
A Solid-State NMR Study of Selenium Substitution into Nanocrystalline Hydroxyapatite

Abstract
:1. Introduction
2. Results and Discussion
2.1. Powder X-ray Diffraction Analysis


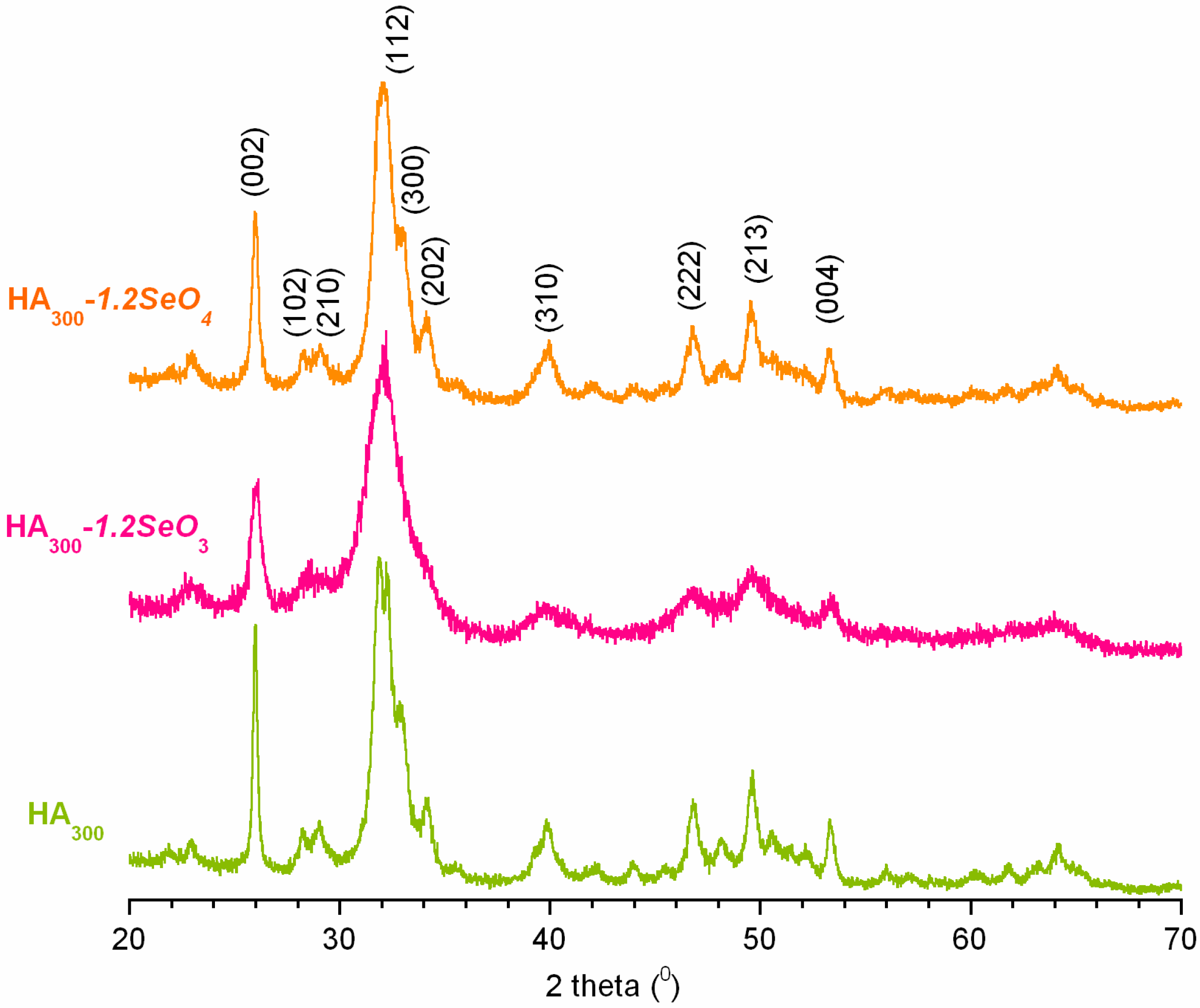{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | HA300 | HA300-1.2SeO3 | HA300-1.2SeO4 |
|---|---|---|---|
| Cell parameters (Å) | a = 9.4287 c = 6.8772 | a = 9.5084 c = 6.8891 | a = 9.4333 c = 6.8879 |
