
Nitric Oxide and Reactive Oxygen Species in the Pathogenesis of Preeclampsia

Abstract
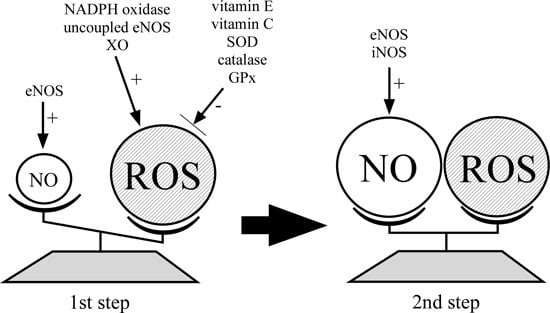
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Oxidative Stress in the Pathogenesis of Preeclampsia (PE)
2.1. Reactive Oxygen Species (ROS)-Producing Enzymes in PE Pathogenesis
2.1.1. NADPH Oxidase
2.1.2. Uncoupled Endothelial Nitric Oxide Synthase (eNOS)
2.1.3. Xanthine Oxidase (XO)
2.2. ROS in the Pathogenesis of PE
3. NO during Normal Pregnancy and in the Pathogenesis of PE
3.1. Role of NO in Physiological Condition during Normal Pregnancy and in the Pathogenesis of PE
4. ONOO− in PE
5. Antioxidant System
5.1. Vitamins (Vitamin C, E)
5.2. Superoxide Dismutase (Mn-SOD, CuZn-SOD, EC-SOD)
5.3. Catalase
5.4. Glutathione Peroxidase (GPx)
6. Conclusions

Acknowledgments
Author Contributions
Abbreviations
| NO | Nitric oxide |
| ROS | Reactive oxygen species |
| PE | Preeclampsia |
| TNFα | Tumor necrosis factor α |
| cGMP | Guanosine 3',5'-cyclic monophosphate |
| PKA | Protein kinase A |
| PKG | Protein kinase G |
| NOS | Nitric oxide synthase |
| nNOS | Neuronal NOS |
| eNOS | Endothelial NOS |
| iNOS | Inducible NOS |
| ONOO− | Peroxynitrite |
| ICAM-1 | Intercellular adhesion molecule 1 |
| LFA-1 | Lymphocyte function-associated antigen 1 |
| MAC-1 | Macrophage-1 antigen |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| XO | Xanthine oxidase |
| AT1 | Angiotensin II receptor subtype 1 |
| AT2 | Angiotensin II receptor subtype 2 |
| FMD | Flow-mediated dilation |
| BH4 | Tetrahydrobiopterin |
| ADMA | Asymmetric dimethyl-l-arginine |
| IFNγ | Interferon γ |
| PGI2 | Prostaglandin I2 |
| GPx | Glutathione peroxidase |
| GSH | Glutathione |
| SOD | Superoxide dismutase |
| Trx | Thioredoxin |
| Mito-T (4) | Mito-Tempol |
Conflicts of Interest
References
- Chappell, L.C.; Enye, S.; Seed, P.; Briley, A.L.; Poston, L.; Shennan, A.H. Adverse perinatal outcomes and risk factors for preeclampsia in women with chronic hypertension: A prospective study. Hypertension 2008, 51, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Group NHBPEPW. National High Blood Pressure Education Program Working Group Report on High Blood Pressure in Pregnancy. Am. J. Obstet. Gynecol. 1990, 163, 1691–1712. [Google Scholar]
- Roberts, J.M.; Taylor, R.N.; Musci, T.J.; Rodgers, G.M.; Hubel, C.A.; McLaughlin, M.K. Preeclampsia: An endothelial cell disorder. Am. J. Obstet. Gynecol. 1989, 161, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.; Lim, K.-H.; Karumanchi, S.A. Circulating angiogenic factors in the pathogenesis and prediction of preeclampsia. Hypertension 2005, 46, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Hubel, C. The two stage model of preeclampsia: Variations on the them. Placenta 2009, 30, 1–6. [Google Scholar]
- Roberts, J.M.; Hubel, C. Is oxidative stress the link in the two-stage model of pre-eclampsia? Lancet 1999, 354, 788–789. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Goldfrad, C.; Carpenter, R.G.; Campbell, S. Transvaginal uterine and umbilical artery Doppler examination of 12–16 weeks and the subsequent development of preeclampsia and intrauterine growth retardation. Ultrasound Obstet. Gynecol. 1997, 9, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Sladek, S.M.; Magness, R.R.; Conrad, K.P. Nitric oxide and pregnancy. Am. J. Physiol. 1997, 272, R441–R463. [Google Scholar] [PubMed]
