
Molecular Mechanisms Underlying Hull-Caryopsis Adhesion/Separation Revealed by Comparative Transcriptomic Analysis of Covered/Naked Barley (Hordeum vulgare L.)

Abstract
:1. Introduction
2. Results
2.1. The RNA-Seq Data Were Highly Reproducible

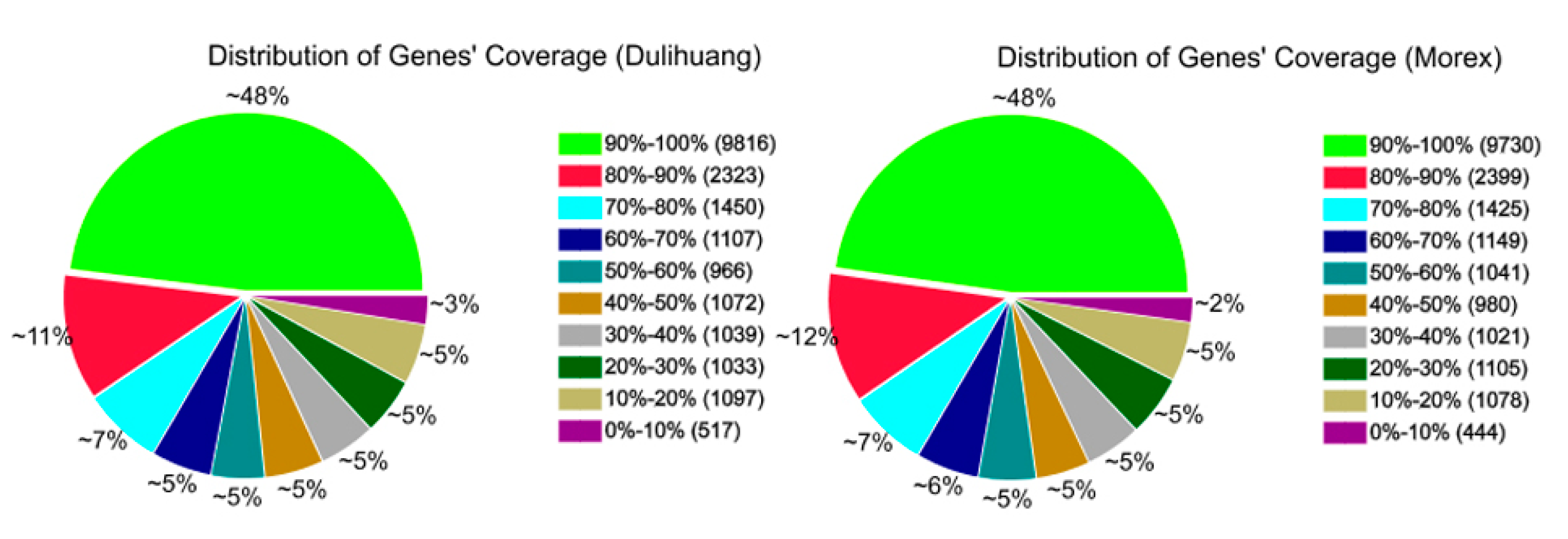{kind=link}
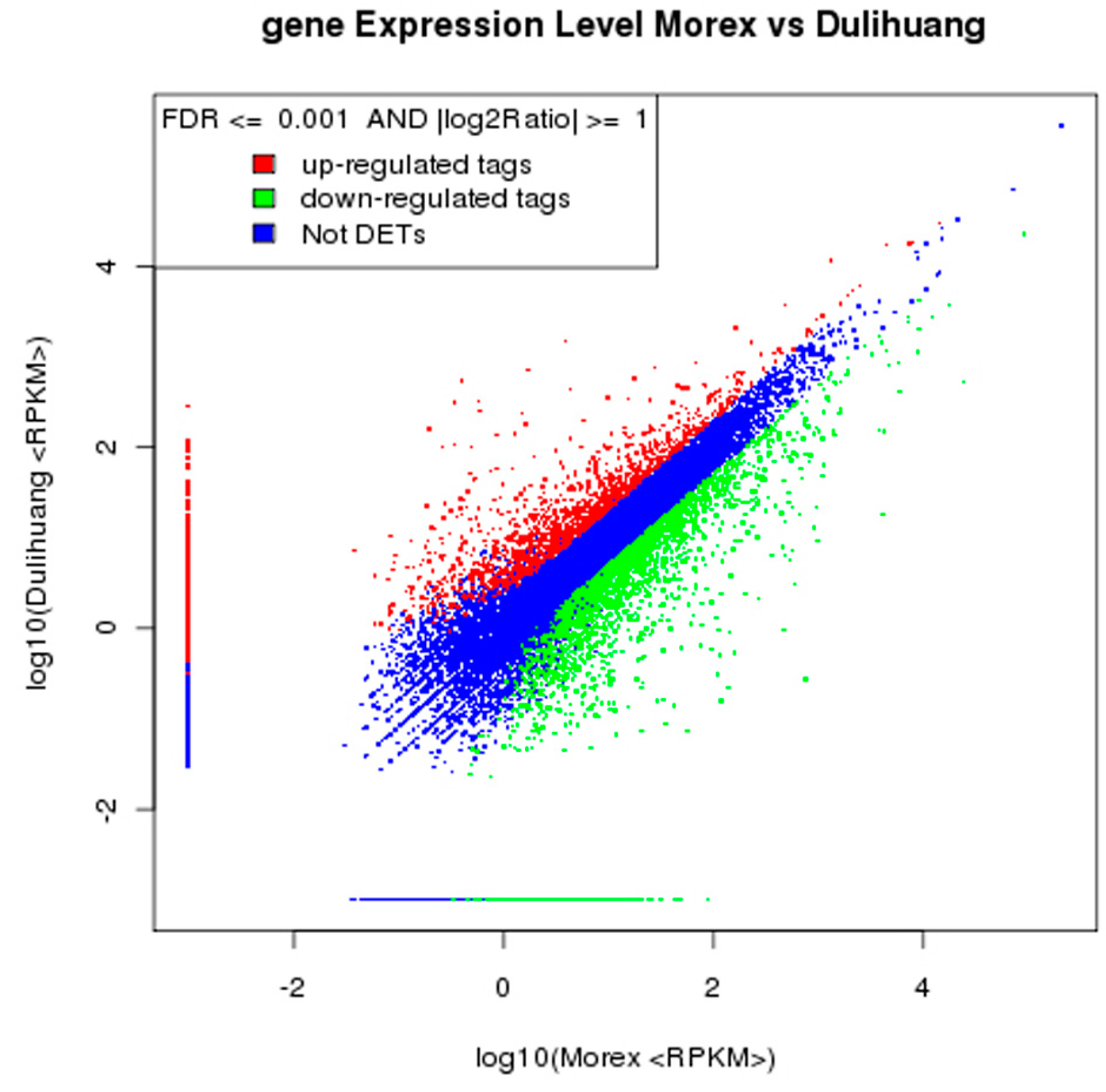{kind=link}
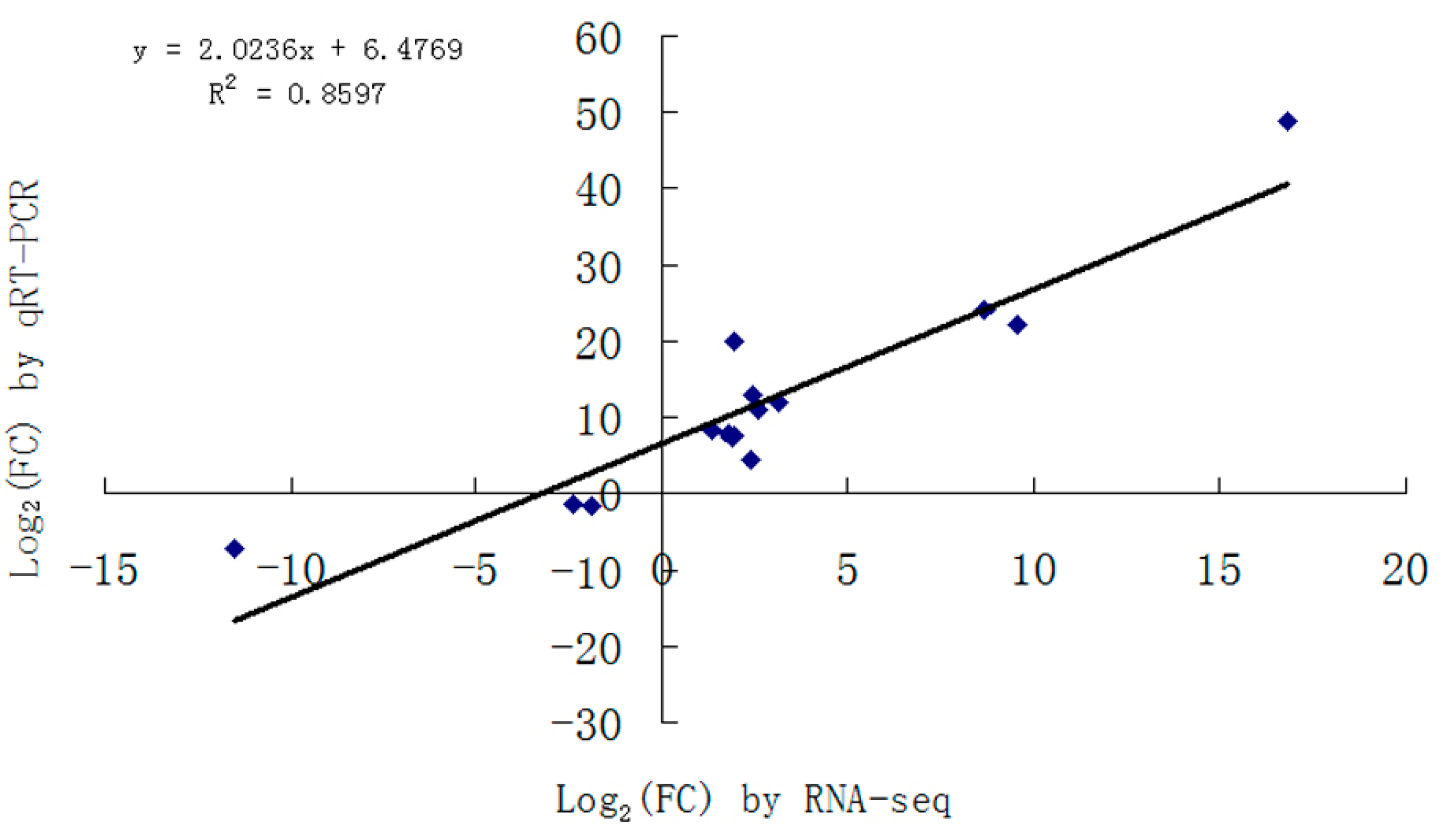{kind=link}
| Map to Genome | Dulihuang Reads Number | Morex Reads Number |
|---|---|---|
| Total clean Reads | 48,428,404 | 48,350,614 |
| Total Base Pairs | 4,358,556,360 | 4,351,555,260 |
| Total Mapped Reads | 26,026,979 | 24,135,038 |
| Perfect match | 12,807,926 | 15,195,341 |
| ≤5 bp mismatch | 13,219,053 | 8,939,697 |
| Unique match | 12,908,475 | 13,718,448 |
| Multi-position match | 13,118,504 | 10,416,590 |
| Total Unmapped Reads | 22,401,425 | 24,215,576 |
| Genome map Rate | 53.74% | 49.92% |
| Gene map Rate | 21.96% | 17.52% |
| Expressed Genes | 20,420 | 20,372 |

2.2. Significantly Differentially Expressed Genes (DEGs) Were Found in the Comparison between Dulihuang and Morex

2.3. Validation of DEGs

2.4. Functional Classification of DEGs
2.5. Differentially Expressed Known Cuticle Genes
3. Discussion
| Gene Name | Gene ID | Plant | Function | Mutant Phenotype | Barley Gene | Log2 Ratio (Morex/Dulihuang) | FDR | |
|---|---|---|---|---|---|---|---|---|
| Cuticle Permeability | Organ Fusion | |||||||
| Nud | GU108424 | Barley | Regulation | No | No | MLOC_59305.1 | +5.13 | 5.78 × 10−18 |
| SHN1 | At1g15360 | Arabidopsis | Regulation | Permeable | Organ fusion | MLOC_12747.2 | −3.12 | 2.48 × 10−10 |
| ATT1 | AT4G00360 | Arabidopsis | Cutin biosynthesis | Permeable | No | MLOC_71597.1 | −2.53 | 2.84 × 10−273 |
| GPAT6 | AT2G38110 | Arabidopsis | Cutin biosynthesis | Permeable | Organ fusion | MLOC_56115.1 | −1.93 | 8.71 × 10−51 |
| GPAT8 | AT4G00400 | Arabidopsis | Cutin biosynthesis | Permeable | Organ fusion | MLOC_19148.1 | −4.38 | 1.36 × 10−5 |
| LACS2 | AT1G49430 | Arabidopsis | Cutin biosynthesis | Permeable | Organ fusion | MLOC_66827.1 | −2.42 | 1.15 × 10−39 |
| LCR | AT2G45970 | Arabidopsis | Cutin biosynthesis | Permeable | Organ fusion | MLOC_75020.2 | −1.37 | 2.27 × 10−7 |
