
Exploiting the Pleiotropic Antioxidant Effects of Established Drugs in Cardiovascular Disease




Abstract
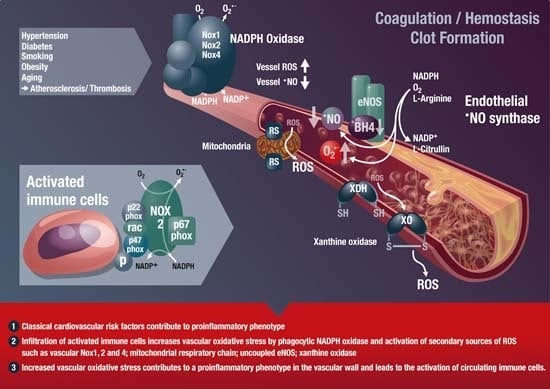
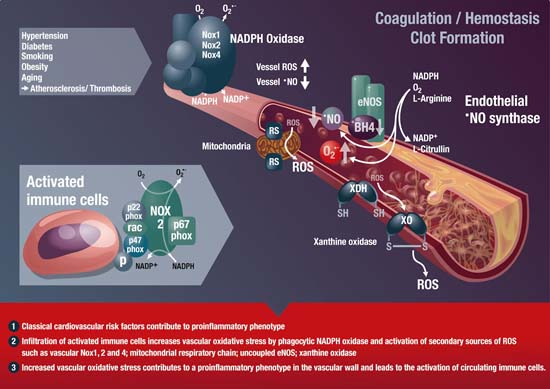
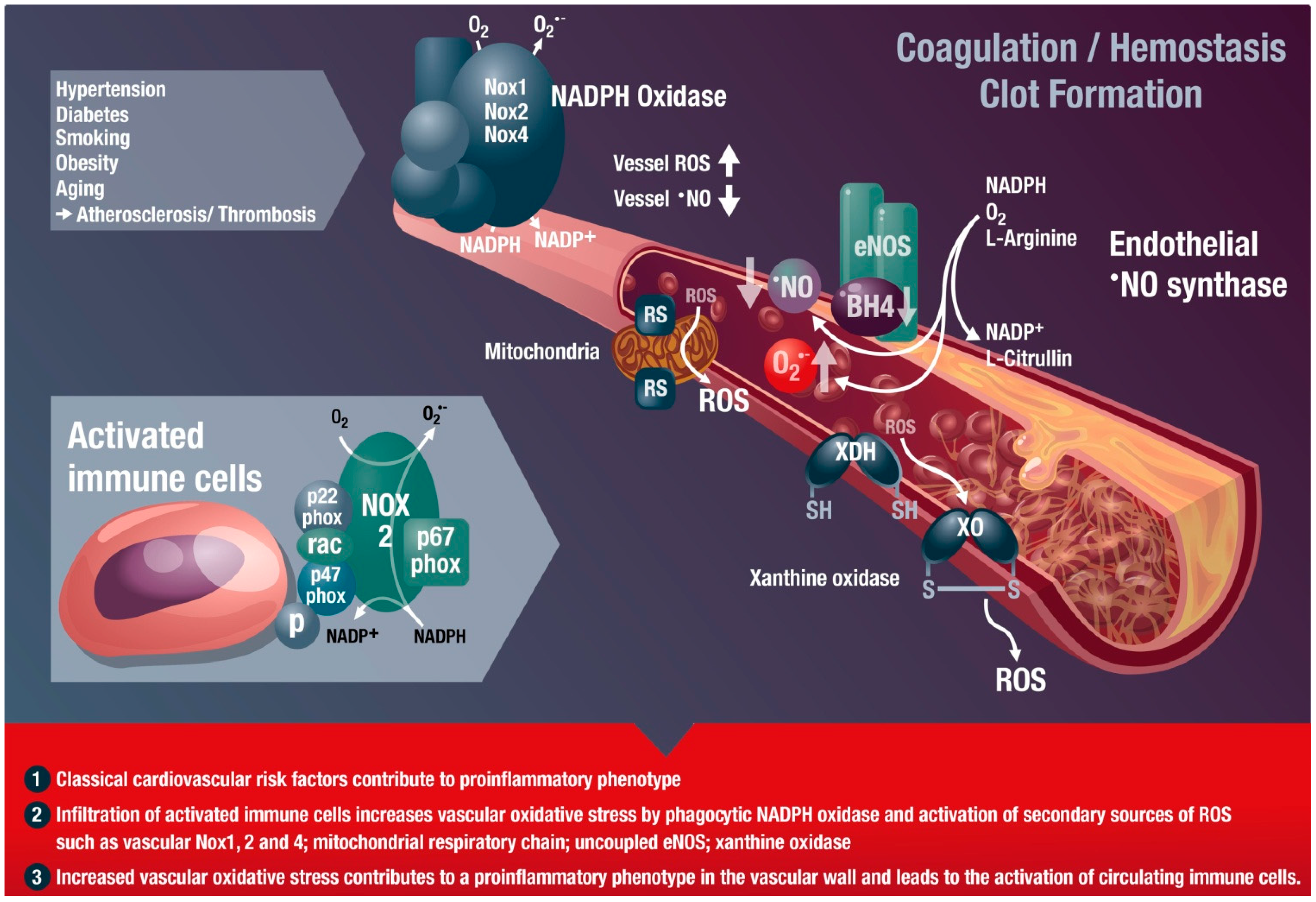
:
{kind=link}
{kind=link}
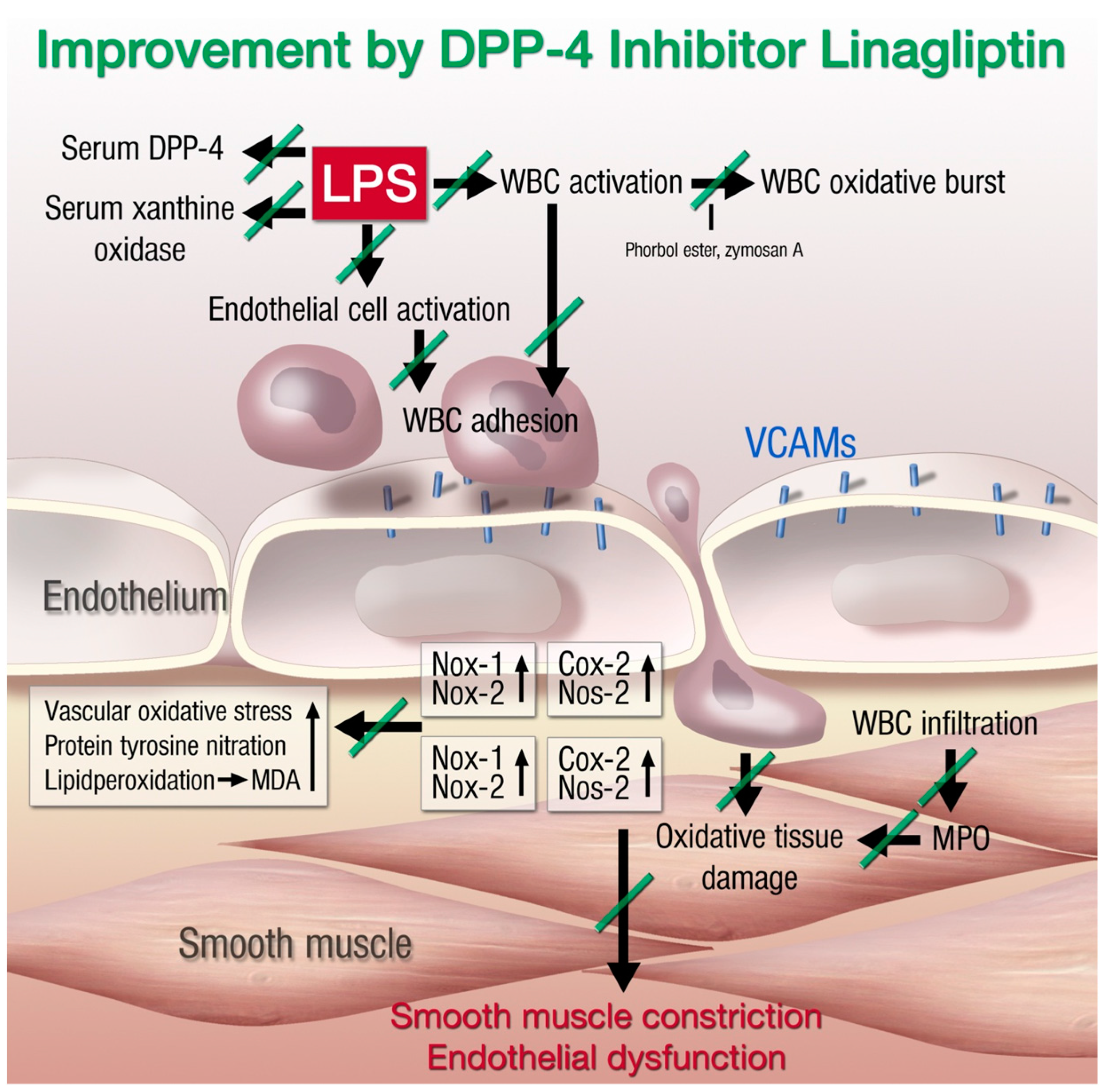{kind=link}
{kind=link}
{kind=link}
1. Oxidative Stress in Cardiovascular Disease and Inflammation
1.1. Introduction
1.2. Inflammation, Oxidative Stress, and Endothelial Dysfunction

1.3. Chronic Autoimmune Diseases Associated with Cardiovascular Disease
2. Classical Antioxidants and New Strategies to Modulate Oxidative Stress
2.1. Classical Antioxidants
2.2. New Antioxidant Strategies
3. Antioxidants 2.0—Pleiotropic Antioxidant Effects of Established Drugs
3.1. Statins, ACE-Inhibitors, and AT1-Receptor Blockers
3.2. Nebivolol, Hydralazine, and Pentaerythrityl Tetranitrate (PETN)
3.3. Gliptins and Glucagon-Like Peptide-1 (GLP-1) Analogs Display Antioxidant and Anti-Inflammatory Properties
3.3.1. Gliptins and GLP-1 in Atherosclerosis
3.3.2. Gliptins and GLP-1 in Sepsis and Chronic Inflammatory Disease



4. Immunomodulation as a Therapeutic Strategy
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part I: Basic mechanisms and in vivo monitoring of ROS. Circulation 2003, 108, 1912–1916. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part II: Animal and human studies. Circulation 2003, 108, 2034–2040. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Munzel, T. Oxidative stress and endothelial dysfunction: Therapeutic implications. Ann. Med. 2011, 43, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.F.; Chen, D.D.; Daiber, A.; Faraci, F.M.; Li, H.; Rembold, C.M.; Laher, I. Free radical biology of the cardiovascular system. Clin. Sci. 2012, 123, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Wenzel, P.; Munzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Oelze, M.; Daub, S.; Steven, S.; Schuff, A.; Kroller-Schon, S.; Hausding, M.; Wenzel, P.; Schulz, E.; Gori, T.; et al. Vascular redox signaling, redox switches in endothelial nitric oxide synthase and endothelial dysfunction. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer-Verlag: Berlin Heidelberg, Germany, 2014; pp. 1177–1211. [Google Scholar]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A.E. eNOS uncoupling in cardiovascular diseases—The role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.; Weyand, C.M. Inflammation, immunity, and hypertension. Hypertension 2011, 57, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Daiber, A.; Ullrich, V.; Mulsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cgmp-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Munzel, T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Schachinger, V.; Britten, M.B.; Zeiher, A.M. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000, 101, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.P.; Lin, F.Y.; Huang, P.H.; Chen, Y.L.; Chen, W.C.; Chen, H.Y.; Huang, Y.C.; Liao, W.L.; Huang, H.C.; Liu, P.L.; et al. Endothelial progenitor cell dysfunction in cardiovascular diseases: Role of reactive oxygen species and inflammation. BioMed Res. Int. 2013, 2013, 845037. [Google Scholar] [CrossRef] [PubMed]
- Assmus, B.; Leistner, D.M.; Schachinger, V.; Erbs, S.; Elsasser, A.; Haberbosch, W.; Hambrecht, R.; Sedding, D.; Yu, J.; Corti, R.; et al. Long-term clinical outcome after intracoronary application of bone marrow-derived mononuclear cells for acute myocardial infarction: Migratory capacity of administered cells determines event-free survival. Eur. Heart J. 2014, 35, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Schachinger, V.; Erbs, S.; Elsasser, A.; Haberbosch, W.; Hambrecht, R.; Holschermann, H.; Yu, J.; Corti, R.; Mathey, D.G.; Hamm, C.W.; et al. Intracoronary bone marrow-derived progenitor cells in acute myocardial infarction. N. Engl. J. Med. 2006, 355, 1210–1221. [Google Scholar] [CrossRef] [PubMed]
- Ohara, Y.; Peterson, T.E.; Harrison, D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Investig. 1993, 91, 2546–2551. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G.; Ohara, Y. Physiologic consequences of increased vascular oxidant stresses in hypercholesterolemia and atherosclerosis: Implications for impaired vasomotion. Am. J. Cardiol. 1995, 75, 75B–81B. [Google Scholar] [CrossRef]
- Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta. 2010, 1797, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 2006, 8, 691–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Knorr, M.; Kossmann, S.; Stratmann, J.; Hausding, M.; Schuhmacher, S.; Karbach, S.H.; Schwenk, M.; Yogev, N.; Schulz, E.; et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 2011, 124, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. Tlr signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- El Assar, M.; Angulo, J.; Rodriguez-Manas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013, 65, 380–401. [Google Scholar] [CrossRef] [PubMed]
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953. [Google Scholar] [CrossRef] [PubMed]
- Kroller-Schon, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 2014, 20, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Ferrebuz, A.; MacGregor, E.G.; Rodriguez-Iturbe, B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J. Am. Soc. Nephrol. 2006, 17, S218–S225. [Google Scholar] [CrossRef] [PubMed]
- Soltesz, P.; Kerekes, G.; Der, H.; Szucs, G.; Szanto, S.; Kiss, E.; Bodolay, E.; Zeher, M.; Timar, O.; Szodoray, P.; et al. Comparative assessment of vascular function in autoimmune rheumatic diseases: Considerations of prevention and treatment. Autoimmun. Rev. 2011, 10, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Colombo, B.M.; Cagnati, P.; Gulli, R.; Spano, F.; Puppo, F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis 2012, 224, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Vena, G.A.; Vestita, M.; Cassano, N. Psoriasis and cardiovascular disease. Dermatol. Ther. 2010, 23, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Hak, A.E.; Karlson, E.W.; Feskanich, D.; Stampfer, M.J.; Costenbader, K.H. Systemic lupus erythematosus and the risk of cardiovascular disease: Results from the nurses’ health study. Arthritis Rheum. 2009, 61, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; Azfar, R.S.; Shin, D.B.; Neimann, A.L.; Troxel, A.B.; Gelfand, J.M. Patients with severe psoriasis are at increased risk of cardiovascular mortality: Cohort study using the general practice research database. Eur. Heart J. 2010, 31, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.J.; Symmons, D.P.; McCarey, D.; Dijkmans, B.A.; Nicola, P.; Kvien, T.K.; McInnes, I.B.; Haentzschel, H.; Gonzalez-Gay, M.A.; Provan, S.; et al. Eular evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann. Rheum. Dis. 2010, 69, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodergren, A.; Karp, K.; Boman, K.; Eriksson, C.; Lundstrom, E.; Smedby, T.; Soderlund, L.; Rantapaa-Dahlqvist, S.; Wallberg-Jonsson, S. Atherosclerosis in early rheumatoid arthritis: Very early endothelial activation and rapid progression of intima media thickness. Arthritis Res. Ther. 2010, 12, R158. [Google Scholar] [CrossRef] [PubMed]
- Balci, D.D.; Balci, A.; Karazincir, S.; Ucar, E.; Iyigun, U.; Yalcin, F.; Seyfeli, E.; Inandi, T.; Egilmez, E. Increased carotid artery intima-media thickness and impaired endothelial function in psoriasis. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, A.; di Meglio, P.; Nestle, F.O. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J. Investig. Dermatol. 2009, 129, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.; Banerjee, S. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med. 2012, 366, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Leonardi, C.; Menter, A.; Ortonne, J.P.; Krueger, J.G.; Kricorian, G.; Aras, G.; Li, J.; Russell, C.B.; Thompson, E.H.; et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med. 2012, 366, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Crispin, J.C.; Tsokos, G.C. IL-17 in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 2010, 943254. [Google Scholar] [CrossRef] [PubMed]