| Crystallite size (nm) | 18 | 9 | 15 |
| Crystallinity index | 0.7 | 0.1 | 0.5 |
| Se content (wt %) | - | 9.64 | 9.55 |
| Ca/(P + Se) | 1.62 | 1.56 | 1.60 |
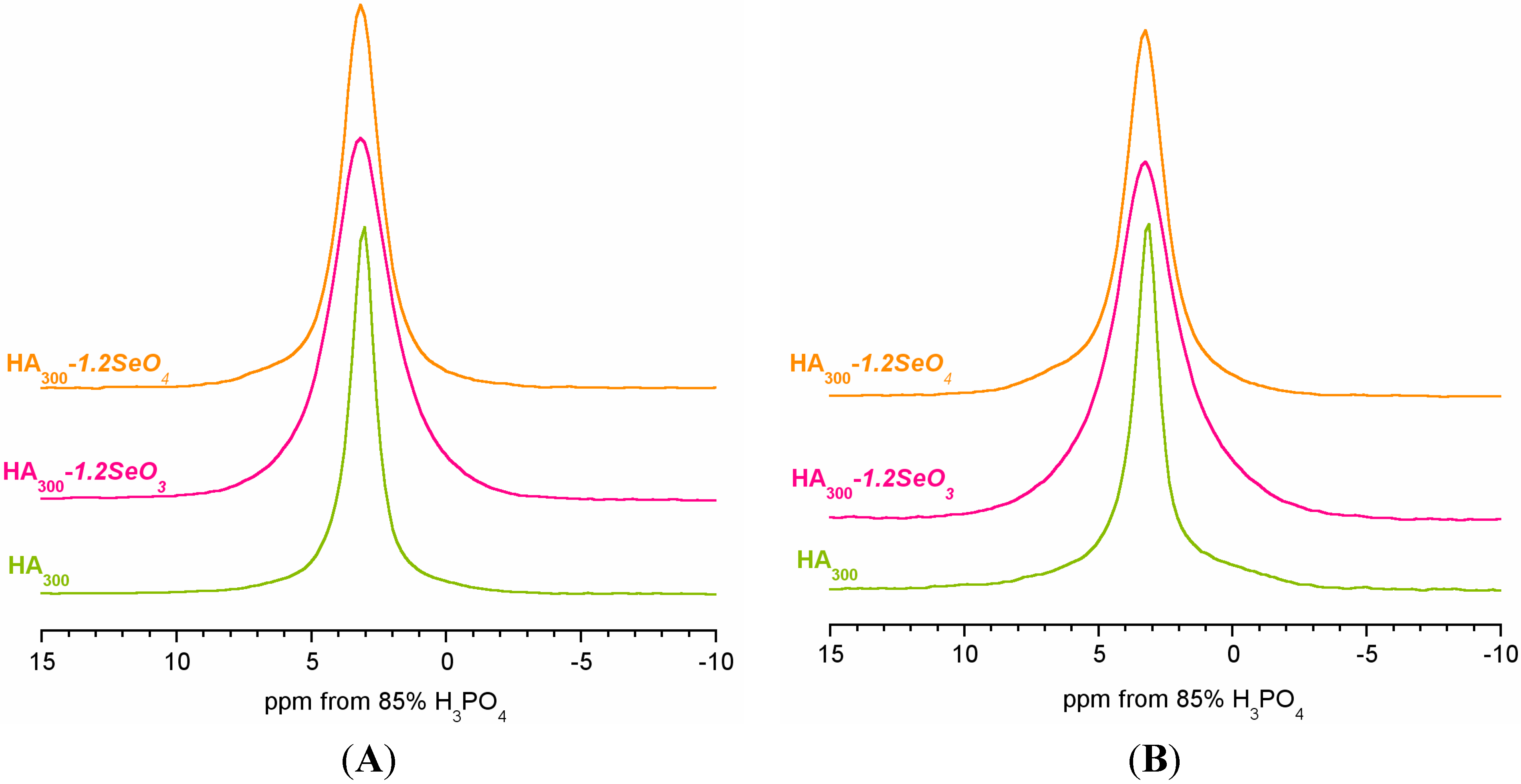
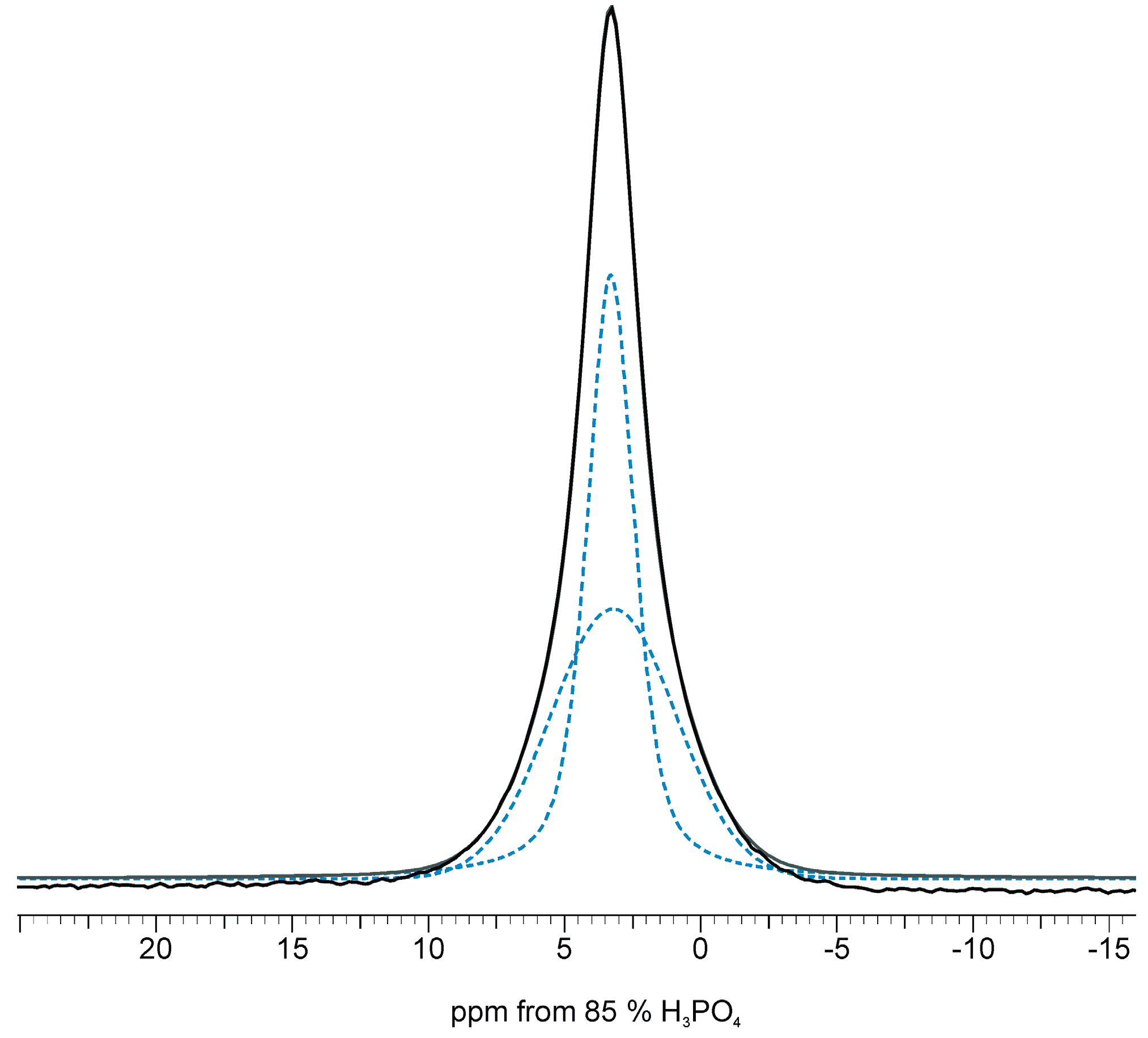
2.2. 31P Solid-State NMR Spectroscopy

| Peak Characteristics | HA300 | HA300-1.2SeO3 | HA300-1.2SeO4 | |||
|---|---|---|---|---|---|---|