| HTH | AT1G72970 | Arabidopsis | Cutin biosynthesis | Permeable | Organ fusion | MLOC_9891.3 | −1.78 | 1.89 × 10−6 |
| FDH1 | AT2G26250 | Arabidopsis | Wax biosynthesis | Permeable | Organ fusion | MLOC_65700.1 | −1.96 | 1.47 × 10−280 |
| WAX2 | AY131334 | Arabidopsis | Unknown | Permeable | Organ fusion | MLOC_51499.1 | −1.05 | 2.16 × 10−6 |
| CER9 | At4g34100 | Arabidopsis | Unknown | No | No | MLOC_36840.2 | +1.10 | 5.29 × 10−46 |
| ACC1 | AT1G36160 | Arabidopsis | Wax biosynthesis | Permeable | Organ fusion | MLOC_37244.1 | −1.63 | 5.61 × 10−47 |
| EIBI1 | AB534898 | Barley | Secretion | Permeable | - | MLOC_62487.1 | −1.63 | 5.43 × 10−86 |
| WBC11 | At1g17840 | Arabidopsis | Secretion | Permeable | Organ fusion | AK355515 | −1.10 | 4.35 × 10−12 |
| MYB96 | At5G62470 | Arabidopsis | Regulation | No | No | MLOC_34885.1 | −1.72 | 5.80 × 10−9 |
| WSD1 | AT5G37300 | Arabidopsis | Wax biosynthesis | No | No | MLOC_74286.1 | −1.86 | 7.78 × 10−15 |
| CUT1 | AT1G68530 | Arabidopsis | Wax biosynthesis | No | No | MLOC_51583.2 | −2.36 | 4.61 × 10−34 |
| FATB | AT1G08510 | Arabidopsis | Fatty acids biosynthesis | No | No | MLOC_76304.1 | −1.21 | 0.00015 |
4. Experimental Section
4.1. Plant Material
4.2. RNA Extraction, cDNA Library Preparation, and Sequencing
4.3. Functional Annotation: Evaluation of Genes from RNA-Seq
4.4. Validation of RNA-Seq by qRT-PCR
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pourkheirandish, M.; Komatsuda, T. The importance of barley genetics and domestication in a global perspective. Ann. Bot. 2007, 100, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Newman, C.W.; Newman, R.K. A brief history of barley foods. Cereal Foods World 2006, 51, 4–7. [Google Scholar] [CrossRef]
- Guo, B. Economic Flora of Qinghai; Qinghai People’s Press: Xining, China, 1987; pp. 529–530. [Google Scholar]
- Sun, L.J.; Lu, W.; Zhang, J.; Zhang, W. Investigation of barley germplasm in China. Genet. Resour. Crop Evolut. 1999, 46, 361–369. [Google Scholar] [CrossRef]
- Harlan, H.V.; Hulton, H. Daily development of kernels of hannchen barley from flowering to maturity, at Aberdeen, Idaho. J. Inst. Brew. 1920, 26, 633–638. [Google Scholar] [CrossRef]