- Choy, E. Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2012, 51 (Suppl. 5), v3–v11. [Google Scholar] [CrossRef] [PubMed]
- Pasceri, V.; Yeh, E.T. A tale of two diseases: Atherosclerosis and rheumatoid arthritis. Circulation 1999, 100, 2124–2126. [Google Scholar] [CrossRef] [PubMed]
- Sesso, H.D.; Christen, W.G.; Bubes, V.; Smith, J.P.; MacFadyen, J.; Schvartz, M.; Manson, J.E.; Glynn, R.J.; Buring, J.E.; Gaziano, J.M. Multivitamins in the prevention of cardiovascular disease in men: The physicians’ health study II randomized controlled trial. JAMA 2012, 308, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Lonn, E.; Bosch, J.; Yusuf, S.; Sheridan, P.; Pogue, J.; Arnold, J.M.; Ross, C.; Arnold, A.; Sleight, P.; Probstfield, J.; et al. Effects of long-term vitamin e supplementation on cardiovascular events and cancer: A randomized controlled trial. JAMA 2005, 293, 1338–1347. [Google Scholar] [PubMed]
- Muntwyler, J.; Hennekens, C.H.; Manson, J.E.; Buring, J.E.; Gaziano, J.M. Vitamin supplement use in a low-risk population of us male physicians and subsequent cardiovascular mortality. Arch. Intern. Med. 2002, 162, 1472–1476. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.; Lonn, E.M.; Yi, Q.; Gerstein, H.C.; Hoogwerf, B.J.; Pogue, J.; Bosch, J.; Dagenais, G.R.; Yusuf, S. Effects of vitamin e on cardiovascular outcomes in people with mild-to-moderate renal insufficiency: Results of the hope study. Kidney Int. 2004, 65, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Dagenais, G.; Pogue, J.; Bosch, J.; Sleight, P. Vitamin e supplementation and cardiovascular events in high-risk patients. The heart outcomes prevention evaluation study investigators. N. Engl. J. Med. 2000, 342, 154–160. [Google Scholar] [PubMed]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.P.; Daiber, A.; Cuadrado, A. Antioxidants in translational medicine. Antioxid. Redox Signal. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Folsom, A.R.; Harnack, L.; Halliwell, B.; Jacobs, D.R., Jr. Does supplemental vitamin C increase cardiovascular disease risk in women with diabetes? Am. J. Clin. Nutr. 2004, 80, 1194–1200. [Google Scholar] [PubMed]
- Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.C.; Ashwood, T.; Wasiewski, W.W.; Emeribe, U.; et al. Nxy-059 for the treatment of acute ischemic stroke. N. Engl. J. Med. 2007, 357, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004, 364, 1219–1228. [Google Scholar] [CrossRef]
- Harris, H.R.; Orsini, N.; Wolk, A. Vitamin C and survival among women with breast cancer: A meta-analysis. Eur. J. Cancer 2014, 50, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Ashor, A.W.; Lara, J.; Mathers, J.C.; Siervo, M. Effect of vitamin C on endothelial function in health and disease: A systematic review and meta-analysis of randomised controlled trials. Atherosclerosis 2014, 235, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Finckh, B.; Albers, S.; Krohn, K.; Kohlschutter, A.; Meinertz, T. Beneficial effects of α-lipoic acid and ascorbic acid on endothelium-dependent, nitric oxide-mediated vasodilation in diabetic patients: Relation to parameters of oxidative stress. Free Radic. Biol. Med. 2001, 31, 53–61. [Google Scholar] [CrossRef]
- Heitzer, T.; Brockhoff, C.; Mayer, B.; Warnholtz, A.; Mollnau, H.; Henne, S.; Meinertz, T.; Munzel, T. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers : Evidence for a dysfunctional nitric oxide synthase. Circ. Res. 2000, 86, E36–E41. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Just, H.; Munzel, T. Antioxidant vitamin C improves endothelial dysfunction in chronic smokers. Circulation 1996, 94, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Rumsey, S.C.; Daruwala, R.; Park, J.B.; Wang, Y. Criteria and recommendations for vitamin C intake. JAMA 1999, 281, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.B.; Li, T.L.; Xiang, H.J.; Chang, Q.; Li, C.L. Impaired endothelial function in the brachial artery after kawasaki disease and the effects of intravenous administration of vitamin C. Pediatr. Infect. Dis. J. 2003, 22, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Schaufele, T.G.; Schlaich, M.P.; Delles, C.; Klingbeil, A.U.; Fleischmann, E.H.; Schmieder, R.E. Impaired basal no activity in patients with glomerular disease and the influence of oxidative stress. Kidney Int. 2006, 70, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Solzbach, U.; Hornig, B.; Jeserich, M.; Just, H. Vitamin C improves endothelial dysfunction of epicardial coronary arteries in hypertensive patients. Circulation 1997, 96, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Guerra, M.; Garcia-Pagan, J.C.; Turnes, J.; Bellot, P.; Deulofeu, R.; Abraldes, J.G.; Bosch, J. Ascorbic acid improves the intrahepatic endothelial dysfunction of patients with cirrhosis and portal hypertension. Hepatology 2006, 43, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Hagel, A.F.; Layritz, C.M.; Hagel, W.H.; Hagel, H.J.; Hagel, E.; Dauth, W.; Kressel, J.; Regnet, T.; Rosenberg, A.; Neurath, M.F.; et al. Intravenous infusion of ascorbic acid decreases serum histamine concentrations in patients with allergic and non-allergic diseases. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Park, J.J.; Ahn, S.K.; Hur, D.G.; Kim, H.Y. Effect of high dose intravenous vitamin C on idiopathic sudden sensorineural hearing loss: A prospective single-blind randomized controlled trial. Eur. Arch. Otorhinolaryngol. 2013, 270, 2631–2636. [Google Scholar] [CrossRef] [PubMed]
- Du, W.D.; Yuan, Z.R.; Sun, J.; Tang, J.X.; Cheng, A.Q.; Shen, D.M.; Huang, C.J.; Song, X.H.; Yu, X.F.; Zheng, S.B. Therapeutic efficacy of high-dose vitamin C on acute pancreatitis and its potential mechanisms. World J. Gastroenterol. 2003, 9, 2565–2569. [Google Scholar] [PubMed]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Pedersen-White, J.; Guo, D.H.; Stallmann-Jorgensen, I.S.; Keeton, D.; Huang, Y.; Shah, Y.; Zhu, H.; Dong, Y. Vitamin D3 supplementation for 16 weeks improves flow-mediated dilation in overweight african-american adults. Am. J. Hypertens. 2011, 24, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Sugden, J.A.; Davies, J.I.; Witham, M.D.; Morris, A.D.; Struthers, A.D. Vitamin D improves endothelial function in patients with type 2 diabetes mellitus and low vitamin D levels. Diabet. Med. 2008, 25, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, M.; Begerow, B.; Minne, H.W.; Nachtigall, D.; Hansen, C. Effects of a short-term vitamin D3 and calcium supplementation on blood pressure and parathyroid hormone levels in elderly women. J. Clin. Endocrinol. Metab. 2001, 86, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Park, C.W.; Shin, Y.S.; Kim, Y.S.; Shin, S.J.; Kim, Y.S.; Choi, E.J.; Chang, Y.S.; Bang, B.K. Calcitriol regresses cardiac hypertrophy and QT dispersion in secondary hyperparathyroidism on hemodialysis. Nephron. Clin. Pract. 2006, 102, c21–29. [Google Scholar] [CrossRef] [PubMed]
- Park, C.W.; Oh, Y.S.; Shin, Y.S.; Kim, C.M.; Kim, Y.S.; Kim, S.Y.; Choi, E.J.; Chang, Y.S.; Bang, B.K. Intravenous calcitriol regresses myocardial hypertrophy in hemodialysis patients with secondary hyperparathyroidism. Am. J. Kidney Dis. 1999, 33, 73–81. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Gluud, L.L.; Nikolova, D.; Whitfield, K.; Wetterslev, J.; Simonetti, R.G.; Bjelakovic, M.; Gluud, C. Vitamin D supplementation for prevention of mortality in adults. Cochrane Database Syst. Rev. 2014, 1, CD007470. [Google Scholar] [PubMed]
- Janssen, H.C.; Samson, M.M.; Verhaar, H.J. Vitamin D deficiency, muscle function, and falls in elderly people. Am. J. Clin. Nutr. 2002, 75, 611–615. [Google Scholar] [PubMed]
- Mozos, I.; Marginean, O. Links between vitamin D deficiency and cardiovascular diseases. BioMed Res. Int. 2015, 2015, 109275. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R. The ambivalence of vitamin E in atherogenesis. Trends Biochem. Sci. 1999, 24, 219–223. [Google Scholar] [CrossRef]
