at 6 months of age in combination with hypothermia [104] |
| Edaravone | Edaravone may inhibit the number of apoptotic neuronal cells and 8-OHdG expression within 48 h after HI insult [105]; Inhibits lipid peroxidation in neonatal HIE rat model [106]; Scavenger that inhibits both lipid and DNA peroxidation [107] |
| Molecules Studied | Possible Effects Related to Neuroprotection |
|---|---|
| Erythropoietin (EPO) | Associated with anti-inflammatory, anti-excitotoxic, anti-oxidative, and anti-apoptotic properties [108,109,110]; Vasogenic and pro-angiogenic functions [109]; Hypoxia-inducible-factor-1 mediates increase in EPO expression [111,112] |
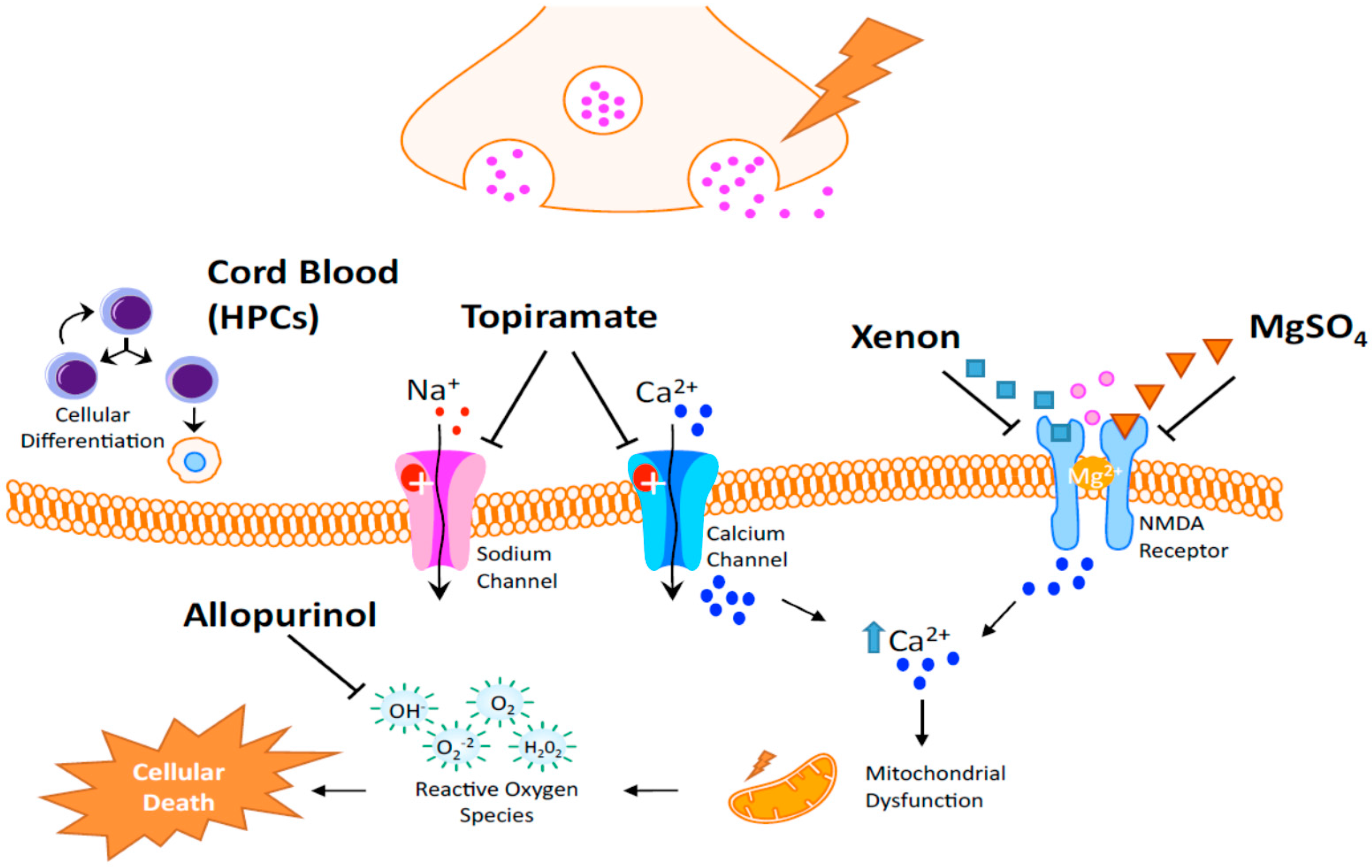
| Allopurinol | Neuroprotection in postnatal day 7 rats after HI [113]; Post hoc analysis revealed a potential benefit in treatment of females [114] |
| Xenon | May trigger neurodegeneration in the developing brain. Thus, the safety of a newborn injured brain is not expected [115]; Neuroprotective in adult rats in transient brain ischemia [115]; Limited protection when given alone but protection for up to 30 days when given in combination with hypothermia (neonatal rodents) [116] |
| Topiramate (TPM) | AMPA and Kainate receptors inhibition [117,118,119]; Blockade of Na channels, high voltage-activated calcium currents, carbonic anhydrase isoenzymes and mitochondrial permeability transition pore [120,121,122,123]; TPM in concert with melatonin decreases infarcted volume and apoptosis in neonatal HI rat model [124]; Pretreatment significantly reduced the brain damage and subsequent cognitive impairments [125] |
| Magnesium Sulfate (MgSO4) | Controversies exist regarding its efficacy in protecting the brain in term infants who may suffer encephalopathy [126] |
| Cord blood | Rich with hematopoietic stem cells [127,128,129,130,131] and neurotrophic factors acting on the following: Immunomodulation, reduction of immune cell infiltration, and the potential to increase neurogenesis and an angiogenesis [132,133] |
4.1. Experimental Translational Studies
4.1.1. Osteopontin
4.1.2. Interferon Beta
4.1.3. c-Jun N-Terminal Kinases
4.1.4. Prophylactic Barbiturates
4.1.5. Melatonin
4.1.6. Edaravone
4.2. Current Clinical Trial Studies
4.2.1. Erythropoietin
4.2.2. Allopurinol
4.2.3. Xenon
4.2.4. Topiramate
4.2.5. Magnesium Sulfate
4.2.6. Stem Cell Therapy and Neonatal HIE
5. Conclusions and Future Perspectives


Author Contributions
Conflicts of Interest
References
- Shetty, J. Neonatal seizures in hypoxic-ischaemic encephalopathy-risks and benefits of anticonvulsant therapy. Dev. Med. Child. Neurol. 2015, 57, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J. Perinatal brain injury: From pathogenesis to neuroprotection. Ment. Retard. Dev. Disabil. Res. Rev. 2001, 7, 56–64. [Google Scholar] [CrossRef]
- Doycheva, D.; Shih, G.; Chen, H.; Applegate, R.; Zhang, J.H.; Tang, J. Granulocyte-colony stimulating factor in combination with stem cell factor confers greater neuroprotection after hypoxic-ischemic brain damage in the neonatal rats than a solitary treatment. Transl. Stroke Res. 2013, 4, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Northington, F.J.; Chavez-Valdez, R.; Martin, L.J. Neuronal cell death in neonatal hypoxia-ischemia. Ann. Neurol. 2011, 69, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.B.; Perez-Polo, J.R. Hypoxia ischemia-mediated cell death in neonatal rat brain. Neurochem. Res. 2008, 33, 2379–2389. [Google Scholar] [CrossRef] [PubMed]
- Badr Zahr, L.K.; Purdy, I. Brain injury in the infant: The old, the new, and the uncertain. J. Perinat. Neonatal Nurs. 2006, 20, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, S. Hypoxic-ischemic encephalopathy and novel strategies for neuroprotection. Clin. Perinat. 2012, 39, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Fathali, N.; Lekic, T.; Zhang, J.H.; Tang, J. Long-term evaluation of granulocyte-colony stimulating factor on hypoxic-ischemic brain damage in infant rats. Intensive Care Med. 2010, 36, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Wayock, C.P.; Meserole, R.L.; Saria, S.; Jennings, J.M.; Huisman, T.A.; Northington, F.J.; Graham, E.M. Perinatal risk factors for severe injury in neonates treated with whole-body hypothermia for encephalopathy. Am. J. Obstet. Gynecol. 2014, 211, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-ischemic encephalopathy: A review for the clinician. JAMA Pediatr. 2015, 169, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Logitharajah, P.; Rutherford, M.A.; Cowan, F.M. Hypoxic-ischemic encephalopathy in preterm infants: Antecedent factors, brain imaging, and outcome. Pediatr. Res. 2009, 66, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, C.; Levene, M. Epidemiology and aetiology of neonatal seizures. Semin. Fetal Neonatal Med. 2013, 18, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Lombroso, C.T. Neonatal seizures: Gaps between the laboratory and the clinic. Epilepsia 2007, 48, 83–106. [Google Scholar] [CrossRef] [PubMed]
- Sheth, R.D. Electroencephalogram confirmatory rate in neonatal seizures. Pediatr. Neurol. 1999, 20, 27–30. [Google Scholar] [CrossRef]
- Silverstein, F.S.; Jensen, F.E. Neonatal seizures. Ann. Neurol. 2007, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Eunson, P. The long-term health, social, and financial burden of hypoxic-ischaemic encephalopathy. Dev. Med. Child. Neurol. 2015, 57, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J. Neonatal encephalopathy: An inadequate term for hypoxic-ischemic encephalopathy. Ann. Neurol. 2012, 72, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.M.; Perlman, M. Follow-up of the term infant after hypoxic-ischemic encephalopathy. Paediatr. Child. Health 2006, 11, 278–282. [Google Scholar] [PubMed]
- Fathali, N.; Khatibi, N.H.; Ostrowski, R.P.; Zhang, J.H. The evolving landscape of neuroinflammation after neonatal hypoxia-ischemia. Acta Neurochir. Suppl. 2011, 111, 93–100. [Google Scholar] [PubMed]
- Wachtel, E.V.; Hendricks-Munoz, K.D. Current management of the infant who presents with neonatal encephalopathy. Curr. Probl. Pediatr. Adolesc. Health Care 2011, 41, 132–153. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.Y.; Ashwal, S. Animal models of perinatal hypoxic-ischemic brain damage. Pediatr. Neurol. 2009, 40, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.A.; Brandon, D.H. Hypoxic ischemic encephalopathy: Pathophysiology and experimental treatments. Newborn Infant Nurs. Rev. 2011, 11, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Baburamani, A.A.; Ek, C.J.; Walker, D.W.; Castillo-Melendez, M. Vulnerability of the developing brain to hypoxic-ischemic damage: Contribution of the cerebral vasculature to injury and repair? Front. Physiol. 2012, 3, 424. [Google Scholar] [CrossRef] [PubMed]