| Narrow | Broad | Narrow | Broad | Narrow | Broad | |
| Chemical shift (ppm) | 3.16 | 3.44 | 3.26 | 3.31 | 3.17 | 3.38 |
| FWHM a (Hz) | 159 | 897 | 332 | 879 | 247 | 867 |
| LF b | 0.5 | 0.0 | 0.6 | 0.1 | 0.6 | 0.0 |
| % of total area | 76 | 24 | 49 | 51 | 66 | 34 |
| Narrow line characteristics | ||||||
| FWHM ratio: CP/BD | 0.95 | 0.83 | 0.92 | |||
| Area ratio c: CP/BD | 0.66 | 0.40 | 0.57 | |||

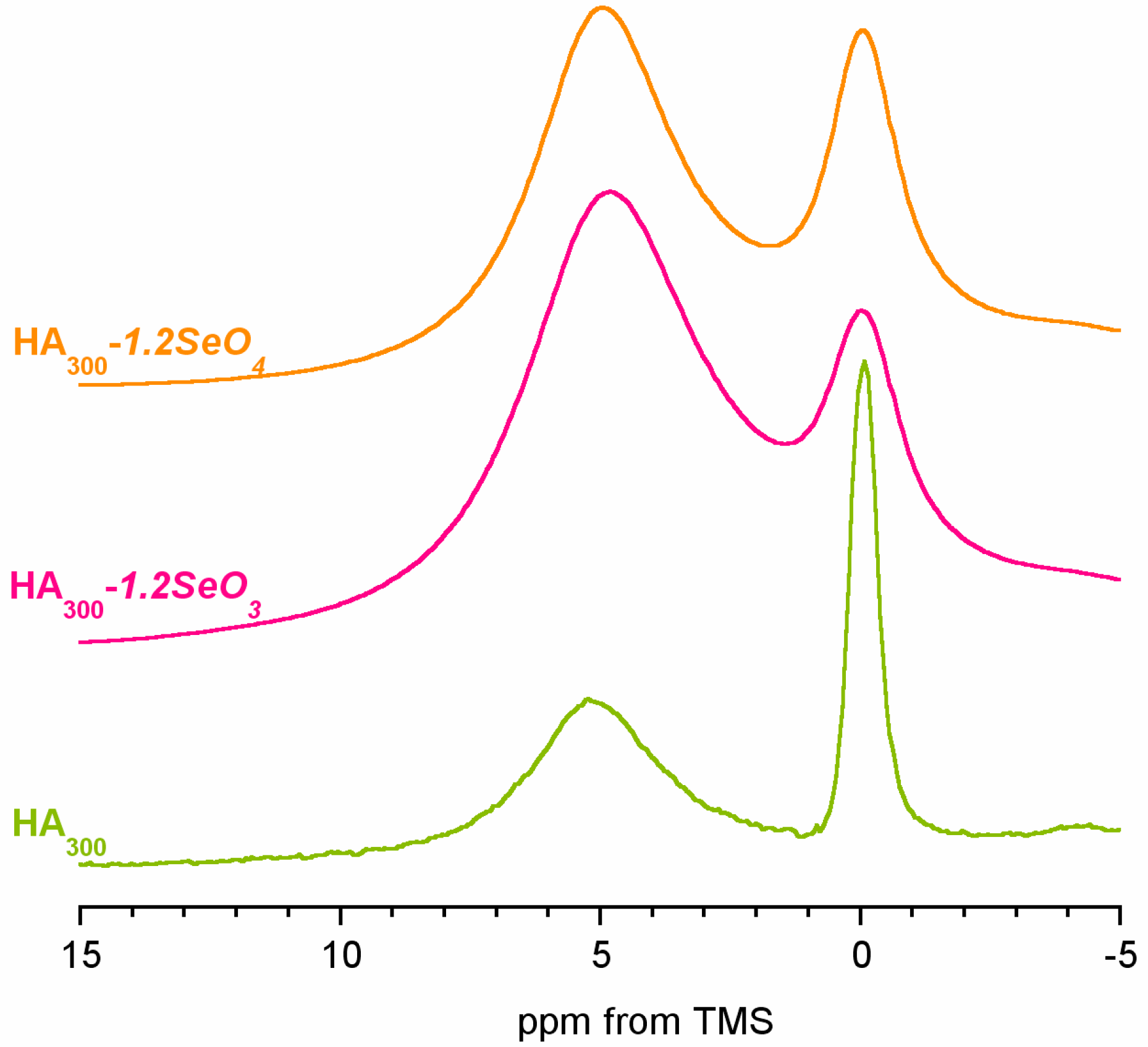
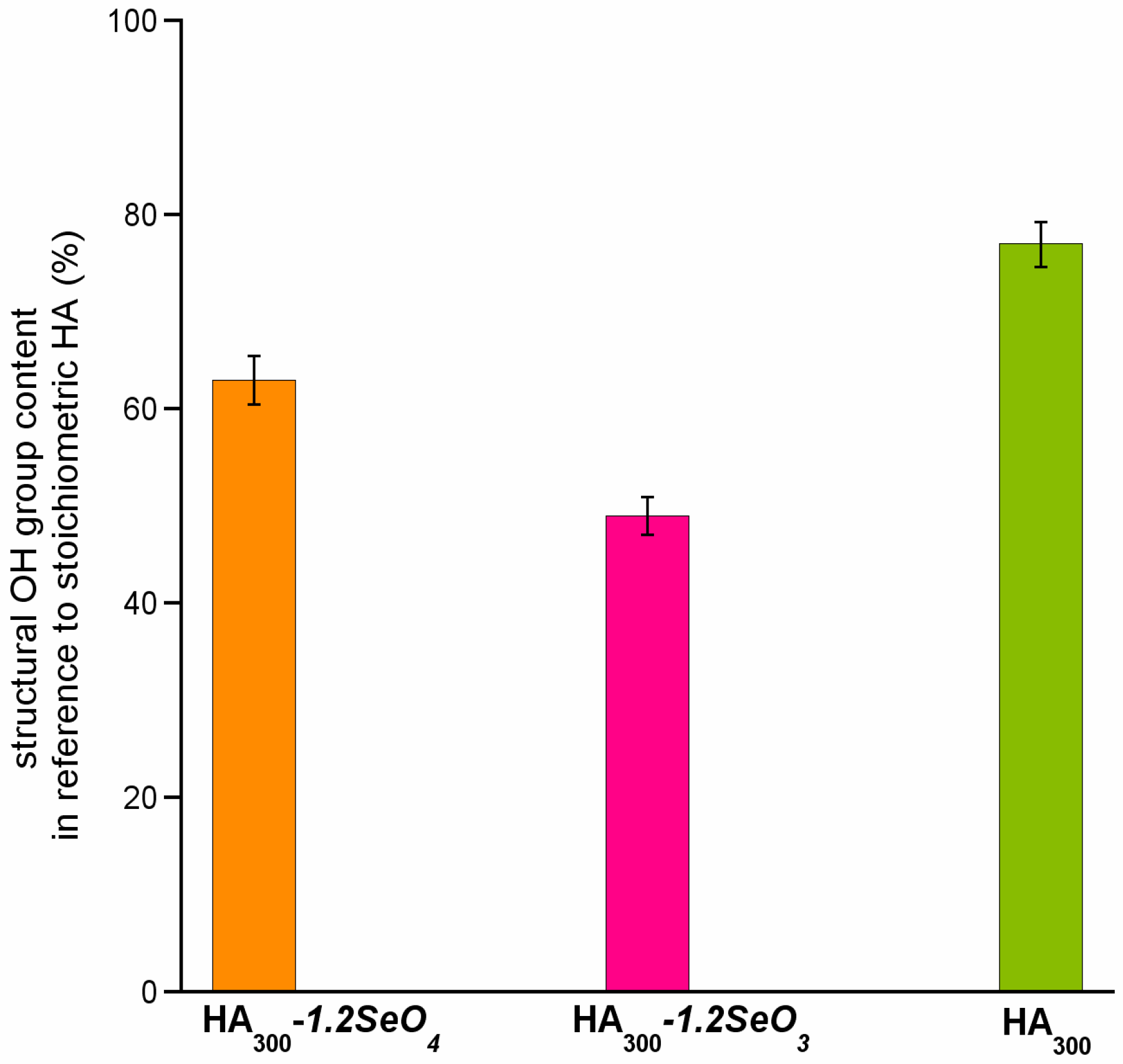
2.3. 1H Solid-State NMR Spectroscopy


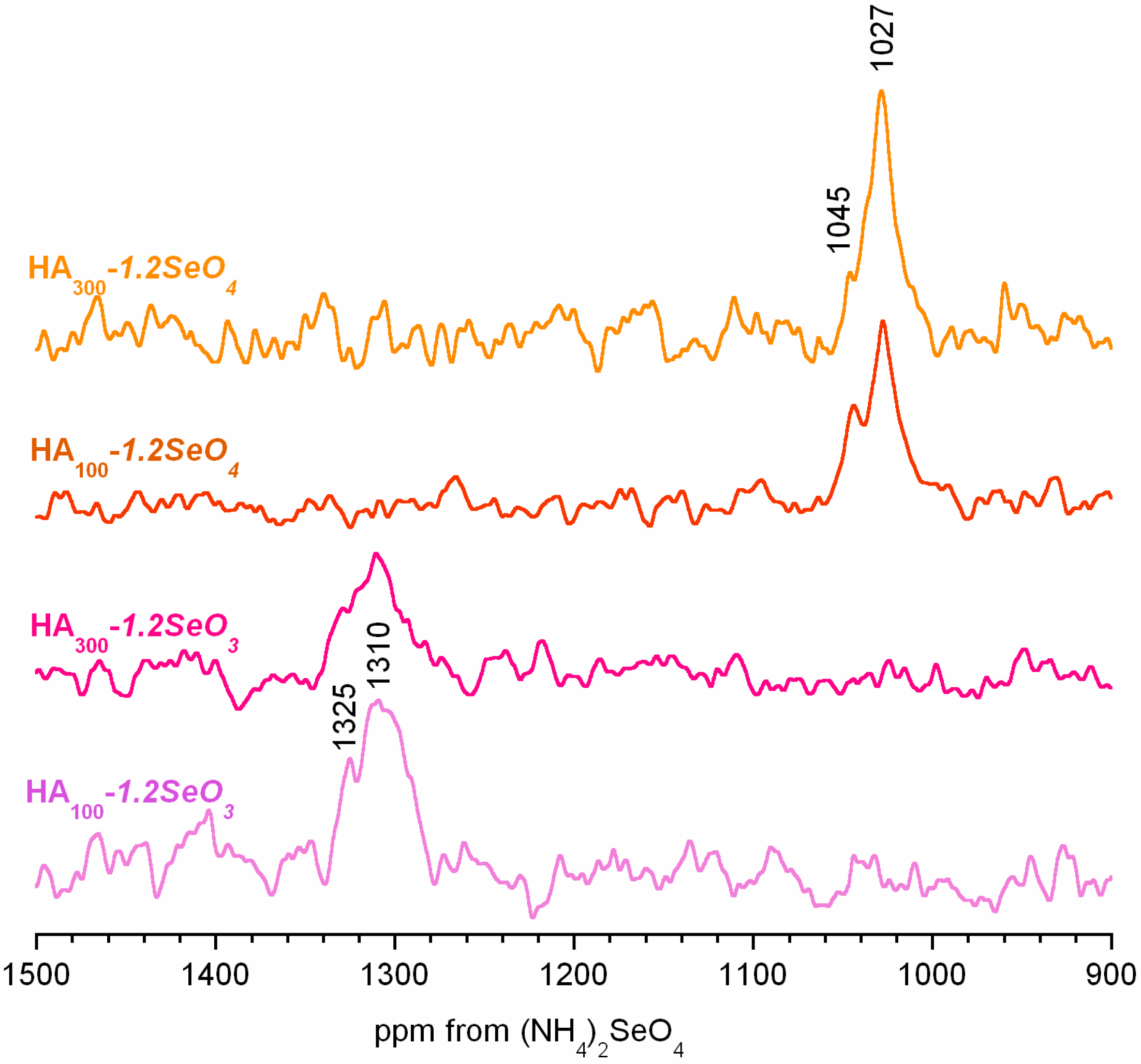
2.4. 77Se Solid-State NMR Spectroscopy

3. Experimental Section
3.1. Preparation of Samples
3.2. Characterization
4. Conclusions
- The content of selenium in HA300-1.2SeO4 and HA300-1.2SeO3 samples is similar and amounts to 9.55% and 9.64%, respectively.
- The analysed samples are nanocrystalline hydroxyapatites and do not contain other crystalline phases.
- The selenite and selenate incorporation has a significant impact on the crystal lattice parameters, crystallite size and crystallinity degree.
- The incorporation of selenium oxyanions causes the decrease of structural hydroxyl groups’ content.
- Selenite and selenate ions are located in the hydroxyapatite crystal interior and on the crystal surface in the surface hydrated layer.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wise, D.L.; Trantolo, D.J.; Lewandrowski, K.-U.; Gresser, J.-D.; Cattaneo, M.V. (Eds.) Biomaterials Engineering and Devices: Human Applications; Humana Press: Totowa, NJ, USA, 2000.
- Planel, J.A. (Ed.) Bone Repair Biomaterials; CRC Press LLC: Boca Raton, FL, USA, 2009.