- Khaw, K.T.; Bingham, S.; Welch, A.; Luben, R.; Wareham, N.; Oakes, S.; Day, N. Relation between plasma ascorbic acid and mortality in men and women in epic-norfolk prospective study: A prospective population study. European prospective investigation into cancer and nutrition. Lancet 2001, 357, 657–663. [Google Scholar] [CrossRef]
- Chen, G.C.; Lu, D.B.; Pang, Z.; Liu, Q.F. Vitamin C intake, circulating vitamin C and risk of stroke: A meta-analysis of prospective studies. J. Am. Heart Assoc. 2013, 2, e000329. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency increase lifestyle-associated vascular disease progression? Evidence based on experimental and clinical studies. Antioxid. Redox Signal. 2013, 19, 2084–2104. [Google Scholar] [CrossRef] [PubMed]
- Ashor, A.W.; Siervo, M.; Lara, J.; Oggioni, C.; Mathers, J.C. Antioxidant vitamin supplementation reduces arterial stiffness in adults: A systematic review and meta-analysis of randomized controlled trials. J. Nutr. 2014, 144, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Verkaart, S.; Visch, H.J.; van der Westhuizen, F.H.; Murphy, M.P.; van den Heuvel, L.W.; Smeitink, J.A.; Willems, P.H. Inhibition of complex I of the electron transport chain causes O2−·-mediated mitochondrial outgrowth. Am. J. Physiol. Cell Physiol. 2005, 288, C1440–C1450. [Google Scholar] [CrossRef] [PubMed]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Esplugues, J.V.; Rocha, M.; Nunez, C.; Bosca, I.; Ibiza, S.; Herance, J.R.; Ortega, A.; Serrador, J.M.; D’Ocon, P.; Victor, V.M. Complex i dysfunction and tolerance to nitroglycerin: An approach based on mitochondrial-targeted antioxidants. Circ. Res. 2006, 99, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cocheme, H.M.; Murphy, M.P.; Dominiczak, A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Miquel, E.; Cassina, A.; Martinez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodriguez-Bottero, S.; Logan, A.; Smith, R.A.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Dashdorj, A.; Jyothi, K.R.; Lim, S.; Jo, A.; Nguyen, M.N.; Ha, J.; Yoon, K.S.; Kim, H.J.; Park, J.H.; Murphy, M.P.; et al. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013, 11, 178. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.K.; Reily, C.; Srivastava, A.; Johnson, M.S.; Ye, Y.; Ulasova, E.; Agarwal, A.; Zinn, K.R.; Murphy, M.P.; Kalyanaraman, B.; et al. Prevention of diabetic nephropathy in Ins2(+/)−(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 2010, 432, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Rajic, Z.; Tovmasyan, A.; Reboucas, J.S.; Ye, X.; Leong, K.W.; Dewhirst, M.W.; Vujaskovic, Z.; Benov, L.; Spasojevic, I. Diverse functions of cationic Mn(III) N-substituted pyridylporphyrins, recognized as SOD mimics. Free Radic. Biol. Med. 2011, 51, 1035–1053. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Tovmasyan, A.; Roberts, E.R.; Vujaskovic, Z.; Leong, K.W.; Spasojevic, I. SOD therapeutics: Latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid. Redox Signal. 2014, 20, 2372–2415. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Hartley, R.C.; Murphy, M.P. Mitochondria-targeted small molecule therapeutics and probes. Antioxid. Redox Signal. 2011, 15, 3021–3038. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Antioxidants as therapies: Can we improve on nature? Free Radic. Biol. Med. 2014, 66, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Shuvaev, V.V.; Christofidou-Solomidou, M.; Bhora, F.; Laude, K.; Cai, H.; Dikalov, S.; Arguiri, E.; Solomides, C.C.; Albelda, S.M.; Harrison, D.G.; et al. Targeted detoxification of selected reactive oxygen species in the vascular endothelium. J. Pharmacol. Exp. Ther. 2009, 331, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.; Hattan, C.M.; Kerns, R.J. Semi-synthetic heparin derivatives: Chemical modifications of heparin beyond chain length, sulfate substitution pattern and N-sulfo/N-acetyl groups. Carbohydr. Res. 2006, 341, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Kleschyov, A.L.; Sen, V.; Golubev, V.; Munnemann, K.; Hinderberger, D.; Lackner, K.J.; Weber, S.; Terekhov, M.; Schreiber, L.M.; Munzel, T. Heparin-polynitroxides: Synthesis and preliminary evaluation as cardiovascular EPR/MR imaging probes and extracellular space-targeted antioxidants. Eur. J. Med. Chem. 2012, 58, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Kleschyov, A.L.; Sen, V.D. Heparin-polynitroxide derivatives: A platform for new diagnostic and therapeutic agents in cardiovascular disease? Future Med. Chem. 2013, 5, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Mikhed, Y.; Gorlach, A.; Knaus, U.G.; Daiber, A. Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol. 2015, 5, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Hayes, P.; Knaus, U.G. Balancing reactive oxygen species in the epigenome: NADPH oxidases as target and perpetrator. Antioxid. Redox Signal. 2013, 18, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Baird, A.M.; O’Byrne, K.J.; Gray, S.G. Reactive oxygen species and reactive nitrogen species in epigenetic modifications. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer-Verlag: Berlin Heidelberg, Germany, 2014; pp. 437–455. [Google Scholar]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thebaud, B.; Husain, A.N.; et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.P.; Keaney, J.F., Jr. Epigenetic control of angiogenesis via DNA methylation. Circulation 2011, 123, 2916–2918. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Ryan, J.J.; Archer, S.L. The role of redox signaling in epigenetics and cardiovascular disease. Antioxid. Redox Signal. 2013, 18, 1920–1936. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, L.; Tan, Y.; Hui, R.; Wang, Y. Hypertensive epigenetics: From DNA methylation to micrornas. J. Hum. Hypertens. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xia, N.; Forstermann, U. Cardiovascular effects and molecular targets of resveratrol. Nitric Oxide 2012, 26, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Pacher, P.; Das, D.K. Microrna signatures of resveratrol in the ischemic heart. Ann. N. Y. Acad. Sci. 2011, 1215, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Farghali, H.; Kutinova Canova, N.; Lekic, N. Resveratrol and related compounds as antioxidants with an allosteric mechanism of action in epigenetic drug targets. Physiol. Res. 2013, 62, 1–13. [Google Scholar] [PubMed]
- Gatz, S.A.; Wiesmuller, L. Take a break—resveratrol in action on DNA. Carcinogenesis 2008, 29, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chai, H.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Clinical use and molecular mechanisms of action of extract of ginkgo biloba leaves in cardiovascular diseases. Cardiovasc. Drug Rev. 2004, 22, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Kleikers, P.W.; Wingler, K.; Hermans, J.J.; Diebold, I.; Altenhofer, S.; Radermacher, K.A.; Janssen, B.; Gorlach, A.; Schmidt, H.H. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. Int. J. Mol. Med. 2012, 90, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Altenhofer, S.; Kleikers, P.W.; Radermacher, K.A.; Scheurer, P.; Rob Hermans, J.J.; Schiffers, P.; Ho, H.; Wingler, K.; Schmidt, H.H. The Nox toolbox: Validating the role of NADPH oxidases in physiology and disease. Cell. Mol. Life Sci. 2012, 69, 2327–2343. [Google Scholar] [CrossRef] [PubMed]
- Radermacher, K.A.; Wingler, K.; Langhauser, F.; Altenhofer, S.; Kleikers, P.; Hermans, J.J.; Hrabe de Angelis, M.; Kleinschnitz, C.; Schmidt, H.H. Neuroprotection after stroke by targeting Nox4 as a source of oxidative stress. Antioxid. Redox Signal. 2013, 18, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Wingler, K.; Hermans, J.J.; Schiffers, P.; Moens, A.; Paul, M.; Schmidt, H.H. Nox1, 2, 4, 5: Counting out oxidative stress. Br. J. Pharmacol. 2011, 164, 866–883. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.E.; Toledo, A.H.; Anaya-Prado, R.; Roach, R.R.; Toledo-Pereyra, L.H. Allopurinol, xanthine oxidase, and cardiac ischemia. J. Investig. Med. 2009, 57, 902–909. [Google Scholar] [PubMed]