- Ginet, V.; Puyal, J.; Clarke, P.G.; Truttmann, A.C. Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am. J. Pathol. 2009, 175, 1962–1974. [Google Scholar] [CrossRef] [PubMed]
- Seevinck, P.R.; Deddens, L.H.; Dijkhuizen, R.M. Magnetic resonance imaging of brain angiogenesis after stroke. Angiogenesis 2010, 13, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.V.; Trescher, W.H.; Ishida, A.; Nakajima, W. Neurobiology of hypoxic-ischemic injury in the developing brain. Pediatr. Res. 2001, 49, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Yang, S.N. Perinatal hypoxic-ischemic encephalopathy. J. Biomed. Biotechnol. 2011, 2011, 609813. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Halamek, L.P. Resuscitation of the Fetus and Newborn, an Issue of Clinics in Perinatology; Elsevier Health Sciences: Philadelphia, PA, USA, 2012. [Google Scholar]
- Juul, S.E.; Ferriero, D.M. Pharmacologic neuroprotective strategies in neonatal brain injury. Clin. Perinatol. 2014, 41, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Lorek, A.; Takei, Y.; Cady, E.B.; Wyatt, J.S.; Penrice, J.; Edwards, A.D.; Peebles, D.; Wylezinska, M.; Owen-Reece, H.; Kirkbride, V.; et al. Delayed ("secondary") cerebral energy failure after acute hypoxia-ischemia in the newborn piglet: Continuous 48-hour studies by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 1994, 36, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, R.C.; Towfighi, J.; Vannucci, S.J. Secondary energy failure after cerebral hypoxia-ischemia in the immature rat. J. Cereb. Blood Flow Metab. 2004, 24, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Laptook, A. The importance of temperature on the neurovascular unit. Early Hum. Dev. 2014, 90, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mittal, R.; Khanna, H.D.; Basu, S. Free radical injury and blood-brain barrier permeability in hypoxic-ischemic encephalopathy. Pediatrics 2008, 122, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Pansiot, J.; Bettati, D.; Strazielle, N.; Ghersi-Egea, J.F.; Damante, G.; Fleiss, B.; Titomanlio, L.; Gressens, P. Blood-brain barrier dysfunction in disorders of the developing brain. Front. Neurosci. 2015, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhao, D.; Wu, S.; Wang, D. Ang-(1–7) exerts protective role in blood-brain barrier damage by the balance of timp-1/mmp-9. Eur. J. Pharmacol. 2015, 748, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, X.; Zhao, Y.; Zhang, L.; Bai, X.; Zhang, J.; Zhao, X.; Chen, L.; Wang, L.; Cui, L. Pretreatment by evodiamine is neuroprotective in cerebral ischemia: Up-regulated pakt, pgsk3beta, down-regulated nf-kappab expression, and ameliorated bbb permeability. Neurochem. Res. 2014, 39, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.W.; Chen, Y.L.; Chuang, S.H.; Wang, J.S.; Jeng, K.C. Protective effect of a sesamin derivative, 3-bis (3-methoxybenzyl) butane-1, 4-diol on ischemic and hypoxic neuronal injury. J. Biomed. Sci. 2014, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Gu, Y.T.; Qin, G.H.; Zhong, L.; Meng, Y.N. Curcumin ameliorates the permeability of the blood-brain barrier during hypoxia by upregulating heme oxygenase-1 expression in brain microvascular endothelial cells. J. Mol. Neurosci. 2013, 51, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Cheon, S.Y.; Lee, W.T.; Park, K.A.; Lee, J.E. The effect of ask1 on vascular permeability and edema formation in cerebral ischemia. Brain Res. 2015, 1595, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; McBride, D.W.; Doycheva, D.; Dixon, B.J.; Krafft, P.R.; Zhang, J.H.; Tang, J. G-csf attenuates neuroinflammation and stabilizes the blood-brain barrier via the pi3k/akt/gsk-3beta signaling pathway following neonatal hypoxia-ischemia in rats. Exp. Neurol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Nemkul, N.; Shereen, A.; Jone, A.; Dunn, R.S.; Lawrence, D.A.; Lindquist, D.; Kuan, C.Y. Therapeutic administration of plasminogen activator inhibitor-1 prevents hypoxic-ischemic brain injury in newborns. J. Neurosci. 2009, 29, 8669–8674. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, S.; Huang, S.F.; Patkar, S.; Gassmann, M.; Ogunshola, O.O. Differential responses of blood-brain barrier associated cells to hypoxia and ischemia: A comparative study. Fluids Barriers CNS 2015, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, H.; Lechpammer, M.; Jensen, F.E.; Warfield, S.K.; Hansen, A.H.; Kosaras, B.; Shevell, M.; Wintermark, P. Increased brain perfusion persists over the first month of life in term asphyxiated newborns treated with hypothermia: Does it reflect activated angiogenesis? Transl. Stroke Res. 2015, 6, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, H.; Boudes, E.; Khoja, Z.; Shevell, M.; Wintermark, P. Angiogenesis dysregulation in term asphyxiated newborns treated with hypothermia. PLoS ONE 2015, 10, 128028. [Google Scholar] [CrossRef] [PubMed]
- Pimentel-Coelho, P.M.; Rosado-de-Castro, P.H.; da Fonseca, L.M.; Mendez-Otero, R. Umbilical cord blood mononuclear cell transplantation for neonatal hypoxic-ischemic encephalopathy. Pediatr. Res. 2012, 71, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Xiong, L.J.; Tong, Y.; Mao, M. The neuroprotective roles of bdnf in hypoxic ischemic brain injury. Biomed. Rep. 2013, 1, 167–176. [Google Scholar] [PubMed]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Guidi, S.; Bianchi, P.; Alstrup, A.K.; Henningsen, K.; Smith, D.F.; Bartesaghi, R. Postnatal neurogenesis in the hippocampal dentate gyrus and subventricular zone of the gottingen minipig. Brain Res. Bull. 2011, 85, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Donega, V.; van Velthoven, C.T.; Nijboer, C.H.; Kavelaars, A.; Heijnen, C.J. The endogenous regenerative capacity of the damaged newborn brain: Boosting neurogenesis with mesenchymal stem cell treatment. J. Cereb. Blood Flow Metab. 2013, 33, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.; Plane, J.M.; Parent, J.M.; Silverstein, F.S. Hypoxic-ischemic injury stimulates subventricular zone proliferation and neurogenesis in the neonatal rat. Pediatr. Res. 2005, 58, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Van Velthoven, C.T.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Mesenchymal stem cell treatment after neonatal hypoxic-ischemic brain injury improves behavioral outcome and induces neuronal and oligodendrocyte regeneration. Brain Behav. Immun. 2010, 24, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lopez, D.; Natarajan, N.; Ashwal, S.; Vexler, Z.S. Mechanisms of perinatal arterial ischemic stroke. J. Cereb. Blood Flow Metab. 2014, 34, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell. Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, H.; Roughton, K.; Wang, X.; Kroemer, G.; Blomgren, K.; Zhu, C. Lithium reduces apoptosis and autophagy after neonatal hypoxia-ischemia. Cell. Death Dis. 2010, 1, 56. [Google Scholar] [CrossRef] [PubMed]