- Chen, Q.; Zhu, C.; Thouas, G.A. Progress and challenges in biomaterials used for bone tissue engineering: Bioactive glasses and elastomeric composites. Prog. Biomater. 2012, 1, 2–22. [Google Scholar] [CrossRef]
- LeGeros, R.Z.; Ito, A.; Ishikawa, K.; Skae, T.; LeGeros, J.P. Foundamentals of hydroxyapatite and related calcium phosphates. In Advanced Biomaterials: Foundamentals, Processing and Applications; Basu, B., Katti, D.S., Kumar, A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Yoshikawa, H.; Myoui, A. Bone tissue engineering with porous hydroxyapatite ceramics. J. Artif. Organs. 2005, 8, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.H.; Shepherd, D.V.; Best, S.M. Substituted hydroxyapatites for bone repair. J. Mater. Sci. Mater. Med. 2012, 23, 2335–2347. [Google Scholar] [CrossRef] [PubMed]
- Ciobanu, C.S.; Massuyeau, F.; Constantin, L.V.; Predoi, D. Structural and physicochemical properties of antibacterial Ag-doped nano-hydroxyapatite synthesized at 100 °C. Nanoscale Res. Lett. 2011, 6, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Kolmas, J.; Jabłoński, M.; Ślósarczyk, A.; Kolodziejski, W. Solid-state NMR study of Mn2+ for Ca2+ substitution in thermally processed hydroxyapatite. J. Am. Ceram. Soc. 2014. [Google Scholar] [CrossRef]
- Elliott, J.C. Studies in Inorganic Chemistry 18: Structure and Chemistry of the Apatites and Other Calcium Orthophosphates; Elsevier: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Webster, T.J.; Massa-Schlueter, E.A.; Smith, J.L.; Slamovich, E.B. Osteoblast response to hydroxyapatite doped with divalent and trivalent cations. Biomaterials 2004, 25, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- De Lima, I.R.; Alves, G.G.; Soriano, C.A.; Campaneli, A.P.; Gasparoto, T.H.; Ramos, E.S., Jr.; de Sena, L.A.; Rossi, A.M.; Granjeiro, J.M. Understanding the impact of divalent cation substitution on hydroxyapatite: An in vitro multiparametric study on biocompatibility. J. Biomed. Mater. Res. A 2011, 98, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.S.; Yoo, D.S.; Chung, Y.C.; Rhee, S.H. Enhanced bioactivity and osteoconductivity of hydroxyapatite through chloride substitution. J. Biomed. Mater. Res. 2014, 102, 455–469. [Google Scholar] [CrossRef]
- Wei, M.; Evans, J.H.; Bostrom, T.; Grondahl, L. Synthesis and characterization of hydroxyapatite, fluoride-substituted hydroxyapatite and fluorapatite. J. Mater. Sci. Mater. Med. 2003, 14, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Gibson, I.R.; Best, S.M.; Bonfield, W. Chemical characterization of silicon-substituted hydroxyapatite. J. Biomed. Mater. Res. 1999, 44, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.L.; Stephens, P.W.; Tang, Y.; Wei, L.; Phillips, B.L.; Parise, J.B.; Reeder, R.J. Arsenate substitution in hydroxylapatite: Structural characterization of the Ca5(PxAs1-xO4)OH solid solution. Am. Mineral. 2009, 94, 666–675. [Google Scholar] [CrossRef]
- Rayman, M.P. The importance of selenium in human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Cao, J.J.; Combs, G.F., Jr. Selenium in bone health: Roles in antioxidant protection and cell proliferation. Nutrients 2013, 5, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Reyes, R.; Egrise, D.; Neve, J.; Pasteels, J.L.; Schoutens, A. Selenium deficiency-induced growth retardation is associated with an impaired bone metabolism and osteopenia. J. Bone Miner. Res. 2001, 16, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.A.; Webster, T.J. Enhanced osteoblast adhesion on nanostructured selenium compacts for anti-cancer orthopedic applications. Int. J. Nanomed. 2008, 3, 391–396. [Google Scholar]
- Kolmas, J.; Olędzka, E.; Sobczak, M.; Nałęcz-Jawecki, G. Nanocrystalline hydroxyapatite doped with selenium oxyanions: A new material for potential biomedical applications. Mater. Sci. Eng. C 2014, 39, 134–142. [Google Scholar] [CrossRef]