- Higgins, P.; Dawson, J.; Lees, K.R.; McArthur, K.; Quinn, T.J.; Walters, M.R. Xanthine oxidase inhibition for the treatment of cardiovascular disease: A systematic review and meta-analysis. Cardiovasc. Ther. 2012, 30, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Jazwa, A.; Cuadrado, A. Targeting heme oxygenase-1 for neuroprotection and neuroinflammation in neurodegenerative diseases. Curr. Drug Targets 2010, 11, 1517–1531. [Google Scholar] [CrossRef] [PubMed]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Hasko, G.; Schmidt, H.H.; Stasch, J.P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Follmann, M.; Griebenow, N.; Hahn, M.G.; Hartung, I.; Mais, F.J.; Mittendorf, J.; Schafer, M.; Schirok, H.; Stasch, J.P.; Stoll, F.; et al. The chemistry and biology of soluble guanylate cyclase stimulators and activators. Angew. Chem. Int. Ed. Engl. 2013, 52, 9442–9462. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Schmidt, P.M.; Stasch, J.P. NO- and haem-independent soluble guanylate cyclase activators. Handb. Exp. Pharmacol. 2009, 191, 309–339. [Google Scholar] [PubMed]
- Lonn, M.E.; Dennis, J.M.; Stocker, R. Actions of “antioxidants” in the protection against atherosclerosis. Free Radic. Biol. Med. 2012, 53, 863–884. [Google Scholar] [CrossRef] [PubMed]
- Drexler, H.; Hornig, B. Endothelial dysfunction in human disease. J. Mol. Cell. Cardiol. 1999, 31, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Warnholtz, A.; Munzel, T. Why do antioxidants fail to provide clinical benefit? Curr. Control. Trials Cardiovasc. Med. 2000, 1, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Porsti, I.; Bara, A.T.; Busse, R.; Hecker, M. Release of nitric oxide by angiotensin-(1–7) from porcine coronary endothelium: Implications for a novel angiotensin receptor. Br. J. Pharmacol. 1994, 111, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Mollnau, H.; Oelze, M.; August, M.; Wendt, M.; Daiber, A.; Schulz, E.; Baldus, S.; Kleschyov, A.L.; Materne, A.; Wenzel, P.; et al. Mechanisms of increased vascular superoxide production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2554–2559. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.C.; Griendling, K.K. NADPH oxidase inhibitors: New antihypertensive agents? J. Cardiovasc. Pharmacol. 2007, 50, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Schmeisser, A.; Cicha, I.; Reiss, C.; Ulbrich, H.; Lindbom, L.; Daniel, W.G.; Garlichs, C.D. Ace inhibition lowers angiotensin-II-induced monocyte adhesion to HUVEC by reduction of p65 translocation and AT1 expression. J. Vasc. Res. 2005, 42, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Caspritz, G.; Alpermann, H.G.; Schleyerbach, R. Influence of the new angiotensin converting enzyme inhibitor ramipril on several models of acute inflammation and the adjuvant arthritis in the rat. Arzneimittelforschung 1986, 36, 1605–1608. [Google Scholar] [PubMed]
- Durik, M.; Seva Pessoa, B.; Roks, A.J. The renin-angiotensin system, bone marrow and progenitor cells. Clin. Sci. 2012, 123, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Gori, T.; Keaney, J.F., Jr.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart. J. 2015. [Google Scholar] [CrossRef]
- Takemoto, M.; Liao, J.K. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Cannon, C.P. Pathological changes in acute coronary syndromes: The role of statin therapy in the modulation of inflammation, endothelial function and coagulation. J. Thromb. Thrombolysis 2004, 18, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.N.; Shishehbor, M.H.; Bhatt, D.L. A review of high-dose statin therapy: Targeting cholesterol and inflammation in atherosclerosis. Eur. Heart J. 2007, 28, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Adam, O.; Laufs, U. Rac1-mediated effects of HMG-CoA reductase inhibitors (statins) in cardiovascular disease. Antioxid. Redox Signal. 2014, 20, 1238–1250. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Daiber, A.; Oelze, M.; Brandt, M.; Closs, E.; Xu, J.; Thum, T.; Bauersachs, J.; Ertl, G.; Zou, M.H.; et al. Mechanisms underlying recoupling of eNOS by HMG-CoA reductase inhibition in a rat model of streptozotocin-induced diabetes mellitus. Atherosclerosis 2008, 198, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Margaritis, M.; Channon, K.M.; Antoniades, C. Statins as regulators of redox state in the vascular endothelium: Beyond lipid lowering. Antioxid. Redox Signal. 2014, 20, 1198–1215. [Google Scholar] [CrossRef] [PubMed]
- Habeos, I.G.; Ziros, P.G.; Chartoumpekis, D.; Psyrogiannis, A.; Kyriazopoulou, V.; Papavassiliou, A.G. Simvastatin activates Keap1/Nrf2 signaling in rat liver. Int. J. Mol. Med. 2008, 86, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Zakkar, M.; Karu, K.; Lidington, E.A.; Hamdulay, S.S.; Boyle, J.J.; Zloh, M.; Bauer, A.; Haskard, D.O.; Evans, P.C.; et al. Induction of the cytoprotective enzyme heme oxygenase-1 by statins is enhanced in vascular endothelium exposed to laminar shear stress and impaired by disturbed flow. J. Biol. Chem. 2009, 284, 18882–18892. [Google Scholar] [CrossRef] [PubMed]
- Hibbert, B.; Simard, T.; Ramirez, F.D.; Pourdjabbar, A.; Raizman, J.E.; Maze, R.; Wilson, K.R.; Hawken, S.; O’Brien, E.R. The effect of statins on circulating endothelial progenitor cells in humans: A systematic review. J. Cardiovasc. Pharmacol. 2013, 62, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Magee, L.A.; Abalos, E.; von Dadelszen, P.; Sibai, B.; Easterling, T.; Walkinshaw, S.; Group, C.S. How to manage hypertension in pregnancy effectively. Br. J. Clin. Pharmacol. 2011, 72, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Anand, I.S.; Tam, S.W.; Rector, T.S.; Taylor, A.L.; Sabolinski, M.L.; Archambault, W.T.; Adams, K.F.; Olukotun, A.Y.; Worcel, M.; Cohn, J.N. Influence of blood pressure on the effectiveness of a fixed-dose combination of isosorbide dinitrate and hydralazine in the african-american heart failure trial. J. Am. Coll. Cardiol. 2007, 49, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.L.; Ziesche, S.; Yancy, C.W.; Carson, P.; Ferdinand, K.; Taylor, M.; Adams, K.; Olukotun, A.Y.; Ofili, E.; Tam, S.W.; et al. Early and sustained benefit on event-free survival and heart failure hospitalization from fixed-dose combination of isosorbide dinitrate/hydralazine: Consistency across subgroups in the african-american heart failure trial. Circulation 2007, 115, 1747–1753. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Tam, S.W.; Anand, I.S.; Taylor, A.L.; Sabolinski, M.L.; Worcel, M. Isosorbide dinitrate and hydralazine in a fixed-dose combination produces further regression of left ventricular remodeling in a well-treated black population with heart failure: Results from A-HeFT. J. Card. Fail. 2007, 13, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Oelze, M.; Coldewey, M.; Kaiser, K.; Huth, C.; Schildknecht, S.; Bachschmid, M.; Nazirisadeh, Y.; Ullrich, V.; Mulsch, A.; et al. Hydralazine is a powerful inhibitor of peroxynitrite formation as a possible explanation for its beneficial effects on prognosis in patients with congestive heart failure. Biochem. Biophys. Res. Commun. 2005, 338, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Mulsch, A.; Hink, U.; Mollnau, H.; Warnholtz, A.; Oelze, M.; Munzel, T. The oxidative stress concept of nitrate tolerance and the antioxidant properties of hydralazine. Am. J. Cardiol. 2005, 96, 25i–36i. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Kurz, S.; Rajagopalan, S.; Thoenes, M.; Berrington, W.R.; Thompson, J.A.; Freeman, B.A.; Harrison, D.G. Hydralazine prevents nitroglycerin tolerance by inhibiting activation of a membrane-bound nadh oxidase. A new action for an old drug. J. Clin. Investig. 1996, 98, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, M.; Sato, M.; Funada, J.; Ohtani, T.; Akutsu, H.; Watanabe, K. Effects of the long-term administration of nicorandil on vascular endothelial function and the progression of arteriosclerosis. J. Cardiovasc. Pharmacol. 2005, 46, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.F. Chemiluminescence investigation of carbon dioxide-enhanced oxidation of dihydralazine sulfate by peroxynitrite and its application to pharmaceutical analysis. Anal. Chim. Acta 2008, 616, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.J.; Tian, Y.M.; Mole, D.R.; Harris, A.L. Novel mechanism of action for hydralazine: Induction of hypoxia-inducible factor-1α, vascular endothelial growth factor, and angiogenesis by inhibition of prolyl hydroxylases. Circ. Res. 2004, 95, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Aslam, S. Cardiovascular disease in dialysis patients: Do some antihypertensive drugs have specific antioxidant effects or is it just blood pressure reduction? Does antioxidant treatment reduce the risk for cardiovascular disease? Curr. Opin. Nephrol. Hypertens. 2008, 17, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Hoenig, M.R.; Bianchi, C.; Sellke, F.W. Hypoxia inducible factor-1 α, endothelial progenitor cells, monocytes, cardiovascular risk, wound healing, cobalt and hydralazine: A unifying hypothesis. Curr. Drug Targets 2008, 9, 422–435. [Google Scholar] [CrossRef] [PubMed]