- Balduini, W.; Carloni, S.; Buonocore, G. Autophagy in hypoxia-ischemia induced brain injury. J. Mater. Fetal Neonatal Med. 2012, 25, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Xiong, L.J.; Tong, Y.; Mao, M. Neuroprotective effect of brain-derived neurotrophic factor mediated by autophagy through the pi3k/akt/mtor pathway. Mol. Med. Rep. 2013, 8, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, S.J.; Hagberg, H. Hypoxia-ischemia in the immature brain. J. Exp. Biol. 2004, 207, 3149–3154. [Google Scholar] [CrossRef] [PubMed]
- Hartung, T.; Daston, G. Are in vitro tests suitable for regulatory use? Toxicol. Sci. 2009, 111, 233–237. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Recent highlights in the development of new antiviral drugs. Curr. Opin. Microbiol. 2005, 8, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Perel, P.; Roberts, I.; Sena, E.; Wheble, P.; Briscoe, C.; Sandercock, P.; Macleod, M.; Mignini, L.E.; Jayaram, P.; Khan, K.S. Comparison of treatment effects between animal experiments and clinical trials: Systematic review. BMJ 2007, 334, 197. [Google Scholar] [CrossRef] [PubMed]
- Hackam, D.G.; Redelmeier, D.A. Translation of research evidence from animals to humans. JAMA 2006, 296, 1731–1732. [Google Scholar] [CrossRef] [PubMed]
- O'Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; van der Worp, B.H.; Howells, D.W. 1026 experimental treatments in acute stroke. Ann. Neurol. 2006, 59, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.V.; Ferriero, D.M.; Vannucci, S.J.; Hagberg, H. Models of cerebral palsy: Which ones are best? J. Child. Neurol. 2005, 20, 984–987. [Google Scholar] [CrossRef] [PubMed]
- Ashwal, S.; Tone, B.; Tian, H.R.; Chong, S.; Obenaus, A. Comparison of two neonatal ischemic injury models using magnetic resonance imaging. Pediatr. Res. 2007, 61, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhuang, L.; Terrando, N.; Wu, X.; Jonhson, M.R.; Maze, M.; Ma, D. A clinically relevant model of perinatal global ischemic brain damage in rats. Brain Res. 2011, 1383, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lai, H.; Xu, H.; Wu, W.; Lai, X.; Ho, G.; Ma, L.; Chen, Y. Impact of perinatal systemic hypoxic-ischemic injury on the brain of male offspring rats: An improved model of neonatal hypoxic-ischemic encephalopathy in early preterm newborns. PLoS ONE 2013, 8, 82502. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, M.; Ohshima, M.; Taguchi, A.; Kasahara, Y.; Ikeda, T.; Matsuyama, T. A novel reproducible model of neonatal stroke in mice: Comparison with a hypoxia-ischemia model. Exp. Neurol. 2013, 247, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Northington, F.J. Brief update on animal models of hypoxic-ischemic encephalopathy and neonatal stroke. ILAR J. 2006, 47, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Clowry, G.J.; Basuodan, R.; Chan, F. What are the best animal models for testing early intervention in cerebral palsy? Front. Neurol. 2014, 5, 258. [Google Scholar] [CrossRef] [PubMed]
- Teo, L.; Bourne, J.A. A reproducible and translatable model of focal ischemia in the visual cortex of infant and adult marmoset monkeys. Brain Pathol. 2014, 24, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Sena, E.; van der Worp, H.B.; Howells, D.; Macleod, M. How can we improve the pre-clinical development of drugs for stroke? Trends Neurosci. 2007, 30, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Van der Worp, H.B.; Howells, D.W.; Sena, E.S.; Porritt, M.J.; Rewell, S.; O'Collins, V.; Macleod, M.R. Can animal models of disease reliably inform human studies? PLoS Med. 2010, 7, 1000245. [Google Scholar] [CrossRef] [PubMed]
- Saver, J.L.; Johnston, K.C.; Homer, D.; Wityk, R.; Koroshetz, W.; Truskowski, L.L.; Haley, E.C. Infarct volume as a surrogate or auxiliary outcome measure in ischemic stroke clinical trials. Stroke 1999, 30, 293–298. [Google Scholar] [CrossRef] [PubMed]
- The National Institute of Neurological Disorders and Stroke (Ninds) rt-pa Stroke Study Group. Effect of intravenous recombinant tissue plasminogen activator on ischemic stroke lesion size measured by computed tomography. Stroke 2000, 31, 2912–2919. [Google Scholar]
- Van der Worp, H.B.; de Haan, P.; Morrema, E.; Kalkman, C.J. Methodological quality of animal studies on neuroprotection in focal cerebral ischaemia. J. Neurol. 2005, 252, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Langley, G.; Evans, T.; Holgate, S.T.; Jones, A. Replacing animal experiments: Choices, chances and challenges. BioEssays 2007, 29, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Sena, E.S.; van der Worp, H.B.; Bath, P.M.; Howells, D.W.; Macleod, M.R. Publication bias in reports of animal stroke studies leads to major overstatement of efficacy. PLoS Biol. 2010, 8, 1000344. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, R.K. Drug delivery systems, cns protection, and the blood brain barrier. Biomed. Res. Int. 2014, 2014, 869269. [Google Scholar] [CrossRef] [PubMed]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx 2005, 2, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Covarrubias, L.; Slosky, L.M.; Thompson, B.J.; Davis, T.P.; Ronaldson, P.T. Transporters at cns barrier sites: Obstacles or opportunities for drug delivery? Curr. Pharm. Des. 2014, 20, 1422–1449. [Google Scholar] [CrossRef] [PubMed]
- Ek, C.J.; D'Angelo, B.; Baburamani, A.A.; Lehner, C.; Leverin, A.L.; Smith, P.L.; Nilsson, H.; Svedin, P.; Hagberg, H.; Mallard, C. Brain barrier properties and cerebral blood flow in neonatal mice exposed to cerebral hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Allegaert, K.; van den Anker, J.N. Clinical pharmacology in neonates: Small size, huge variability. Neonatology 2014, 105, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.A. Neonatal drug development. Early Hum. Dev. 2011, 87, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Dabliz, R.; Levine, S. Medication safety in neonates. Am. J. Perinatol. 2012, 29, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Lehr, V.T.; Lieh-Lai, M.; Koo, W.; Ward, R.M.; Rieder, M.J.; van den Anker, J.N.; Reeves, J.H.; Mathew, M.; Lulic-Botica, M.; et al. An algorithm to detect adverse drug reactions in the neonatal intensive care unit. J. Clin. Pharmacol. 2013, 53, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Tuleu, C. 'Formulating better medicines for children'—Still paving the road. Int. J. Pharm. 2012, 435, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Van den Anker, J.; Allegaert, K. Clinical pharmacology in neonates and young infants: The benefit of a population-tailored approach. Exp. Rev. Clin. Pharmacol. 2012, 5, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Choonara, I. Who wants safer medicines for children. Arch. Dis. Child. 2008, 93, 456–457. [Google Scholar] [CrossRef] [PubMed]