- Gaines, R.; Bechtel, D.; Pomeranz, Y. A microscopic study on the development of a layer in barley that causes hull-caryopsis adherence. Cereal Chem. 1985, 62, 35–40. [Google Scholar]
- Taketa, S.; Kikuchi, S.; Awayama, T.; Yamamoto, S.; Ichii, M.; Kawasaki, S. Monophyletic origin of naked barley inferred from molecular analyses of a marker closely linked to the naked caryopsis gene (nud). Theor. Appl. Genet. 2004, 10, 1236–1242. [Google Scholar]
- Taketa, S.; Awayama, T.; Amano, S.; Sakurai, Y.; Ichii, M. High-resolution mapping of the nud locus controlling the naked caryopsis in barley. Plant Breed. 2006, 125, 337–342. [Google Scholar] [CrossRef]
- Taketa, S.; Amano, S.; Tsujino, Y.; Sato, T.; Saisho, D.; Kakeda, K.; Nomura, M.; Suzuki, T.; Matsumoto, T.; Sato, K.; et al. Barley grain with adhering hulls is controlled by an ERF family transcription factor gene regulating a lipid biosynthesis pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 4062–4067. [Google Scholar] [CrossRef] [PubMed]
- Franckowiack, J.; Konishi, T. Naked caryopsis. Barley Genet. Newsl. 1997, 26, 51–52. [Google Scholar]
- Samuels, L.; Kunst, L.; Jetter, R. Sealing plant surfaces: Cuticular wax formation by epidermal cells. Annu. Rev. Plant Biol. 2008, 59, 683–707. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.; Faure, J.D. Role of very-long-chain fatty acids in plant development, when chain length does matter. Comptes Rendus Biol. 2010, 333, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Yeats, T.H.; Rose, J.K. The formation and function of plant cuticles. Plant Physiol. 2013, 163, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Bernard, A.; Joubes, J. Arabidopsis cuticular waxes: Advances in synthesis, export and regulation. Prog. Lipid Res. 2013, 52, 110–129. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, U.; Lundqvist, A. Mutagen specificity in barley for 1580 eceriferum mutants localized to 79 loci. Hereditas 1988, 108, 1–12. [Google Scholar] [CrossRef]
- Von Wettstein-Knowles, P. Molecular genetics of lipid synthesisi in barley. In Barley Genetics VI; Munksgaard Intl Publ: Copenhagen, Denmark, 1992; pp. 753–771. [Google Scholar]