- Hayashi, T.; Yamada, K.; Esaki, T.; Kuzuya, M.; Satake, S.; Ishikawa, T.; Hidaka, H.; Iguchi, A. Estrogen increases endothelial nitric oxide by a receptor-mediated system. Biochem. Biophys. Res. Commun. 1995, 214, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Murthy, K.S. Signaling for contraction and relaxation in smooth muscle of the gut. Annu. Rev. Physiol. 2006, 68, 345–374. [Google Scholar] [CrossRef] [PubMed]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [PubMed]
- Baker, P.N.; Davidge, S.T.; Roberts, J.M. Plasma from women with preeclampsia increases endothelial cell nitric oxide production. Hypertension 1995, 26, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Farrow, K.N.; Lakshminrusimha, S.; Reda, W.J.; Wedgwood, S.; Czech, L.; Gugino, S.F.; Davis, J.M.; Russell, J.A.; Steinhorn, R.H. Superoxide dismutase restores eNOS expression and function in resistance pulmonary arteries from neonatal lambs with persistent pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L979–L987. [Google Scholar] [CrossRef]
- Hubel, C. Oxidative stress in the pathogenesis of preeclampsia. Exp. Biol. Med. 1999, 222, 222–235. [Google Scholar] [CrossRef]
- Laresgoiti-Servitje, E. A leading role for the immune system in the pathophysiology of preeclampsia. J. Leukoc. Biol. 2013, 94, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Chekir, C.; Nakatsuka, M.; Noguchi, S.; Konishi, H.; Kamada, Y.; Sasaki, A.; Hao, L.; Hiramatsu, Y. Accumulation of advanced glycation end products in women with preeclampsia: Possible involvement of placental oxidative and nitrative stress. Placenta 2006, 27, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Abe, E.; Matsubara, K.; Ochi, H.; Ito, M.; Oka, K.; Kameda, K. Elevated levels of adhesion molecules derived from leukocytes and endothelial cells in patients with pregnancy-induced hypertension. Hypertens. Pregnancy 2003, 22, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Greer, I.A.; Haddad, N.G.; Dawes, J.; Johnstone, F.; Calder, A. Neutrophil activation in pregnancy-induced hypertension. Br. J. Obstet. Gynaecol. 1989, 96, 978–982. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.T.; Linner, J.G.; Siemsen, D.; Dratz, E.; Buescher, E.; Jesaitis, A. Immunocytochemical detection of lipid peroxidation in phagosomes of human neutrophils: Correlation with expression of flavocytochrome b. J. Leukoc. Biol. 1995, 57, 415–421. [Google Scholar] [PubMed]
- Lee, V.M.; Quinn, P.A.; Jennings, S.C.; Ng, L.L. Neutrophil activation and production of reactive oxygen species in preeclampsia. J. Hypertens. 2003, 21, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Gielis, J.F.; Lin, J.Y.; Wingler, K.; van Schil, P.E.; Schmidt, H.H.; Moens, A.L. Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radic. Biol. Med. 2011, 50, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Quinn, P.A.; Jennings, S.C.; Ng, L.L. NADPH oxidase activity in preeclampsia with immortalized lymphoblasts used as models. Hypertension 2003, 41, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Matsubara, Y.; Hyodo, S.; Katayama, T.; Ito, M. Role of nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. J. Obstet. Gynaecol. Res. 2010, 36, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Gutterman, D.D. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2023–H2031. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Förstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Sorescu, D.; Szöcs, K.; Yin, Q.; Akers, M.; Zhang, Y.; Grant, S.; Lambeth, J.; Griendling, K. Novel gp91phox homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Gurovich, A.N.; Avery, J.C.; Holtgrieve, N.B.; Braith, R.W. Flow-mediated dilation is associated with endothelial oxidative stress in human venous endothelial cells. Vasc. Med. 2014, 19, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M. How we learned to say NO. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, H.; Matsuoka, H.; Cooke, J.P.; Usui, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Endogenous nitric oxide synthase inhibitor: A novel marker of atherosclerosis. Circulation 1999, 99, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Sydow, K.; Münzel, T. ADMA and oxidative stress. Atheroscler 2003, 4, 41–51. [Google Scholar] [CrossRef]
- Mitchell, B.M.; Cook, L.G.; Danchuk, S.; Puschett, J.B. Uncoupled endothelial nitric oxide synthase and oxidative stress in a rat model of pregnancy-induced hypertension. AJH 2007, 20, 1297–1304. [Google Scholar] [PubMed]
- Beetsch, J.W.; Park, T.S.; Dugan, L.L.; Shah, A.R.; Gidday, J.M. Xanthine oxidase-derived superoxide causes reoxygenation injury of ischemic cerebral endothelial cells. Brain Res. 1998, 786, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am. J. Physiol. 1988, 255, H1269–H1275. [Google Scholar] [PubMed]
- Patetsios, P.; Song, M.; Shutze, W.P.; Pappas, C.; Rodino, W.; Ramirez, J.A.; Panetta, T.F. Identification of uric acid and xanthine oxidase in atherosclerotic plaque. Am. J. Cardiol. 2001, 88, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Corte, E.D.; Stirpe, F. The regulation of rat liver xanthine oxidase. Involvement of thiol groups in the conversion of the enzyme activity from dehydrogenase (type D) into oxidase (type O) and purification of the enzyme. Biochem. J. 1972, 126, 739–745. [Google Scholar] [PubMed]
- Many, A.; Hubel, C.A.; Fisher, S.J.; Roberts, J.M.; Zhou, Y. Invasive cytotrophoblasts manifest evidence of oxidative stress in preeclampsia. Am. J. Pathol. 2000, 156, 321–331. [Google Scholar] [CrossRef]
- Myatt, L. Review: Reactive oxygen and nitrogen species and functional adaptation of the placenta. Placenta 2010, 31, S66–S69. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C.; Ranheim, T.; Khoury, J.; Henriksen, T. Increased contents of phospholipids, cholesterol, and lipid peroxides in decidua basalis in women with preeclampsia. Am. J. Obstet. Gynecol. 1999, 180, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Roggensack, A.M.; Zhang, Y.; Davidge, S.T. Evidence for peroxynitrite formation in the vasculature of women with preeclampsia. Hypertension 1999, 33, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Aris, A.; Benali, S.; Ouellet, A.; Moutquin, J.M.; Leblanc, S. Potential biomarkers of preeclampsia: Inverse correlation between hydrogen peroxide and nitric oxide early in maternal circulation and at term in placenta of women with preeclampsia. Placenta 2009, 324, 324–327. [Google Scholar]
- Madazli, R.; Benian, A.; Aydin, S.; Uzun, H.; Tolun, N. The plasma and placental levels of malondialdehyde, glutathione and superoxide dismutase in preeclampsia. J. Obstet. Gynaecol. 2002, 22, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Molvarec, A.; Rigó, J., Jr.; Lázár, L.; Balogh, K.; Makó, V.; Cervenak, L.; Mézes, M.; Prohászka, Z. Increased serum heat-shock protein 70 levels reflect systemic inflammation, oxidative stress and hepatocellular injury in preeclampsia. Cell Stress Chaperones 2009, 14, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Abe, E.; Ochi, H.; Kusanagi, Y.; Ito, M. Changes in serum concentrations of tumor necrosis factor α and adhesion molecules in normal pregnant women and those with pregnancy-induced hypertension. J. Obstet. Gynaecol. Res. 2003, 29, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Takeda-Matsubara, Y.; Matsubara, K.; Ochi, H.; Ito, M.; Iwai, M.; Horiuchi, M. Expression of endothelial angiotensin II receptor mRNA in pregnancy-induced hypertension. Am. J. Hypertens. 