- Salunke, S.; Giacoia, G.; Tuleu, C. The step (safety and toxicity of excipients for paediatrics) database. Part 1—A need assessment study. Int. J. Pharm. 2012, 435, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Nahata, M.C. Safety of “inert” additives or excipients in paediatric medicines. Arch. Dis. child. Fetal Neonatal Ed. 2009, 94, 392–393. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.A.; Storme, T. European study for neonatal excipient exposure (esnee). Eur. J. Hosp. Pharm. 2012, 19, 67. [Google Scholar] [CrossRef]
- Johnston, M.V.; Fatemi, A.; Wilson, M.A.; Northington, F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol. 2011, 10, 372–382. [Google Scholar] [CrossRef]
- O'Regan, A.; Berman, J.S. Osteopontin: A key cytokine in cell-mediated and granulomatous inflammation. Int. J. Exp. Pathol. 2000, 81, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Chabot, S.; Williams, G.; Yong, V.W. Microglial production of tnf-alpha is induced by activated T lymphocytes. Involvement of vla-4 and inhibition by interferonbeta-1b. J. Clin. Investig. 1997, 100, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. Jnk signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed]
- Nijboer, C.H.; Heijnen, C.J.; Groenendaal, F.; van Bel, F.; Kavelaars, A. Alternate pathways preserve tumor necrosis factor-alpha production after nuclear factor-kappab inhibition in neonatal cerebral hypoxia-ischemia. Stroke 2009, 40, 3362–3368. [Google Scholar] [CrossRef] [PubMed]
- Nijboer, C.H.; Bonestroo, H.J.; Zijlstra, J.; Kavelaars, A.; Heijnen, C.J. Mitochondrial jnk phosphorylation as a novel therapeutic target to inhibit neuroinflammation and apoptosis after neonatal ischemic brain damage. Neurobiol. Dis. 2013, 54, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Meyn, D.F., Jr.; Ness, J.; Ambalavanan, N.; Carlo, W.A. Prophylactic phenobarbital and whole-body cooling for neonatal hypoxic-ischemic encephalopathy. J. Pediatr. 2010, 157, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Alconada, D.; Alvarez, A.; Arteaga, O.; Martinez-Ibarguen, A.; Hilario, E. Neuroprotective effect of melatonin: A novel therapy against perinatal hypoxia-ischemia. Int. J. Mol. Sci. 2013, 14, 9379–9395. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Perrone, S.; Buonocore, G.; Longini, M.; Proietti, F.; Balduini, W. Melatonin protects from the long-term consequences of a neonatal hypoxic-ischemic brain injury in rats. J. Pineal. Res. 2008, 44, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.J.; Faulkner, S.; Fleiss, B.; Bainbridge, A.; Andorka, C.; Price, D.; Powell, E.; Lecky-Thompson, L.; Thei, L.; Chandrasekaran, M.; et al. Melatonin augments hypothermic neuroprotection in a perinatal asphyxia model. Brain 2013, 136, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Aly, H.; Elmahdy, H.; El-Dib, M.; Rowisha, M.; Awny, M.; El-Gohary, T.; Elbatch, M.; Hamisa, M.; El-Mashad, A.R. Melatonin use for neuroprotection in perinatal asphyxia: A randomized controlled pilot study. J. Perinatol. 2015, 35, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, Y.; Miyazawa, T.; Nonoyama, S.; Goto, Y.; Itoh, M. Edaravone inhibits DNA peroxidation and neuronal cell death in neonatal hypoxic-ischemic encephalopathy model rat. Pediatr. Res. 2009, 65, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Noor, J.I.; Ueda, Y.; Ikeda, T.; Ikenoue, T. Edaravone inhibits lipid peroxidation in neonatal hypoxic-ischemic rats: An in vivo microdialysis study. Neurosci. Lett. 2007, 414, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Yang, Z.J.; Carter, E.L.; Martin, L.J.; Koehler, R.C. Striatal neuroprotection from neonatal hypoxia-ischemia in piglets by antioxidant treatment with euk-134 or edaravone. Dev. Neurosci. 2011, 33, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Villa, P.; Bigini, P.; Mennini, T.; Agnello, D.; Laragione, T.; Cagnotto, A.; Viviani, B.; Marinovich, M.; Cerami, A.; Coleman, T.R.; et al. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J. Exp. Med. 2003, 198, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Z.; Wang, Y.; Zhang, R.; Chopp, M. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke 2004, 35, 1732–1737. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.E. Hypothermia plus erythropoietin for neonatal neuroprotection? Commentary on Fan et al. and Fang et al. Pediatr. Res. 2013, 73, 10–11. [Google Scholar]
- Bernaudin, M.; Marti, H.H.; Roussel, S.; Divoux, D.; Nouvelot, A.; MacKenzie, E.T.; Petit, E. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 1999, 19, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Chang, Y.S.; Vexler, Z.S.; Ferriero, D.M. Hypoxia-inducible factor 1alpha and erythropoietin upregulation with deferoxamine salvage after neonatal stroke. Exp. Neurol. 2005, 195, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.; Towfighi, J.; Roberts, R.L.; Heitjan, D.F. Allopurinol administered after inducing hypoxia-ischemia reduces brain injury in 7-day-old rats. Pediatr. Res. 1993, 33, 405–411. [Google Scholar] [PubMed]
- Kaandorp, J.J.; Benders, M.J.; Schuit, E.; Rademaker, C.M.; Oudijk, M.A.; Porath, M.M.; Oetomo, S.B.; Wouters, M.G.; van Elburg, R.M.; Franssen, M.T.; et al. Maternal allopurinol administration during suspected fetal hypoxia: A novel neuroprotective intervention? A multicentre randomised placebo controlled trial. Arch. Dis. Child. Fetal Neonatal Ed. 2015, 100, 216–223. [Google Scholar] [CrossRef] [PubMed]
- David, H.N.; Haelewyn, B.; Rouillon, C.; Lecoq, M.; Chazalviel, L.; Apiou, G.; Risso, J.J.; Lemaire, M.; Abraini, J.H. Neuroprotective effects of xenon: A therapeutic window of opportunity in rats subjected to transient cerebral ischemia. FASEB J. 2008, 22, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Hossain, M.; Chow, A.; Arshad, M.; Battson, R.M.; Sanders, R.D.; Mehmet, H.; Edwards, A.D.; Franks, N.P.; Maze, M. Xenon and hypothermia combine to provide neuroprotection from neonatal asphyxia. Ann. Neurol. 2005, 58, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Follett, P.L.; Deng, W.; Dai, W.; Talos, D.M.; Massillon, L.J.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: A protective role for topiramate. J. Neurosci. 2004, 24, 4412–4420. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.M.; Banerjee, M.; Rogawski, M.A. Topiramate selectively protects against seizures induced by atpa, a glur5 kainate receptor agonist. Neuropharmacology 2004, 46, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.; Tibayan, F.D.; Simpson, J.N.; Jensen, F.E. Nbqx or topiramate treatment after perinatal hypoxia-induced seizures prevents later increases in seizure-induced neuronal injury. Epilepsia 2004, 45, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Zona, C.; Ciotti, M.T.; Avoli, M. Topiramate attenuates voltage-gated sodium currents in rat cerebellar granule cells. Neurosci. Lett. 1997, 231, 123–126. [Google Scholar] [CrossRef]