- Brehm, B.R.; Wolf, S.C.; Bertsch, D.; Klaussner, M.; Wesselborg, S.; Schuler, S.; Schulze-Osthoff, K. Effects of nebivolol on proliferation and apoptosis of human coronary artery smooth muscle and endothelial cells. Cardiovasc. Res. 2001, 49, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, A.; Pluteanu, F.; Flonta, M.L.; Badila, E.; Dorobantu, M.; Popov, D. The cellular mechanisms involved in the vasodilator effect of nebivolol on the renal artery. Eur. J. Pharmacol. 2005, 508, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Broeders, M.A.; Doevendans, P.A.; Bekkers, B.C.; Bronsaer, R.; van Gorsel, E.; Heemskerk, J.W.; Egbrink, M.G.; van Breda, E.; Reneman, R.S.; van Der Zee, R. Nebivolol: A third-generation β-blocker that augments vascular nitric oxide release: Endothelial β2—adrenergic receptor—Mediated nitric oxide production. Circulation 2000, 102, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Tzemos, N.; Lim, P.O.; MacDonald, T.M. Nebivolol reverses endothelial dysfunction in essential hypertension: A randomized, double-blind, crossover study. Circulation 2001, 104, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Mollnau, H.; Schulz, E.; Daiber, A.; Baldus, S.; Oelze, M.; August, M.; Wendt, M.; Walter, U.; Geiger, C.; Agrawal, R.; et al. Nebivolol prevents vascular nos III uncoupling in experimental hyperlipidemia and inhibits NADPH oxidase activity in inflammatory cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Oelze, M.; Daiber, A.; Brandes, R.P.; Hortmann, M.; Wenzel, P.; Hink, U.; Schulz, E.; Mollnau, H.; von Sandersleben, A.; Kleschyov, A.L.; et al. Nebivolol inhibits superoxide formation by NADPH oxidase and endothelial dysfunction in angiotensin II-treated rats. Hypertension 2006, 48, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Pasini, A.F.; Garbin, U.; Nava, M.C.; Stranieri, C.; Davoli, A.; Sawamura, T.; Cascio, V.L.; Cominacini, L. Nebivolol decreases oxidative stress in essential hypertensive patients and increases nitric oxide by reducing its oxidative inactivation. J. Hypertens. 2005, 23, 589–596. [Google Scholar] [CrossRef]
- Sorrentino, S.A.; Doerries, C.; Manes, C.; Speer, T.; Dessy, C.; Lobysheva, I.; Mohmand, W.; Akbar, R.; Bahlmann, F.; Besler, C.; et al. Nebivolol exerts beneficial effects on endothelial function, early endothelial progenitor cells, myocardial neovascularization, and left ventricular dysfunction early after myocardial infarction beyond conventional β1-blockade. J. Am. Coll. Cardiol. 2011, 57, 601–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munzel, T.; Daiber, A.; Mulsch, A. Explaining the phenomenon of nitrate tolerance. Circ. Res. 2005, 97, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Daiber, A. Non-hemodynamic effects of organic nitrates and the distinctive characteristics of pentaerithrityl tetranitrate. Am. J. Cardiovasc. Drugs. 2009, 9, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Meinertz, T.; Tebbe, U.; Schneider, H.T.; Stalleicken, D.; Wargenau, M.; Gori, T.; Klingmann, I.; Investigators, C.S. Efficacy of the long-acting nitro vasodilator pentaerithrityl tetranitrate in patients with chronic stable angina pectoris receiving anti-anginal background therapy with β-blockers: A 12-week, randomized, double-blind, placebo-controlled trial. Eur. Heart. J. 2014, 35, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Schnorbus, B.; Schiewe, R.; Ostad, M.A.; Medler, C.; Wachtlin, D.; Wenzel, P.; Daiber, A.; Munzel, T.; Warnholtz, A. Effects of pentaerythritol tetranitrate on endothelial function in coronary artery disease: Results of the penta study. Clin. Res. Cardiol. 2010, 99, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Schuhmacher, S.; Wenzel, P.; Schulz, E.; Oelze, M.; Mang, C.; Kamuf, J.; Gori, T.; Jansen, T.; Knorr, M.; Karbach, S.; et al. Pentaerythritol tetranitrate improves angiotensin II-induced vascular dysfunction via induction of heme oxygenase-1. Hypertension 2010, 55, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Oelze, M.; Coldewey, M.; Hortmann, M.; Seeling, A.; Hink, U.; Mollnau, H.; Stalleicken, D.; Weiner, H.; Lehmann, J.; et al. Heme oxygenase-1: A novel key player in the development of tolerance in response to organic nitrates. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Oberle, S.; Abate, A.; Grosser, N.; Hemmerle, A.; Vreman, H.J.; Dennery, P.A.; Schneider, H.T.; Stalleicken, D.; Schroder, H. Endothelial protection by pentaerithrityl trinitrate: Bilirubin and carbon monoxide as possible mediators. Exp. Biol. Med. (Maywood) 2003, 228, 529–534. [Google Scholar] [PubMed]
- Oberle, S.; Abate, A.; Grosser, N.; Vreman, H.J.; Dennery, P.A.; Schneider, H.T.; Stalleicken, D.; Schroder, H. Heme oxygenase-1 induction may explain the antioxidant profile of pentaerythrityl trinitrate. Biochem. Biophys. Res. Commun. 2002, 290, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Schuhmacher, S.; Oelze, M.; Bollmann, F.; Kleinert, H.; Otto, C.; Heeren, T.; Steven, S.; Hausding, M.; Knorr, M.; Pautz, A.; et al. Vascular dysfunction in experimental diabetes is improved by pentaerithrityl tetranitrate but not isosorbide-5-mononitrate therapy. Diabetes 2011, 60, 2608–2616. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, M.; Balz, V.; Adams, V.; Dao, V.T.; Bas, M.; Suvorava, T.; Kojda, G. Pharmacological induction of vascular extracellular superoxide dismutase expression in vivo. J. Cell. Mol. Med. 2009, 13, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Hacker, A.; Muller, S.; Meyer, W.; Kojda, G. The nitric oxide donor pentaerythritol tetranitrate can preserve endothelial function in established atherosclerosis. Br. J. Pharmacol. 2001, 132, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Flierl, U.; Fraccarollo, D.; Widder, J.D.; Micka, J.; Neuser, J.; Bauersachs, J.; Schafer, A. The nitric oxide donor pentaerythritol tetranitrate reduces platelet activation in congestive heart failure. PLoS ONE 2015, 10, e0123621. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Fraccarollo, D.; Thum, S.; Schultheiss, M.; Daiber, A.; Wenzel, P.; Munzel, T.; Ertl, G.; Bauersachs, J. Differential effects of organic nitrates on endothelial progenitor cells are determined by oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Wiebking, V.; Ertl, G.; Bauersachs, J. Organic nitrates differentially modulate circulating endothelial progenitor cells and endothelial function in patients with symptomatic coronary artery disease. Antioxid. Redox Signal. 2011, 15, 925–931. [Google Scholar] [CrossRef] [PubMed]