- Lundqvist, U.; Franckowiack, J.D. Diversity of barley mutants. In Diversity in Barley (Hordeum Vulgare), 1st ed.; Elsevier Science B.V.: Amsterdam, The Netherlands, 2003; pp. 77–96. [Google Scholar]
- International Barley Genome Sequencing, C.; Mayer, K.F.; Waugh, R.; Brown, J.W.; Schulman, A.; Langridge, P.; Platzer, M.; Fincher, G.B.; Muehlbauer, G.J.; Sato, K.; et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar]
- Chen, X.; Long, H.; Gao, P.; Deng, G.; Pan, Z.; Liang, J.; Tang, Y.; Tashi, N.; Yu, M. Transcriptome assembly and analysis of Tibetan Hulless Barley (Hordeum vulgare L. var. nudum) developing grains, with emphasis on quality properties. PLoS ONE 2014, 9, e98144. [Google Scholar] [CrossRef] [PubMed]
- Freeman, P.; Palmer, G. The structure of the pericarp and testa of barley. J. Inst. Brew. 1984, 90, 88–94. [Google Scholar] [CrossRef]
- Kannangara, R.; Branigan, C.; Liu, Y.; Penfield, T.; Rao, V.; Mouille, G.; Höfte, H.; Pauly, M.; Riechmann, J.L.; Broun, P. The transcription factor WIN1/SHN1 regulates Cutin biosynthesis in Arabidopsis thaliana. Plant Cell 2007, 19, 1278–1294. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Mark Goodwin, S.; Xiao, Y.; Sun, Z.; Baker, D.; Tang, X.; Jenks, M.A.; Zhou, J.-M. Arabidopsis CYP86A2 represses Pseudomonas syringae type III genes and is required for cuticle development. EMBO J. 2004, 23, 2903–2913. [Google Scholar] [CrossRef] [PubMed]
- Wellesen, K. Functional analysis of the LACERATA gene of Arabidopsis provides evidence for different roles of fatty acid omega -hydroxylation in development. Proc. Natl. Acad. Sci. USA 2001, 98, 9694–9699. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Pollard, M.; Li-Beisson, Y.; Beisson, F.; Feig, M.; Ohlrogge, J. A distinct type of glycerol-3-phosphate acyltransferase with sn-2 preference and phosphatase activity producing 2-monoacylglycerol. Proc. Natl. Acad. Sci. USA 2010, 107, 12040–12045. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, J.; Shockey, J.; Browse, J. The acyl-CoA synthetase encoded by LACS2 is essential for normal cuticle development in Arabidopsis. Plant Cell 2004, 16, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Kurdyukov, S.; Faust, A.; Trenkamp, S.; Bär, S.; Franke, R.; Efremova, N.; Tietjen, K.; Schreiber, L.; Saedler, H.; Yephremov, A. Genetic and biochemical evidence for involvement of HOTHEAD in the biosynthesis of long-chain alpha-, omega-dicarboxylic fatty acids and formation of extracellular matrix. Planta 2006, 224, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Yephremov, A.; Wisman, E.; Huijser, P.; Huijser, C.; Wellesen, K.; Saedler, H. Characterization of the FIDDLEHEAD gene of Arabidopsis reveals a link between adhesion response and cell differentiation in the epidermis. Plant Cell 1999, 11, 2187–2201. [Google Scholar] [CrossRef] [PubMed]
- Voisin, D.; Nawrath, C.; Kurdyukov, S.; Franke, R.B.; Reina-Pinto, J.J.; Efremova, N.; Will, I.; Schreiber, L.; Yephremov, A. Dissection of the complex phenotype in cuticular mutants of Arabidopsis reveals a role of SERRATE as a mediator. PLoS Genet. 2009, 5, e1000703. [Google Scholar] [CrossRef] [PubMed]
- Nawrath, C.; Schreiber, L.; Franke, R.B.; Geldner, N.; Reina-Pinto, J.J.; Kunst, L. Apoplastic diffusion barriers in Arabidopsis. Arabidopsis Book 2013, 11, e0167. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Goodwin, S.M.; Boroff, V.L.; Liu, X.; Jenks, M.A. Cloning and characterization of the WAX2 gene of Arabidopsis involved in cuticle membrane and wax production. Plant Cell 2003, 15, 1170–1185. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhao, H.; Des Marais, D.L.; Parsons, E.P.; Wen, X.; Xu, X.; Bangarusamy, D.K.; Wang, G.; Rowland, O.; Juenger, T.; et al. Arabidopsis ECERIFERUM9 involvement in cuticle formation and maintenance of plant water status. Plant Physiol. 2012, 159, 930–944. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhao, H.; Parsons, E.P.; Xu, C.; Kosma, D.K.; Xu, X.; Chao, D.; Lohrey, G.; Bangarusamy, D.K.; Wang, G.; et al. The glossyhead1 allele of ACC1 reveals a principal role for multidomain acetyl-coenzyme A carboxylase in the biosynthesis of cuticular waxes by Arabidopsis. Plant Physiol. 