- Costa, C.; Martella, G.; Picconi, B.; Prosperetti, C.; Pisani, A.; di Filippo, M.; Pisani, F.; Bernardi, G.; Calabresi, P. Multiple mechanisms underlying the neuroprotective effects of antiepileptic drugs against in vitro ischemia. Stroke 2006, 37, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Dodgson, S.J.; Shank, R.P.; Maryanoff, B.E. Topiramate as an inhibitor of carbonic anhydrase isoenzymes. Epilepsia 2000, 41, 35–39. [Google Scholar] [CrossRef]
- Kudin, A.P.; Debska-Vielhaber, G.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. The mechanism of neuroprotection by topiramate in an animal model of epilepsy. Epilepsia 2004, 45, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Ozyener, F.; Cetinkaya, M.; Alkan, T.; Goren, B.; Kafa, I.M.; Kurt, M.A.; Koksal, N. Neuroprotective effects of melatonin administered alone or in combination with topiramate in neonatal hypoxic-ischemic rat model. Restor. Neurol. Neurosci. 2012, 30, 435–444. [Google Scholar] [PubMed]
- Noh, M.R.; Kim, S.K.; Sun, W.; Park, S.K.; Choi, H.C.; Lim, J.H.; Kim, I.H.; Kim, H.J.; Kim, H.; Eun, B.L. Neuroprotective effect of topiramate on hypoxic ischemic brain injury in neonatal rats. Exp. Neurol. 2006, 201, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, R.; Bennet, L.; Groenendaal, F.; Lear, C.A.; Tan, S.; van Bel, F.; Juul, S.E.; Robertson, N.J.; Mallard, C.; Gunn, A.J. Magnesium is not consistently neuroprotective for perinatal hypoxia-ischemia in term-equivalent models in preclinical studies: A systematic review. Dev. Neurosci. 2014, 36, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Tagin, M.; Shah, P.S.; Lee, K.S. Magnesium for newborns with hypoxic-ischemic encephalopathy: A systematic review and meta-analysis. J. Perinatol. 2013, 33, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Diamandis, T.; Borlongan, C.V. One, two, three steps toward cell therapy for stroke. Stroke 2015, 46, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gomez, J.A.; Lu, J.Q.; Velasco, I.; Rivera, S.; Zoghbi, S.S.; Liow, J.S.; Musachio, J.L.; Chin, F.T.; Toyama, H.; Seidel, J.; et al. Persistent dopamine functions of neurons derived from embryonic stem cells in a rodent model of parkinson disease. Stem Cells 2007, 25, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with als can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.S.; Cleren, C.; Singh, S.K.; Yang, L.; Beal, M.F.; Goldman, S.A. Functional engraftment of human es cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat. Med. 2006, 12, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.; Guzman, R.; Daadi, M.; Steinberg, G.K. Cell transplantation therapy for stroke. Stroke 2007, 38, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Cotten, M.; Tan, S.; Kurtzberg, J.; Cairo, M.S. Rescuing the neonatal brain from hypoxic injury with autologous cord blood. Bone Marrow Transplant. 2013, 48, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Denhardt, D.T.; Noda, M.; O'Regan, A.W.; Pavlin, D.; Berman, J.S. Osteopontin as a means to cope with environmental insults: Regulation of inflammation, tissue remodeling, and cell survival. J. Clin. Investig. 2001, 107, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Mazzali, M.; Kipari, T.; Ophascharoensuk, V.; Wesson, J.A.; Johnson, R.; Hughes, J. Osteopontin-a molecule for all seasons. QJM: Int. J. Med. 2002, 95, 3–13. [Google Scholar] [CrossRef]
- van Velthoven, C.T.; Heijnen, C.J.; van Bel, F.; Kavelaars, A. Osteopontin enhances endogenous repair after neonatal hypoxic-ischemic brain injury. Stroke 2011, 42, 2294–2301. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ma, Q.; Suzuki, H.; Hartman, R.; Tang, J.; Zhang, J.H. Osteopontin reduced hypoxia-ischemia neonatal brain injury by suppression of apoptosis in a rat pup model. Stroke 2011, 42, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Bonestroo, H.J.; Nijboer, C.H.; van Velthoven, C.T.; van Bel, F.; Heijnen, C.J. The neonatal brain is not protected by osteopontin peptide treatment after hypoxia-ischemia. Dev. Neurosci. 2015, 37, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Veldhuis, W.B.; Floris, S.; van der Meide, P.H.; Vos, I.M.; de Vries, H.E.; Dijkstra, C.D.; Bar, P.R.; Nicolay, K. Interferon-beta prevents cytokine-induced neutrophil infiltration and attenuates blood-brain barrier disruption. J. Cereb. Blood Flow Metab. 2003, 23, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Maier, C.M.; Yu, F.; Nishi, T.; Lathrop, S.J.; Chan, P.H. Interferon-beta fails to protect in a model of transient focal stroke. Stroke 2006, 37, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Boehm, I.; Oakley, A.; Ketterman, A.J.; Barr, R.K. Targeting the jnk mapk cascade for inhibition: Basic science and therapeutic potential. Biochim. Biophys. Acta Prot. Proteom. 2004, 1697, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Gallagher, E. From jnk to pay dirt: Jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 2005, 57, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Nijboer, C.H.; van der Kooij, M.A.; van Bel, F.; Ohl, F.; Heijnen, C.J.; Kavelaars, A. Inhibition of the jnk/ap-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury. Brain Behav. Immun. 2010, 24, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Kaandorp, J.J.; van den Broek, M.P.; Benders, M.J.; Oudijk, M.A.; Porath, M.M.; Bambang Oetomo, S.; Wouters, M.G.; van Elburg, R.; Franssen, M.T.; Bos, A.F.; et al. Rapid target allopurinol concentrations in the hypoxic fetus after maternal administration during labour. Arch. Dis. Child. Fetal Aneonatal Ed. 2014, 99, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Merchant, N.M.; Azzopardi, D.V.; Hawwa, A.F.; McElnay, J.C.; Middleton, B.; Arendt, J.; Arichi, T.; Gressens, P.; Edwards, A.D. Pharmacokinetics of melatonin in preterm infants. Br. J. Clin. Pharmacol. 2013, 76, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Shea, K.L.; Palanisamy, A. What can you do to protect the newborn brain? Curr. Opin. Anaesthesiol. 2015, 28, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Noor, J.I.; Ikeda, T.; Mishima, K.; Aoo, N.; Ohta, S.; Egashira, N.; Iwasaki, K.; Fujiwara, M.; Ikenoue, T. Short-term administration of a new free radical scavenger, edaravone, is more effective than its long-term administration for the treatment of neonatal hypoxic-ischemic encephalopathy. Stroke 2005, 36, 2468–2474. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.E.; Yachnis, A.T.; Rojiani, A.M.; Christensen, R.D. Immunohistochemical localization of erythropoietin and its receptor in the developing human brain. Pediatr. Dev. Pathol. 1999, 2, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.E.; McPherson, R.J.; Farrell, F.X.; Jolliffe, L.; Ness, D.J.; Gleason, C.A. Erytropoietin concentrations in cerebrospinal fluid of nonhuman primates and fetal sheep following high-dose recombinant erythropoietin. Neonatology 2004, 85, 138–144. [Google Scholar]