- Kolmas, J.; Kolodziejski, W. Concentration of hydroxyl groups in dental apatites: A solid-state 1H NMR study using inverse 31P→1H cross-polarization. Chem. Comm. 2007, 4390–4392. [Google Scholar] [CrossRef]
- Nakata, K.; Takashi, K.; Numako, C.; Onoki, T.; Nakahira, A. Synthesis and characterization of silicon-doped hydroxyapatite. Mater. Trans. 2009, 50, 1046–1049. [Google Scholar] [CrossRef]
- Landi, E.; Tampieri, A.; Celotti, G.; Spiro, S. Densification behavior and mechanisms of synthetic hydroxyapatites. J. Eur. Ceram. Soc. 2000, 20, 2377–2387. [Google Scholar] [CrossRef]
- Kolodziejski, W. Solid-state NMR of bone. Top. Curr. Chem. 2005, 246, 235–270. [Google Scholar] [PubMed]
- Aue, W.P.; Rouffose, A.H.; Roberts, J.E.; Glimcher, M.J.; Griffin, R.G. Solid-State 31P Nuclear Magnetic Resonance Studies of Synthetic Solid Phases of Calcium Phosphates. Biochemistry 1984, 23, 6110–6114. [Google Scholar] [CrossRef] [PubMed]
- Pajchel, L.; Kolodziejski, W. Solid-state MAS NMR, TEM, and TGA studies of structural hydroxyl groups and water in nanocrystalline apatites prepared by dry milling. J. Nanopart. Res. 2013, 15, 1868. [Google Scholar] [CrossRef] [PubMed]
- Kaflak, A.; Chmielewski, D.; Górecki, A.; Ślósarczyk, A.; Kolodziejski, W. Efficiency of 1H→31P cross-polarization in bone apatite and its mineral standards. Solid State Nucl. Magn. Reson. 2006, 29, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Kolmas, J.; Jaklewicz, A.; Zima, A.; Bucko, M.; Paszkiewicz, Z.; Lis, J.; Ślósarczyk, A.; Kolodziejski, W. Incorporation of carbonate and magnesium ions into synthetic hydroxyapatite: The effect on physicochemical properties. J. Mol. Struct. 2011, 987, 40–50. [Google Scholar] [CrossRef]
- Kaflak-Hachulska, A.; Samoson, A.; Kolodziejski, W. 1H MAS and 1H→31P CP/MAS NMR study of human bone mineral. Calcif. Tissue Int. 2003, 73, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Yesinowski, J.P.; Eckert, H. Hydrogen environments in calcium phosphates: 1H MAS NMR at high spinning speeds. J. Am. Chem. Soc. 1987, 19, 6274–6282. [Google Scholar] [CrossRef]
- Kaflak, A.; Kolodziejski, W. Complementary information on water and hydroxyl groups in nanocrystalline carbonated hydroxyapatites from TGA, NMR and IR measurements. J. Mol. Struct. 2011, 990, 263–270. [Google Scholar] [CrossRef]
- Yoder, C.H.; Pasteris, J.D.; Worcester, K.N.; Schermerhorn, D.V. Structural water in carbonated hydroxylapatite and fluorapatite: Confirmation by solid state 2H NMR. Calcif. Tissue Int. 2012, 90, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.M.; Knight, F.R.; Hua, G.; Ferrara, J.S.; Hogan, S.W.L.; Woolins, J.D.; Ashbrook, S.E. 77Se solid-state NMR of inorganic and organoselenium systems: A combined experimental and computational study. J. Phys. Chem. C 2011, 115, 10859–10872. [Google Scholar] [CrossRef]
- Kemp, T.F.; Wong, A.; Smith, M.E.; Bishop, P.T.; Carthey, N. A natural abundance 77Se solid-state NMR study of inorganic compounds. Solid State Nucl. Magn. Reson. 2008, 34, 224–227. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolmas, J.; Kuras, M.; Oledzka, E.; Sobczak, M. A Solid-State NMR Study of Selenium Substitution into Nanocrystalline Hydroxyapatite. Int. J. Mol. Sci. 2015, 16, 11452-11464. https://doi.org/10.3390/ijms160511452
Kolmas J, Kuras M, Oledzka E, Sobczak M. A Solid-State NMR Study of Selenium Substitution into Nanocrystalline Hydroxyapatite. International Journal of Molecular Sciences. 2015; 16(5):11452-11464. https://doi.org/10.3390/ijms160511452
Chicago/Turabian StyleKolmas, Joanna, Marzena Kuras, Ewa Oledzka, and Marcin Sobczak. 2015. "A Solid-State NMR Study of Selenium Substitution into Nanocrystalline Hydroxyapatite" International Journal of Molecular Sciences 16, no. 5: 11452-11464. https://doi.org/10.3390/ijms160511452