- DiFabio, J.M.; Thomas, G.R.; Zucco, L.; Kuliszewski, M.A.; Bennett, B.M.; Kutryk, M.J.; Parker, J.D. Nitroglycerin attenuates human endothelial progenitor cell differentiation, function, and survival. J. Pharmacol. Exp. Ther. 2006, 318, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Pautz, A.; Rauschkolb, P.; Schmidt, N.; Art, J.; Oelze, M.; Wenzel, P.; Forstermann, U.; Daiber, A.; Kleinert, H. Effects of nitroglycerin or pentaerithrityl tetranitrate treatment on the gene expression in rat hearts: Evidence for cardiotoxic and cardioprotective effects. Physiol. Genom. 2009, 38, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Siuda, D.; Xia, N.; Reifenberg, G.; Daiber, A.; Munzel, T.; Forstermann, U.; Li, H. Maternal treatment of spontaneously hypertensive rats with pentaerythritol tetranitrate reduces blood pressure in female offspring. Hypertension 2015, 65, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Abbott, C.A.; Yu, D.M.; Woollatt, E.; Sutherland, G.R.; McCaughan, G.W.; Gorrell, M.D. Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP) IV homolog, DPP8. Eur. J. Biochem. 2000, 267, 6140–6150. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Rao, X.; Rajagopalan, S. An emerging role of dipeptidyl peptidase 4 (DPP4) beyond glucose control: Potential implications in cardiovascular disease. Atherosclerosis 2013, 226, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Buhling, F.; Junker, U.; Reinhold, D.; Neubert, K.; Jager, L.; Ansorge, S. Functional role of CD26 on human B lymphocytes. Immunol. Lett. 1995, 45, 47–51. [Google Scholar] [CrossRef]
- Buhling, F.; Kunz, D.; Reinhold, D.; Ulmer, A.J.; Ernst, M.; Flad, H.D.; Ansorge, S. Expression and functional role of dipeptidyl peptidase IV (CD26) on human natural killer cells. Nat. Immun. 1994, 13, 270–279. [Google Scholar] [PubMed]
- Gorrell, M.D. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clin. Sci. 2005, 108, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.E.; Ward, P.E. Dipeptidyl(amino)peptidase iv and post proline cleaving enzyme in cultured endothelial and smooth muscle cells. Adv. Exp. Med. Biol. 1989, 247A, 305–311. [Google Scholar]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Lund, P.K.; Goodman, R.H.; Dee, P.C.; Habener, J.F. Pancreatic preproglucagon cdna contains two glucagon-related coding sequences arranged in tandem. Proc. Natl. Acad. Sci. USA 1982, 79, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.E.; Egan, J.M. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol. Ther. 2007, 113, 546–593. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A. Incretin-based therapies for type 2 diabetes mellitus: Properties, functions, and clinical implications. Am. J. Med. 2011, 124, S3–18. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, P.L.; Drucker, D.J. Structure-function of the glucagon receptor family of G protein-coupled receptors: The glucagon, GIP, GLP-1, and GLP-2 receptors. Recept. Channels 2002, 8, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Mentlein, R.; Gallwitz, B.; Schmidt, W.E. Dipeptidyl-peptidase iv hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur. J. Biochem. 1993, 214, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, T.J.; McIntosh, C.H.; Pederson, R.A. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 1995, 136, 3585–3596. [Google Scholar] [PubMed]
- International Diabetes Federation Guideline Development Group. Global guideline for type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 104, 1–52. [Google Scholar]
- Matsubara, J.; Sugiyama, S.; Sugamura, K.; Nakamura, T.; Fujiwara, Y.; Akiyama, E.; Kurokawa, H.; Nozaki, T.; Ohba, K.; Konishi, M.; et al. A dipeptidyl peptidase-4 inhibitor, des-fluoro-sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. J. Am. Coll. Cardiol. 2012, 59, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Shah, Z.; Kampfrath, T.; Deiuliis, J.A.; Zhong, J.; Pineda, C.; Ying, Z.; Xu, X.; Lu, B.; Moffatt-Bruce, S.; Durairaj, R.; et al. Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation 2011, 124, 2338–2349. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Shinohara, M.; Tanimoto, N.; Kumagai, C.; Hashimoto, K. Sitagliptin, a dipeptidyl peptidase-iv inhibitor, improves psoriasis. Dermatology 2012, 224, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Kern, M.; Kloting, N.; Niessen, H.G.; Thomas, L.; Stiller, D.; Mark, M.; Klein, T.; Bluher, M. Linagliptin improves insulin sensitivity and hepatic steatosis in diet-induced obesity. PLoS ONE 2012, 7, e38744. [Google Scholar] [CrossRef] [PubMed]
- Darsalia, V.; Ortsater, H.; Olverling, A.; Darlof, E.; Wolbert, P.; Nystrom, T.; Klein, T.; Sjoholm, A.; Patrone, C. The DPP-4 inhibitor linagliptin counteracts stroke in the normal and diabetic mouse brain: A comparison with glimepiride. Diabetes 2013, 62, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Shih, C.M.; Tsao, N.W.; Lin, Y.W.; Huang, P.H.; Wu, S.C.; Lee, A.W.; Kao, Y.T.; Chang, N.C.; Nakagami, H.; et al. Dipeptidyl peptidase-4 inhibitor improves neovascularization by increasing circulating endothelial progenitor cells. Br. J. Pharmacol. 2012, 167, 1506–1519. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Krankel, N.; Kuhlenthal, S.; Israel, L.; Remm, F.; Fischer, C.; Herbach, N.; Speer, T.; Grabmaier, U.; Laskowski, A.; et al. Short-term inhibition of DPP-4 enhances endothelial regeneration after acute arterial injury via enhanced recruitment of circulating progenitor cells. Int. J. Cardiol. 2014, 177, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Boscaro, E.; Albiero, M.; Menegazzo, L.; Frison, V.; de Kreutzenberg, S.; Agostini, C.; Tiengo, A.; Avogaro, A. The oral dipeptidyl peptidase-4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes: Possible role of stromal-derived factor-1α. Diabetes care 2010, 33, 1607–1609. [Google Scholar] [CrossRef] [PubMed]
- Xiao-Yun, X.; Zhao-Hui, M.; Ke, C.; Hong-Hui, H.; Yan-Hong, X. Glucagon-like peptide-1 improves proliferation and differentiation of endothelial progenitor cells via upregulating vegf generation. Med. Sci. Monit. 2011, 17, BR35–BR41. [Google Scholar] [CrossRef] [PubMed]
- Oeseburg, H.; de Boer, R.A.; Buikema, H.; van der Harst, P.; van Gilst, W.H.; Sillje, H.H. Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence through the activation of protein kinase a. Arterioscler. Thromb. Vasc. biol. 2010, 30, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Akarte, A.S.; Srinivasan, B.P.; Gandhi, S.; Sole, S. Chronic DPP-IV inhibition with PKF-275-055 attenuates inflammation and improves gene expressions responsible for insulin secretion in streptozotocin induced diabetic rats. Eur. J. Pharm. Sci. 2012, 47, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Chinda, K.; Palee, S.; Surinkaew, S.; Phornphutkul, M.; Chattipakorn, S.; Chattipakorn, N. Cardioprotective effect of dipeptidyl peptidase-4 inhibitor during ischemia-reperfusion injury. Int. J. Cardiol. 2013, 167, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Inthachai, T.; Lekawanvijit, S.; Kumfu, S.; Apaijai, N.; Pongkan, W.; Chattipakorn, S.C.; Chattipakorn, N. Dipeptidyl peptidase-4 inhibitor improves cardiac function by attenuating adverse cardiac remodelling in rats with chronic myocardial infarction. Exp. Physiol. 2015, 100, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Morimoto, K.; Hasegawa, T.; Sasaki, N.; Yamashita, T.; Hirata, K.; Okita, Y.; Okada, K. Orally administered dipeptidyl peptidase-4 inhibitor (alogliptin) prevents abdominal aortic aneurysm formation through an antioxidant effect in rats. J. Vasc. Surg. 2014, 59, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Abdelsalam, R.M.; Safar, M.M. Neuroprotective effects of vildagliptin in rat rotenone parkinson’s disease model: Role of RAGE-NFκB and Nrf2-antioxidant signaling pathways. J. Neurochem. 2015, 133, 700–707. [Google Scholar] [CrossRef] [PubMed]
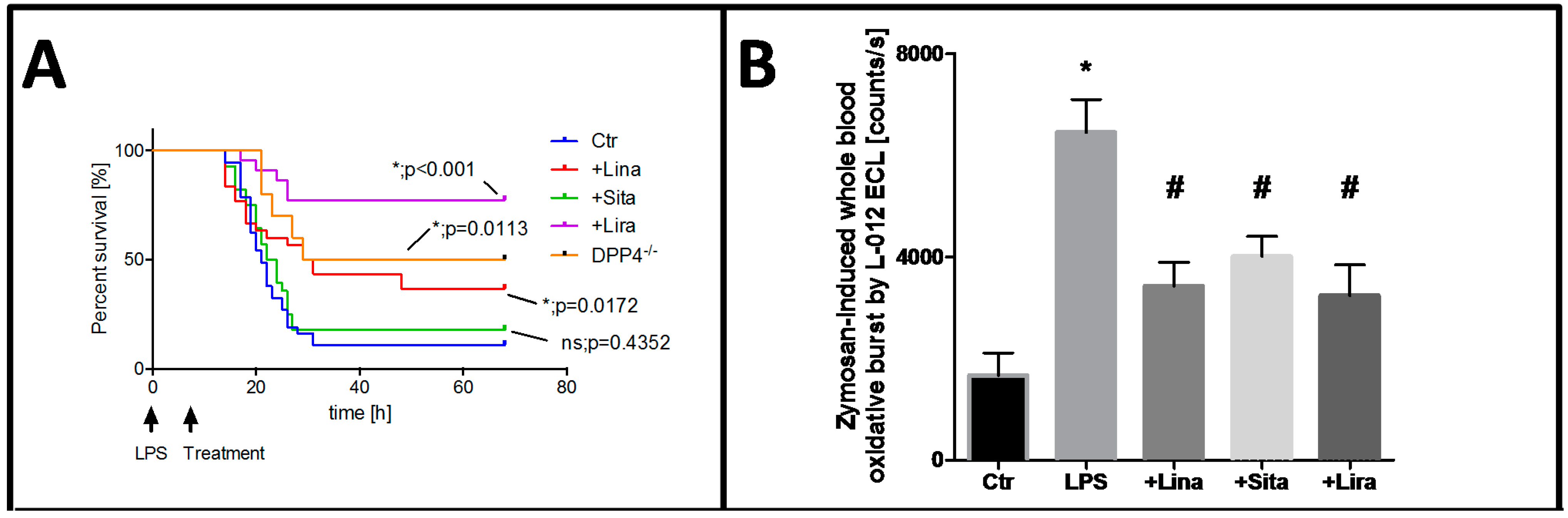
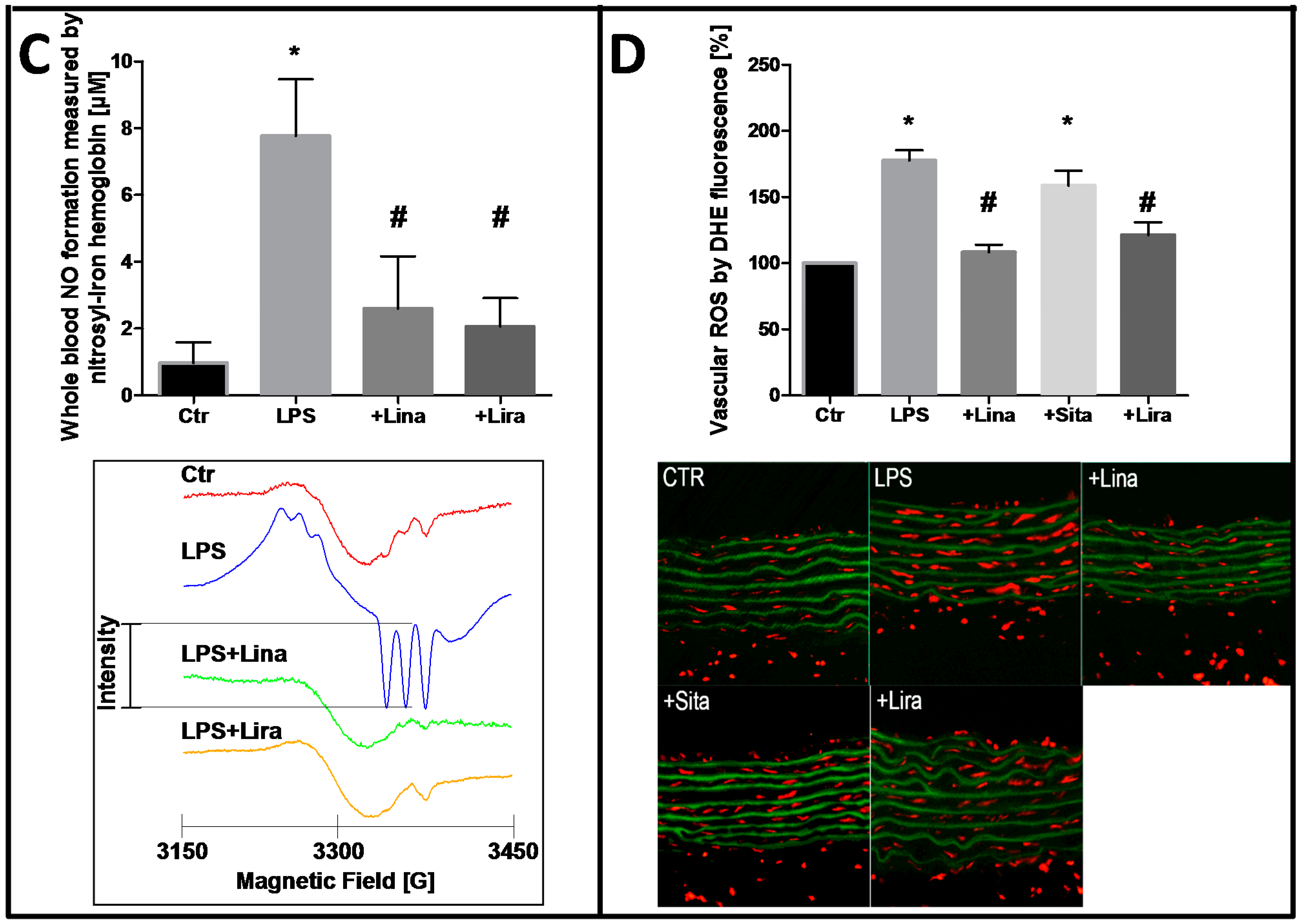
- Steven, S.; Hausding, M.; Kroller-Schon, S.; Mader, M.; Mikhed, Y.; Stamm, P.; Zinssius, E.; Pfeffer, A.; Welschof, P.; Agdauletova, S.; et al. Gliptin and GLP-1 analog treatment improves survival and vascular inflammation/dysfunction in animals with lipopolysaccharide-induced endotoxemia. Basic Res. Cardiol. 2015, 110, 6. [Google Scholar] [CrossRef] [PubMed]