- Zhu, C.; Kang, W.; Xu, F.; Cheng, X.; Zhang, Z.; Jia, L.; Ji, L.; Guo, X.; Xiong, H.; Simbruner, G.; et al. Erythropoietin improved neurologic outcomes in newborns with hypoxic-ischemic encephalopathy. Pediatrics 2009, 124, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Plateel, M.; Teissier, E.; Cecchelli, R. Hypoxia dramatically increases the nonspecific transport of blood-borne proteins to the brain. J. Neurochem. 1997, 68, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Statler, P.A.; McPherson, R.J.; Bauer, L.A.; Kellert, B.A.; Juul, S.E. Pharmacokinetics of high-dose recombinant erythropoietin in plasma and brain of neonatal rats. Pediatr. Res. 2007, 61, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Xenocostas, A.; Cheung, W.K.; Farrell, F.; Zakszewski, C.; Kelley, M.; Lutynski, A.; Crump, M.; Lipton, J.H.; Kiss, T.L.; Lau, C.Y.; et al. The pharmacokinetics of erythropoietin in the cerebrospinal fluid after intravenous administration of recombinant human erythropoietin. Eur J. Clin. Pharmacol. 2005, 61, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Bauer, L.A.; Ballard, R.A.; Ferriero, D.M.; Glidden, D.V.; Mayock, D.E.; Chang, T.; Durand, D.J.; Song, D.; Bonifacio, S.L.; et al. Erythropoietin for neuroprotection in neonatal encephalopathy: Safety and pharmacokinetics. Pediatrics 2012, 130, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Kumral, A.; Uysal, N.; Tugyan, K.; Sonmez, A.; Yilmaz, O.; Gokmen, N.; Kiray, M.; Genc, S.; Duman, N.; Koroglu, T.F.; et al. Erythropoietin improves long-term spatial memory deficits and brain injury following neonatal hypoxia-ischemia in rats. Behav. Brain Res. 2004, 153, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.F.; Abel, R.; Almli, C.R.; Mu, D.; Wendland, M.; Ferriero, D.M. Erythropoietin sustains cognitive function and brain volume after neonatal stroke. Dev. Neurosci. 2009, 31, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Rogers, E.E.; Bonifacio, S.L.; Glass, H.C.; Juul, S.E.; Chang, T.; Mayock, D.E.; Durand, D.J.; Song, D.; Barkovich, A.J.; Ballard, R.A.; et al. Erythropoietin and hypothermia for hypoxic-ischemic encephalopathy. Pediatr. Neurol. 2014, 51, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, S. Outcomes of hypoxic-ischemic encephalopathy in neonates treated with hypothermia. Clin. Perinatol. 2014, 41, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Elmahdy, H.; El-Mashad, A.R.; El-Bahrawy, H.; El-Gohary, T.; El-Barbary, A.; Aly, H. Human recombinant erythropoietin in asphyxia neonatorum: Pilot trial. Pediatrics 2010, 125, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Kawabata, H.; Tcherniamtchouk, O.; Huynh, T.; Black, K.L.; Koeffler, H.P. Glioblastoma multiforme cells: Expression of erythropoietin receptor and response to erythropoietin. Int. J. Oncol. 2007, 31, 1193–1198. [Google Scholar] [PubMed]
- Digicaylioglu, M.; Lipton, S.A. Erythropoietin-mediated neuroprotection involves cross-talk between jak2 and nf-kappab signalling cascades. Nature 2001, 412, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Chu, K.; Sinn, D.I.; Jung, K.H.; Kim, E.H.; Kim, S.J.; Kim, J.M.; Ko, S.Y.; Kim, M.; Roh, J.K. Erythropoietin reduces perihematomal inflammation and cell death with enos and stat3 activations in experimental intracerebral hemorrhage. J. Neurochem. 2006, 96, 1728–1739. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Lee, S.M.; Lee, J.W.; Son, D.J.; Moon, D.C.; Yoon, D.Y.; Hong, J.T. Erk-mediated production of neurotrophic factors by astrocytes promotes neuronal stem cell differentiation by erythropoietin. Biochem. Biophys. Res. Commun. 2006, 339, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Peeters-Scholte, C.; Braun, K.; Koster, J.; Kops, N.; Blomgren, K.; Buonocore, G.; van Buul-Offers, S.; Hagberg, H.; Nicolay, K.; van Bel, F.; et al. Effects of allopurinol and deferoxamine on reperfusion injury of the brain in newborn piglets after neonatal hypoxia-ischemia. Pediatr. Res. 2003, 54, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Benders, M.J.; Bos, A.F.; Rademaker, C.M.; Rijken, M.; Torrance, H.L.; Groenendaal, F.; van Bel, F. Early postnatal allopurinol does not improve short term outcome after severe birth asphyxia. Arch. Dis. Child. Fetal Neonatal Ed. 2006, 91, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Kaandorp, J.J.; van Bel, F.; Veen, S.; Derks, J.B.; Groenendaal, F.; Rijken, M.; Roze, E.; Venema, M.M.; Rademaker, C.M.; Bos, A.F.; et al. Long-term neuroprotective effects of allopurinol after moderate perinatal asphyxia: Follow-up of two randomised controlled trials. Arch. Dis. Child. Fetal Neonatal Ed. 2012, 97, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Dworschak, M. Pharmacologic neuroprotection-is xenon the light at the end of the tunnel? Crit. Care Med. 2008, 36, 2477–2479. [Google Scholar] [CrossRef] [PubMed]
- Istaphanous, G.K.; Loepke, A.W. General anesthetics and the developing brain. Curr. Opin. Anaesthesiol. 2009, 22, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, C.; Thoresen, M.; Tucker, A.; Aquilina, K.; Chakkarapani, E.; Dingley, J. Xenon and hypothermia combine additively, offering long-term functional and histopathologic neuroprotection after neonatal hypoxia/ischemia. Stroke 2008, 39, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, M.; Hobbs, C.E.; Wood, T.; Chakkarapani, E.; Dingley, J. Cooling combined with immediate or delayed xenon inhalation provides equivalent long-term neuroprotection after neonatal hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2009, 29, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Dingley, J.; Tooley, J.; Liu, X.; Scull-Brown, E.; Elstad, M.; Chakkarapani, E.; Sabir, H.; Thoresen, M. Xenon ventilation during therapeutic hypothermia in neonatal encephalopathy: A feasibility study. Pediatrics 2014, 133, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.J.; Tan, S.; Groenendaal, F.; van Bel, F.; Juul, S.E.; Bennet, L.; Derrick, M.; Back, S.A.; Valdez, R.C.; Northington, F.; et al. Which neuroprotective agents are ready for bench to bedside translation in the newborn infant? J. Pediatr. 2012, 160, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Cilio, M.R.; Ferriero, D.M. Synergistic neuroprotective therapies with hypothermia. Semin. Fetal Neonatal Med. 2010, 15, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Parmeggiani, L. Topiramate and its clinical applications in epilepsy. Expert Opin. Pharmacother. 2006, 7, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Shank, R.P.; Gardocki, J.F.; Streeter, A.J.; Maryanoff, B.E. An overview of the preclinical aspects of topiramate: Pharmacology, pharmacokinetics, and mechanism of action. Epilepsia 2000, 41, 3–9. [Google Scholar] [CrossRef]