2003, 16, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.P.; Mittal, C.K.; Katsuki, S.; Murad, F. Nitric oxide activates guanylate cyclase and increases guanosine 3':5'-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. USA 1977, 74, 3203–3207. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, L.; Matys, T.; Chabielska, E.; Buczko, W.; Malinski, T. Angiotensin II AT1 receptor antagonists inhibit platelet adhesion and aggregation by nitric oxide release. Hypertension 2002, 40, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298 Pt 2, 249–258. [Google Scholar] [PubMed]
- Li, H.; Förstermann, U. Nitric oxide in the pathogenesis of vascular disease. J. Pathol. 2000, 190, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Thiemermann, C. Nitric oxide and septic shock. Gen. Pharmacol. 1997, 29, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Aires, R.D.; Capettini, L.S.; Silva, J.F.; Rodrigues-Machado Mda, G.; Pinho, V.; Teixeira, M.M.; Cortes, S.F.; Lemos, V.S. Paraquat poisoning induces TNF-α-dependent iNOS/NO mediated hyporesponsiveness of the aorta to vasoconstrictors in rats. PLoS One 2013, 8, e73562. [Google Scholar] [CrossRef] [PubMed]
- Kilbourn, R.G.; Traber, D.L.; Szabo, C. Nitric oxide and shock. Dis. Mon. 1997, 43, 277–348. [Google Scholar] [CrossRef] [PubMed]
- Cannon, P.J.; Yang, X.; Szabolcs, M.J.; Ravalli, S.; Sciacca, R.R.; Michler, R.E. The role of inducible nitric oxide synthase in cardiac allograft rejection. Cardiovasc. Res. 1998, 28, 6–15. [Google Scholar] [CrossRef]
- Leal, E.C.; Manivannan, A.; Hosoya, K.; Terasaki, T.; Cunha-Vaz, J.; Ambrósio, A.F.; Forrester, J.V. Inducible nitric oxide synthase isoform is a key mediator of leukostasis and blood-retinal barrier breakdown in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5257–5265. [Google Scholar] [CrossRef]
- Kone, B.C.; Baylis, C. Biosynthesis and homeostatic roles of nitric oxide in the normal kidney. Am. J. Physiol. 1997, 272, F561–F578. [Google Scholar] [PubMed]
- Carey, R.M.; Jin, X.; Wang, Z.Q.; Siragy, H.M. Nitric oxide: A physiological mediator of the type 2 (AT2) angiotensin receptor. Acta Physiol. Scand. 2000, 168, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.; Oeckler, R.; Li, X.; Du, L.; Traganos, F.; Zhao, X.; Burke-Wolin, T. Angiotensin II stimulates nitric oxide production in pulmonary artery endothelium via the type 2 receptor. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L559–L568. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.D.; Palmer, R.M.; Moncada, S. Role of endotheliumderived nitric oxide in the regulation of blood pressure. Proc. Natl. Acad. Sci. USA 1989, 86, 3375–3378. [Google Scholar] [CrossRef] [PubMed]
- Kakui, K.; Sagawan, N.; Itoh, H.; Yura, S.; Korita, D.; Takemura, M.; Nuamah, M.A.; Fujii, S. Expression of nitric oxide synthase isoforms in the human placenta is not altered by labor. Endocr. J. 2003, 50, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Myatt, L.; Eis, A.L.W.; Brockman, D.E.; Kossenjans, W.; Greer, I.; Lyall, F. Inducible (type II) nitric oxide synthase in human placental villous tissue of normotensive, preeclamptic and intrauterine growth-restricted pregnancy. Placenta 1997, 18, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Nevzati, E.; Shafighi, M.; Bakhtian, K.D.; Treiber, H.; Fandino, J.; Fathi, A.R. Estrogen induces nitric oxide production via nitric oxide synthase activation in endothelial cells. Acta Neurochir. Suppl. 2015, 120, 141–145. [Google Scholar] [PubMed]