- Kroller-Schon, S.; Knorr, M.; Hausding, M.; Oelze, M.; Schuff, A.; Schell, R.; Sudowe, S.; Scholz, A.; Daub, S.; Karbach, S.; et al. Glucose-independent improvement of vascular dysfunction in experimental sepsis by dipeptidyl-peptidase 4 inhibition. Cardiovasc. Res. 2012, 96, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Ardestani, A.; Dharmadhikari, G.; Laue, S.; Schumann, D.M.; Kerr-Conte, J.; Pattou, F.; Klein, T.; Maedler, K. The DPP-4 inhibitor linagliptin restores β-cell function and survival in human isolated islets through GLP-1 stabilization. J. Clin. Endocrinol. Metab. 2013, 98, E1163–E1172. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.R.; Barbieri, M.; Marfella, R.; Paolisso, G. Reduction of oxidative stress and inflammation by blunting daily acute glucose fluctuations in patients with type 2 diabetes: Role of dipeptidyl peptidase-iv inhibition. Diabetes Care 2012, 35, 2076–2082. [Google Scholar] [CrossRef] [PubMed]
- Batchuluun, B.; Inoguchi, T.; Sonoda, N.; Sasaki, S.; Inoue, T.; Fujimura, Y.; Miura, D.; Takayanagi, R. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis 2014, 232, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, A.; Oyama, J.; Komoda, H.; Asaka, M.; Komatsu, A.; Sakuma, M.; Kodama, K.; Sakamoto, Y.; Kotooka, N.; Hirase, T.; et al. The glucagon-like peptide 1 analog liraglutide reduces TNF-α-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis 2012, 221, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, I.; Tweedie, D.; Li, Y.; Greig, N.H. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: An emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br. J. Pharmacol. 2012, 166, 1586–1599. [Google Scholar] [CrossRef] [PubMed]
- Fisslthaler, B.; Fleming, I. Activation and signaling by the amp-activated protein kinase in endothelial cells. Circ. Res. 2009, 105, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Balteau, M.; van Steenbergen, A.; Timmermans, A.D.; Dessy, C.; Behets-Wydemans, G.; Tajeddine, N.; Castanares-Zapatero, D.; Gilon, P.; Vanoverschelde, J.L.; Horman, S.; et al. AMPK activation by glucagon-like peptide-1 prevents NADPH oxidase activation induced by hyperglycemia in adult cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1120–H1133. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Huguet, J.; Centelles, J.J.; Franco, R. Expression of ecto-adenosine deaminase and CD26 in human T cells triggered by the TCR-CD3 complex. Possible role of adenosine deaminase as costimulatory molecule. J. Immunol. 1995, 155, 4630–4643. [Google Scholar] [PubMed]
- Kameoka, J.; Tanaka, T.; Nojima, Y.; Schlossman, S.F.; Morimoto, C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science 1993, 261, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Dopheide, J.F.; Scheer, M.; Doppler, C.; Obst, V.; Stein, P.; Vosseler, M.; Abegunewardene, N.; Gori, T.; Munzel, T.; Daiber, A.; et al. Change of walking distance in intermittent claudication: Impact on inflammation, oxidative stress and mononuclear cells: A pilot study. Clin. Res. Cardiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dopheide, J.F.; Obst, V.; Doppler, C.; Radmacher, M.C.; Scheer, M.; Radsak, M.P.; Gori, T.; Warnholtz, A.; Fottner, C.; Daiber, A.; et al. Phenotypic characterisation of pro-inflammatory monocytes and dendritic cells in peripheral arterial disease. Thromb. Haemost. 2012, 108, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Dopheide, J.F.; Doppler, C.; Scheer, M.; Obst, V.; Radmacher, M.C.; Radsak, M.P.; Gori, T.; Warnholtz, A.; Fottner, C.; Munzel, T.; et al. Critical limb ischaemia is characterised by an increased production of whole blood reactive oxygen species and expression of TREM-1 on neutrophils. Atherosclerosis 2013, 229, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Burgmaier, M.; Liberman, A.; Mollmann, J.; Kahles, F.; Reith, S.; Lebherz, C.; Marx, N.; Lehrke, M. Glucagon-like peptide-1 (GLP-1) and its split products GLP-1(9-37) and GLP-1(28-37) stabilize atherosclerotic lesions in apoe−/− mice. Atherosclerosis 2013, 231, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, T.; Liu, H.; Welungoda, I.; Hu, Y.; Widdop, R.E.; Knudsen, L.B.; Simpson, R.W.; Dear, A.E. A GLP-1 receptor agonist liraglutide inhibits endothelial cell dysfunction and vascular adhesion molecule expression in an apoe−/− mouse model. Diabetes Vasc. Dis. Res. 2011, 8, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Buldak, L.; Labuzek, K.; Buldak, R.J.; Machnik, G.; Boldys, A.; Okopien, B. Exenatide (a GLP-1 agonist) improves the antioxidative potential of in vitro cultured human monocytes/macrophages. Naunyn Schmiedebergs Arch. Pharmacol. 2015, in press. [Google Scholar]
- Arakawa, M.; Mita, T.; Azuma, K.; Ebato, C.; Goto, H.; Nomiyama, T.; Fujitani, Y.; Hirose, T.; Kawamori, R.; Watada, H. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 2010, 59, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Erdogdu, O.; Nathanson, D.; Sjoholm, A.; Nystrom, T.; Zhang, Q. Exendin-4 stimulates proliferation of human coronary artery endothelial cells through eNOS-, PKA- and PI3K/Akt-dependent pathways and requires GLP-1 receptor. Mol. Cell. Endocrinol. 2010, 325, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Ban, K.; Noyan-Ashraf, M.H.; Hoefer, J.; Bolz, S.S.; Drucker, D.J.; Husain, M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 2008, 117, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Green, B.D.; Hand, K.V.; Dougan, J.E.; McDonnell, B.M.; Cassidy, R.S.; Grieve, D.J. GLP-1 and related peptides cause concentration-dependent relaxation of rat aorta through a pathway involving katp and camp. Arch. Biochem. Biophys. 2008, 478, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, Z.; Pineda, C.; Kampfrath, T.; Maiseyeu, A.; Ying, Z.; Racoma, I.; Deiuliis, J.; Xu, X.; Sun, Q.; Moffatt-Bruce, S.; et al. Acute DPP-4 inhibition modulates vascular tone through GLP-1 independent pathways. Vascul. Pharmacol. 2011, 55, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lu, L.; Guo, Y.; Lin, F.; Chen, H.; Chen, W.; Chen, M. Effect of glucagon-like peptide-1 on high-glucose-induced oxidative stress and cell apoptosis in human endothelial cells and its underlying mechanism. J. Cardiovasc. Pharmacol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.M.; Artigas, A.; Phillips, G.S.; Rhodes, A.; Beale, R.; Osborn, T.; Vincent, J.L.; Townsend, S.; Lemeshow, S.; Dellinger, R.P. Outcomes of the surviving sepsis campaign in intensive care units in the USA and europe: A prospective cohort study. Lancet Infect. Dis. 2012, 12, 919–924. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T. Disseminated intravascular coagulation: A review for the internist. Intern. Emerg. Med. 2013, 8, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Boerma, E.C.; van der Voort, P.H.; Spronk, P.E.; Ince, C. Relationship between sublingual and intestinal microcirculatory perfusion in patients with abdominal sepsis. Crit. Care Med. 2007, 35, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Bone, R.C. Sepsis syndrome. New insights into its pathogenesis and treatment. Infect. Dis. Clin. N. Am. 1991, 5, 793–805. [Google Scholar]
- Nguyen, H.B.; Rivers, E.P.; Knoblich, B.P.; Jacobsen, G.; Muzzin, A.; Ressler, J.A.; Tomlanovich, M.C. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit. Care Med. 2004, 32, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Gkaliagkousi, E.; Ferro, A. Nitric oxide signalling in the regulation of cardiovascular and platelet function. Front. Biosci. 2011, 16, 1873–1897. [Google Scholar] [CrossRef]
- Huet, O.; Dupic, L.; Harrois, A.; Duranteau, J. Oxidative stress and endothelial dysfunction during sepsis. Front. Biosci. 2011, 16, 1986–1995. [Google Scholar] [CrossRef]
- Krotz, F.; Sohn, H.Y.; Pohl, U. Reactive oxygen species: Players in the platelet game. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1988–1996. [Google Scholar] [CrossRef] [PubMed]
- Heyworth, P.G.; Cross, A.R.; Curnutte, J.T. Chronic granulomatous disease. Curr. Opin. Immunol. 2003, 15, 578–584. [Google Scholar] [CrossRef]
- Nathens, A.B.; Neff, M.J.; Jurkovich, G.J.; Klotz, P.; Farver, K.; Ruzinski, J.T.; Radella, F.; Garcia, I.; Maier, R.V. Randomized, prospective trial of antioxidant supplementation in critically ill surgical patients. Ann. Surg. 2002, 236, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Domenighetti, G.; Suter, P.M.; Schaller, M.D.; Ritz, R.; Perret, C. Treatment with N-acetylcysteine during acute respiratory distress syndrome: A randomized, double-blind, placebo-controlled clinical study. J. Crit. Care 1997, 12, 177–182. [Google Scholar] [CrossRef]