- Angehagen, M.; Ronnback, L.; Hansson, E.; Ben-Menachem, E. Topiramate reduces ampa-induced Ca2+ transients and inhibits glur1 subunit phosphorylation in astrocytes from primary cultures. J. Neurochem. 2005, 94, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Sfaello, I.; Baud, O.; Arzimanoglou, A.; Gressens, P. Topiramate prevents excitotoxic damage in the newborn rodent brain. Neurobiol. Dis. 2005, 20, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Zeevalk, G.D.; Nicklas, W.J. Evidence that the loss of the voltage-dependent Mg2+ block at the n-methyl-d-aspartate receptor underlies receptor activation during inhibition of neuronal metabolism. J. Neurochem. 1992, 59, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, J.; Romani, A.M.; Valentin-Torres, A.M.; Luciano, A.A.; Ramirez Kitchen, C.M.; Funderburg, N.; Mesiano, S.; Bernstein, H.B. Magnesium decreases inflammatory cytokine production: A novel innate immunomodulatory mechanism. J. Immunol. 2012, 188, 6338–6346. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.J.; Marro, P.J.; McGowan, J.E.; Mishra, O.P.; Delivoria-Papadopoulos, M. Protective effect of mgso4 infusion on nmda receptor binding characteristics during cerebral cortical hypoxia in the newborn piglet. Brain Res. 1994, 644, 144–149. [Google Scholar] [CrossRef]
- Shokry, M.; Elsedfy, G.O.; Bassiouny, M.M.; Anmin, M.; Abozid, H. Effects of antenatal magnesium sulfate therapy on cerebral and systemic hemodynamics in preterm newborns. Acta Obstet. Gynecol. Scand. 2010, 89, 801–806. [Google Scholar] [CrossRef] [PubMed]
- de Haan, H.H.; Gunn, A.J.; Williams, C.E.; Heymann, M.A.; Gluckman, P.D. Magnesium sulfate therapy during asphyxia in near-term fetal lambs does not compromise the fetus but does not reduce cerebral injury. Am. J. Obstet. Gynecol. 1997, 176, 18–27. [Google Scholar] [CrossRef]
- Penrice, J.; Amess, P.N.; Punwani, S.; Wylezinska, M.; Tyszczuk, L.; D'Souza, P.; Edwards, A.D.; Cady, E.B.; Wyatt, J.S.; Reynolds, E.O. Magnesium sulfate after transient hypoxia-ischemia fails to prevent delayed cerebral energy failure in the newborn piglet. Pediatr. Res. 1997, 41, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, K.; Cox, P.; Mehmet, H.; Penrice, J.; Amess, P.N.; Cady, E.B.; Wyatt, J.S.; Edwards, A.D. Magnesium sulfate treatment after transient hypoxia-ischemia in the newborn piglet does not protect against cerebral damage. Pediatr. Res. 2000, 48, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Schira, J.; Gasis, M.; Estrada, V.; Hendricks, M.; Schmitz, C.; Trapp, T.; Kruse, F.; Kogler, G.; Wernet, P.; Hartung, H.P.; et al. Significant clinical, neuropathological and behavioural recovery from acute spinal cord trauma by transplantation of a well-defined somatic stem cell from human umbilical cord blood. Brain 2012, 135, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Escolar, M.L.; Poe, M.D.; Provenzale, J.M.; Richards, K.C.; Allison, J.; Wood, S.; Wenger, D.A.; Pietryga, D.; Wall, D.; Champagne, M.; et al. Transplantation of umbilical-cord blood in babies with infantile krabbe's disease. N. Engl. J. Med. 2005, 352, 2069–2081. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Allison, J.; McLaughlin, C.; Sledge, L.; Waters-Pick, B.; Wease, S.; Kurtzberg, J. Differences in quality between privately and publicly banked umbilical cord blood units: A pilot study of autologous cord blood infusion in children with acquired neurologic disorders. Transfusion 2010, 50, 1980–1987. [Google Scholar] [CrossRef] [PubMed]
- Cotten, C.M.; Murtha, A.P.; Goldberg, R.N.; Grotegut, C.A.; Smith, P.B.; Goldstein, R.F.; Fisher, K.A.; Gustafson, K.E.; Waters-Pick, B.; Swamy, G.K.; et al. Feasibility of autologous cord blood cells for infants with hypoxic-ischemic encephalopathy. J. Pediatr. 2014, 164, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V.; Weiss, M.D. Baby steps: A giant leap for cell therapy in neonatal brain injury. Pediatr. Res. 2011, 70, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Gonzales-Portillo, G.S.; Reyes, S.; Aguirre, D.; Pabon, M.M.; Borlongan, C.V. Stem cell therapy for neonatal hypoxic-ischemic encephalopathy. Front. Neurol. 2014, 5, 147. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, S.; Ikeda, T.; Date, I.; Shingo, T.; Yasuhara, T.; Mishima, K.; Aoo, N.; Harada, K.; Egashira, N.; Iwasaki, K.; et al. Implantation of encapsulated glial cell line-derived neurotrophic factor-secreting cells prevents long-lasting learning impairment following neonatal hypoxic-ischemic brain insult in rats. Am. J. Obstet. Gynecol. 2005, 192, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Zalewska, T.; Jaworska, J.; Ziemka-Nalecz, M. Current and experimental pharmacological approaches in neonatal hypoxic- ischemic encephalopathy. Curr. Pharm. Des. 2015, 21, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Douglas-Escobar, M.; Weiss, M.D. Biomarkers of hypoxic-ischemic encephalopathy in newborns. Front. Neurol. 2012, 3, 144. [Google Scholar] [CrossRef] [PubMed]
- Bennet, L.; Booth, L.; Gunn, A.J. Potential biomarkers for hypoxic-ischemic encephalopathy. Semin. Fetal Neonatal Med. 2010, 15, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Middelanis, J.; Wasielewski, B.; Neuhoff, S.; Roth-Haerer, A.; Gantert, M.; Dinse, H.R.; Dermietzel, R.; Jensen, A. Spastic paresis after perinatal brain damage in rats is reduced by human cord blood mononuclear cells. Pediatr. Res. 2006, 59, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Geissler, M.; Dinse, H.R.; Neuhoff, S.; Kreikemeier, K.; Meier, C. Human umbilical cord blood cells restore brain damage induced changes in rat somatosensory cortex. PLoS ONE 2011, 6, 20194. [Google Scholar] [CrossRef] [PubMed]
- Pimentel-Coelho, P.M.; Magalhães, E.S.; Lopes, L.M.; deAzevedo, L.C.; Santiago, M.F.; Mendez-Otero, R. Human cord blood transplantation in a neonatal rat model of hypoxic-ischemic brain damage: Functional outcome related to neuroprotection in the striatum. Stem Cells Dev. 2010, 19, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Hara, K.; Maki, M.; Xu, L.; Yu, G.; Ali, M.M.; Masuda, T.; Yu, S.J.; Bae, E.K.; Hayashi, T.; et al. Mannitol facilitates neurotrophic factor up-regulation and behavioural recovery in neonatal hypoxic-ischaemic rats with human umbilical cord blood grafts. J. Cell. Mol. Med. 2010, 14, 914–921. [Google Scholar] [CrossRef] [PubMed]