2011, 157, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Komatsuda, T.; Ma, J.F.; Nawrath, C.; Pourkheirandish, M.; Tagiri, A.; Hu, Y.G.; Sameri, M.; Li, X.; Zhao, X.; et al. An ATP-binding cassette subfamily G full transporter is essential for the retention of leaf water in both wild barley and rice. Proc. Natl. Acad. Sci. USA 2011, 108, 12354–12359. [Google Scholar] [CrossRef] [PubMed]
- Bird, D.; Beisson, F.; Brigham, A.; Shin, J.; Greer, S.; Jetter, R.; Kunst, L.; Wu, X.; Yephremov, A.; Samuels, L. Characterization of Arabidopsis ABCG11/WBC11, an ATP binding cassette (ABC) transporter that is required for cuticular lipid secretion. Plant J. 2007, 52, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Sieber, P.; Schorderet, M.; Ryser, U.; Buchala, A.; Kolattukudy, P.; Métraux, J.P.; Nawrath, C. Transgenic Arabidopsis plants expressing a fungal cutinase show alterations in the structure and properties of the cuticle and postgenital organ fusions. Plant Cell 2000, 12, 721–738. [Google Scholar] [CrossRef] [PubMed]
- Seo, P.J.; Lee, S.B.; Suh, M.C.; Park, M.J.; Go, Y.S.; Park, C.M. The MYB96 transcription factor regulates cuticular wax biosynthesis under drought conditions in Arabidopsis. Plant Cell 2011, 23, 1138–1152. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, R.; Xiong, H.; Wang, A.; Chen, G. Molecular Mechanisms Underlying Hull-Caryopsis Adhesion/Separation Revealed by Comparative Transcriptomic Analysis of Covered/Naked Barley (Hordeum vulgare L.). Int. J. Mol. Sci. 2015, 16, 14181-14193. https://doi.org/10.3390/ijms160614181
Duan R, Xiong H, Wang A, Chen G. Molecular Mechanisms Underlying Hull-Caryopsis Adhesion/Separation Revealed by Comparative Transcriptomic Analysis of Covered/Naked Barley (Hordeum vulgare L.). International Journal of Molecular Sciences. 2015; 16(6):14181-14193. https://doi.org/10.3390/ijms160614181
Chicago/Turabian StyleDuan, Ruijun, Huiyan Xiong, Aidong Wang, and Guoxiong Chen. 2015. "Molecular Mechanisms Underlying Hull-Caryopsis Adhesion/Separation Revealed by Comparative Transcriptomic Analysis of Covered/Naked Barley (Hordeum vulgare L.)" International Journal of Molecular Sciences 16, no. 6: 14181-14193. https://doi.org/10.3390/ijms160614181