- Sprung, C.L.; Annane, D.; Keh, D.; Moreno, R.; Singer, M.; Freivogel, K.; Weiss, Y.G.; Benbenishty, J.; Kalenka, A.; Forst, H.; et al. Hydrocortisone therapy for patients with septic shock. N. Engl. J. Med. 2008, 358, 111–124. [Google Scholar] [CrossRef] [PubMed]
- National Heart, L.; Blood Institute, A.C.T.N.; Truwit, J.D.; Bernard, G.R.; Steingrub, J.; Matthay, M.A.; Liu, K.D.; Albertson, T.E.; Brower, R.G.; Shanholtz, C.; et al. Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N. Engl. J. Med. 2014, 370, 2191–2200. [Google Scholar]
- Ku, H.C.; Chen, W.P.; Su, M.J. GLP-1 signaling preserves cardiac function in endotoxemic fischer 344 and DPP4-deficient rats. Naunyn Schmiedebergs Arch. Pharmacol. 2010, 382, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Amori, R.E.; Lau, J.; Pittas, A.G. Efficacy and safety of incretin therapy in type 2 diabetes: Systematic review and meta-analysis. JAMA 2007, 298, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Parlevliet, E.T.; Geerling, J.J.; van der Tuin, S.J.; Zhang, H.; Bieghs, V.; Jawad, A.H.; Shiri-Sverdlov, R.; Bot, I.; de Jager, S.C.; et al. Exendin-4 decreases liver inflammation and atherosclerosis development simultaneously by reducing macrophage infiltration. Br. J. Pharmacol. 2014, 171, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.A.; Choi, Y.K.; Jung, G.S.; Seo, H.Y.; Kim, H.S.; Jang, B.K.; Kim, J.G.; Lee, I.K.; Kim, M.K.; Park, K.G. Sitagliptin attenuates methionine/choline-deficient diet-induced steatohepatitis. Diabetes Res. Clin. Pract. 2014, 105, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.Y.; Schuppan, D. AMPK regulates macrophage polarization in adipose tissue inflammation and NASH. J. Hepatol. 2013, 58, 619–621. [Google Scholar] [CrossRef] [PubMed]
- Iwaya, H.; Fujii, N.; Hagio, M.; Hara, H.; Ishizuka, S. Contribution of dipeptidyl peptidase IV to the severity of dextran sulfate sodium-induced colitis in the early phase. Biosci. Biotechnol. Biochem. 2013, 77, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Schade, J.; Schmiedl, A.; Kehlen, A.; Veres, T.Z.; Stephan, M.; Pabst, R.; von Horsten, S. Airway-specific recruitment of T cells is reduced in a CD26-deficient F344 rat substrain. Clin. Exp. Immunol. 2009, 158, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Viby, N.E.; Isidor, M.S.; Buggeskov, K.B.; Poulsen, S.S.; Hansen, J.B.; Kissow, H. Glucagon-like peptide-1 (GLP-1) reduces mortality and improves lung function in a model of experimental obstructive lung disease in female mice. Endocrinology 2013, 154, 4503–4511. [Google Scholar] [CrossRef] [PubMed]
- Busso, N.; Wagtmann, N.; Herling, C.; Chobaz-Peclat, V.; Bischof-Delaloye, A.; So, A.; Grouzmann, E. Circulating CD26 is negatively associated with inflammation in human and experimental arthritis. Am. J. Pathol. 2005, 166, 433–442. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E. C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med. 2005, 352, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wang, Q.; Guo, C.; Wang, X.; Cao, X.; Shi, Y.; Gao, F.; Ma, C.; Zhang, L. IL-17 induces apoptosis of vascular endothelial cells: A potential mechanism for human acute coronary syndrome. Clin. Immunol. 2011, 141, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.; Closhen, D.; Croxford, A.; White, R.; Kulig, P.; Pietrowski, E.; Bechmann, I.; Becher, B.; Luhmann, H.J.; Waisman, A.; et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010, 24, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Roussel, L.; Houle, F.; Chan, C.; Yao, Y.; Berube, J.; Olivenstein, R.; Martin, J.G.; Huot, J.; Hamid, Q.; Ferri, L.; et al. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J. Immunol. 2010, 184, 4531–4537. [Google Scholar] [CrossRef] [PubMed]
- Hoch, N.E.; Guzik, T.J.; Chen, W.; Deans, T.; Maalouf, S.A.; Gratze, P.; Weyand, C.; Harrison, D.G. Regulation of t-cell function by endogenously produced angiotensin II. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R208–R216. [Google Scholar] [CrossRef] [PubMed]
- Geiger, H. T-cells in angiotensin-II-induced vascular damage. Nephrol. Dial. Transplant. 2008, 23, 1107–1108. [Google Scholar] [PubMed]
- Micha, R.; Imamura, F.; Wyler von Ballmoos, M.; Solomon, D.H.; Hernan, M.A.; Ridker, P.M.; Mozaffarian, D. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. Am. J. Cardiol. 2011, 108, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, H.; Sanada, S.; Ogai, A.; Minamino, T.; Takashima, S.; Asakura, M.; Ogita, H.; Shinozaki, Y.; Mori, H.; Node, K.; et al. Methotrexate and MX-68, a new derivative of methotrexate, limit infarct size via adenosine-dependent mechanisms in canine hearts. J. Cardiovasc. Pharmacol. 2004, 43, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.M.; Lueneberg, M.E.; da Silva, R.L.; Fattah, T.; Mascia Gottschall, C.A. Rationale and design of the tethys trial: The effects of methotrexate therapy on myocardial infarction with ST-segment elevation. Cardiology 2013, 126, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Pradhan, A.D.; Solomon, D.H.; Paynter, N.; Macfadyen, J.; Zaharris, E.; Gupta, M.; Clearfield, M.; Libby, P.; Hasan, A.A.; et al. Rationale and design of the cardiovascular inflammation reduction trial: A test of the inflammatory hypothesis of atherothrombosis. Am. Heart J. 2013, 166, 199–207 e115. [Google Scholar] [CrossRef] [PubMed]
- Elhage, R.; Maret, A.; Pieraggi, M.T.; Thiers, J.C.; Arnal, J.F.; Bayard, F. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation 1998, 97, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Messas, E.; Levine, R.A.; Graves, D.T.; Amar, S. Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein e heterozygote model: Pharmacotherapeutic implications. Circulation 2004, 110, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Howard, C.P.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T.; Group, C.P.I. Effects of interleukin-1β inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: A phase IIB randomized, placebo-controlled trial. Circulation 2012, 126, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Sinning, C.; Post, F.; Warnholtz, A.; Schulz, E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann. Med. 2008, 40, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart. J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zalewski, A.; Liu, Y.; Mazurek, T.; Cowan, S.; Martin, J.L.; Hofmann, S.M.; Vlassara, H.; Shi, Y. Diabetes-induced oxidative stress and low-grade inflammation in porcine coronary arteries. Circulation 2003, 108, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Steinhubl, S.R. Why have antioxidants failed in clinical trials? Am. J. Cardiol. 2008, 101, 14D–19D. [Google Scholar] [CrossRef] [PubMed]
- Warnholtz, A.; Genth-Zotz, S.; Munzel, T. Should treatment of sepsis include statins? Circulation 2005, 111, 1735–1737. [Google Scholar] [CrossRef] [PubMed]
- Merx, M.W.; Liehn, E.A.; Janssens, U.; Lutticken, R.; Schrader, J.; Hanrath, P.; Weber, C. HMG-CoA reductase inhibitor simvastatin profoundly improves survival in a murine model of sepsis. Circulation 2004, 109, 2560–2565. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Pasupuleti, V.; Rothberg, M.B. Statin therapy and mortality from sepsis: A meta-analysis of randomized trials. Am. J. Med. 2015, 128, 410–417 e411. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steven, S.; Münzel, T.; Daiber, A. Exploiting the Pleiotropic Antioxidant Effects of Established Drugs in Cardiovascular Disease. Int. J. Mol. Sci. 2015, 16, 18185-18223. https://doi.org/10.3390/ijms160818185
Steven S, Münzel T, Daiber A. Exploiting the Pleiotropic Antioxidant Effects of Established Drugs in Cardiovascular Disease. International Journal of Molecular Sciences. 2015; 16(8):18185-18223. https://doi.org/10.3390/ijms160818185
Chicago/Turabian StyleSteven, Sebastian, Thomas Münzel, and Andreas Daiber. 2015. "Exploiting the Pleiotropic Antioxidant Effects of Established Drugs in Cardiovascular Disease" International Journal of Molecular Sciences 16, no. 8: 18185-18223. https://doi.org/10.3390/ijms160818185