- De Paula, S.; Vitola, A.S.; Greggio, S.; de Paula, D.; Mello, P.B.; Lubianca, J.M.; Xavier, L.L.; Fiori, H.H.; Dacosta, J.C. Hemispheric brain injury and behavioral deficits induced by severe neonatal hypoxia-ischemia in rats are not attenuated by intravenous administration of human umbilical cord blood cells. Pediatr. Res. 2009, 65, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Greggio, S.; de Paula, S.; Azevedo, P.N.; Venturin, G.T.; Dacosta, J.C. Intra-arterial transplantation of human umbilical cord blood mononuclear cells in neonatal hypoxic-ischemic rats. Life Sci. 2014, 96, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, Y.; Wang, X. Umbilical cord blood cells regulate the differentiation of endogenous neural stem cells in hypoxic ischemic neonatal rats via the hedgehog signaling pathway. Brain Res. 2014, 1560, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Matsukawa, N.; Yu, G.; Xu, L.; Mays, R.W.; Kovach, J.; Deans, R.J.; Hess, D.C.; Carroll, J.E.; Borlongan, C.V. Behavioral and histological characterization of intrahippocampal grafts of human bone marrow-derived multipotent progenitor cells in neonatal rats with hypoxic-ischemic injury. Cell. Transplant. 2006, 15, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Hara, K.; Maki, M.; Mays, R.W.; Deans, R.J.; Hess, D.C.; Carroll, J.E.; Borlongan, C.V. Intravenous grafts recapitulate the neurorestoration afforded by intracerebrally delivered multipotent adult progenitor cells in neonatal hypoxic-ischemic rats. J. Cereb. Blood Flow Metab. 2008, 28, 1804–1810. [Google Scholar] [CrossRef] [PubMed]
- Van Velthoven, C.T.J.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Repeated mesenchymal stem cell treatment after neonatal hypoxia-ischemia has distinct effects on formation and maturation of new neurons and oligodendrocytes leading to restoration of damage, corticospinal motor tract activity, and sensorimotor function. J. Neurosci. 2010, 30, 9603–9611. [Google Scholar] [CrossRef] [PubMed]
- Van Velthoven, C.T.J.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Nasal administration of stem cells: A promising novel route to treat neonatal ischemic brain damage. Pediatr. Res. 2010, 68, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Donega, V.; Nijboer, C.H.; van Tilborg, G.; Dijkhuizen, R.M.; Kavelaars, A.; Heijnen, C.J. Intranasally administered mesenchymal stem cells promote a regenerative niche for repair of neonatal ischemic brain injury. Exp. Neurol. 2014, 261, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Van Velthoven, C.T.; Braccioli, L.; Willemen, H.L.; Kavelaars, A.; Heijnen, C.J. Therapeutic potential of genetically modified mesenchymal stem cells after neonatal hypoxic-ischemic brain damage. Mol. Ther. 2014, 22, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, X.; Zhang, X.; Chen, R.; Sun, T.; Fok, K.L.; Dong, J.; Tsang, L.L.; Yi, S.; Ruan, Y.; et al. Dedifferentiation-reprogrammed mesenchymal stem cells with improved therapeutic potential. Stem Cells 2011, 29, 2077–2089. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Ahn, S.Y.; Im, G.H.; Sung, D.K.; Park, Y.R.; Choi, S.H.; Choi, S.J.; Chang, Y.S.; Oh, W.; Lee, J.H.; et al. Human umbilical cord blood-derived mesenchymal stem cell transplantation attenuates severe brain injury by permanent middle cerebral artery occlusion in newborn rats. Pediatr. Res. 2012, 72, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Sung, S.I.; Ahn, S.Y.; Yoo, H.S.; Sung, D.K.; Im, G.H.; Choi, S.J.; Chang, Y.S. Hypothermia augments neuroprotective activity of mesenchymal stem cells for neonatal hypoxic-ischemic encephalopathy. PLoS ONE 2015, 10, 120893. [Google Scholar]
- Xia, G.; Hong, X.; Chen, X.; Lan, F.; Zhang, G.; Liao, L. Intracerebral transplantation of mesenchymal stem cells derived from human umbilical cord blood alleviates hypoxic ischemic brain injury in rat neonates. J. Perinat. Med. 2010, 38, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Q.; Li, W.; Nie, D.; Chen, W.; Xu, C.; Yi, X.; Shi, J.; Tian, M.; Qin, J.; et al. Therapeutic effect of human umbilical cord mesenchymal stem cells on neonatal rat hypoxic-ischemic encephalopathy. J. Neurosci. Res. 2014, 92, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Kim, B.I.; Jo, C.H.; Choi, C.W.; Kim, E.-K.; Kim, H.-S.; Yoon, K.-S.; Choi, J.-H. Mesenchymal stem-cell transplantation for hypoxic-ischemic brain injury in neonatal rat model. Pediatr. Res. 2010, 67, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, M.; Yamamoto, A.; Kako, E.; Kaneko, N.; Matsubara, K.; Sakai, K.; Sawamoto, K.; Ueda, M. Human dental pulp-derived stem cells protect against hypoxic-ischemic brain injury in neonatal mice. Stroke 2013, 44, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Yang, Y.; Wang, Q.; Yao, Y.; Zhang, X.; He, X. Intraventricular injection of human dental pulp stem cells improves hypoxic-ischemic brain damage in neonatal rats. PLoS ONE 2013, 8, 66748. [Google Scholar] [CrossRef] [PubMed]
- Park, K.I.; Teng, Y.D.; Snyder, E.Y. The injured brain interacts reciprocally with neural stem cells supported by scaffolds to reconstitute lost tissue. Nat. Biotechnol. 2002, 20, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Park, K.I.; Himes, B.T.; Stieg, P.E.; Tessler, A.; Fischer, I.; Snyder, E.Y. Neural stem cells may be uniquely suited for combined gene therapy and cell replacement: Evidence from engraftment of Neurotrophin-3-expressing stem cells in hypoxic-ischemic brain injury. Exp. Neurol. 2006, 199, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Daadi, M.M.; Davis, A.S.; Arac, A.; Li, Z.; Maag, A.-L.; Bhatnagar, R.; Jiang, K.; Sun, G.; Wu, J.C.; Steinberg, G.K. Human neural stem cell grafts modify microglial response and enhance axonal sprouting in neonatal hypoxic-ischemic brain injury. Stroke 2010, 41, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Ashwal, S.; Ghosh, N.; Turenius, C.I.; Dulcich, M.; Denham, C.M.; Tone, B.; Hartman, R.; Snyder, E.Y.; Obenaus, A. Reparative effects of neural stem cells in neonatal rats with hypoxic-ischemic injury are not influenced by host sex. Pediatr. Res. 2014, 75, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-X.; Ma, S.-M.; Zhang, P.; Fan, Z.-C.; Xiong, M.; Cheng, G.-Q.; Yang, Y.; Qiu, Z.-L.; Zhou, W.-H.; Li, J. Neuroprotective effects of oligodendrocyte progenitor cell transplantation in premature rat brain following hypoxic-ischemic injury. PLoS ONE 2015, 10, 115997. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dixon, B.J.; Reis, C.; Ho, W.M.; Tang, J.; Zhang, J.H. Neuroprotective Strategies after Neonatal Hypoxic Ischemic Encephalopathy. Int. J. Mol. Sci. 2015, 16, 22368-22401. https://doi.org/10.3390/ijms160922368
Dixon BJ, Reis C, Ho WM, Tang J, Zhang JH. Neuroprotective Strategies after Neonatal Hypoxic Ischemic Encephalopathy. International Journal of Molecular Sciences. 2015; 16(9):22368-22401. https://doi.org/10.3390/ijms160922368
Chicago/Turabian StyleDixon, Brandon J., Cesar Reis, Wing Mann Ho, Jiping Tang, and John H. Zhang. 2015. "Neuroprotective Strategies after Neonatal Hypoxic Ischemic Encephalopathy" International Journal of Molecular Sciences 16, no. 9: 22368-22401. https://doi.org/10.3390/ijms160922368