- Buhimschi, I.; Yallampalli, C.; Dong, Y.L.; Garfield, R.E. Invovement of a nitric oxide-cyclic guanosine monophosphate pathway in control of human uterine contractility during pregnancy. Am. J. Obstet. Gynecol. 1995, 172, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- Cockell, A.P.; Poston, L. Flow-mediated vasodilatation is enhanced in normal pregnancy but reduced in preeclampsia. Hypertension 1997, 30, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Davenpeck, K.L.; Gauthier, T.W.; Lefer, A.M. Inhibition of endothelial-derived nitric oxide promotes P-selectin expression and actions in the rat microcirculation. Gastroenterology 1994, 107, 1050–1058. [Google Scholar] [PubMed]
- Gauthier, T.W.; Scalia, R.; Murohara, T.; Guo, J.P.; Lefer, A.M. Nitric oxide protects against leukocyte-endothelium interactions in the early stages of hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Szarka, A.; Rigó, J.; Lázár, L.; Beko, G.; Molvarec, A. Circulating cytokines, chemokines and adhesion molecules in normal pregnancy and preeclampsia determined by multiplex suspension array. BMC Immunol. 2010, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Abe, E.; Matsubara, K.; Oka, K.; Kusanagi, Y.; Ito, M. Cytokine regulation of intercellular adhesion molecule-1 expression on trophoblasts in preeclampsia. Gynecol. Obstet. Investig. 2008, 66, 27–33. [Google Scholar] [CrossRef]
- Kerr, S.; Brosnan, M.J.; McIntyre, M.; Reid, J.; Dominiczak, A.; Hamilton, C. Superoxide anion production is increased in a model of genetic hypertension. Hypertension 1999, 33, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, P.T.; Flavahan, S.; Hassanain, H.; Mitra, S.; Holland, S.; Goldschmidt-Clermont, P.J.; Flavahan, N.A. Redox signaling of the arteriolar myogenic response. Circ. Res. 2001, 89, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Tschudi, M.R.; Mesaros, S.; LuÈscher, T.F.; Malinski, T. Direct in situ measurement of nitric oxide in mesenteric resistance arteries. Increased decomposition by superoxide in hypertension. Hypertension 1996, 27, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Dong, Y.; Zhang, M.; Cui, M.Z.; Cohen, R.A.; Riek, U.; Neumann, D.; Schlattner, U.; Zou, M.H. Activation of protein kinase C ζ by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J. Biol. Chem. 2006, 281, 6366–6375. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, V.; Bachschmid, M. Superoxide as a messenger of endothelial function. Biochem. Biophys. Res. Commun. 2000, 278, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.A.; Dinauer, M.C.; Rosen, H.; Chait, A.; Heinecke, J.W.; LeBoeuf, R.C. Impaired superoxide production due to a deficiency in phagocyte NADPH oxidase fails to inhibit atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Kampfrath, T.; Thurston, G.; Farrar, B.; Lippmann, M.; Wang, A.; Sun, Q.; Chen, L.C.; Rajagopalan, S. Ambient particulates alter vascular function through induction of reactive oxygen and nitrogen species. Toxicol. Sci. 2009, 111, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.P.; Skepper, J.N.; Burton, G.J. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef] [PubMed]
- Hubel, C.A.; Kagan, V.; Kisin, E.R.; McLaughlin, M.K.; Roberts, J.M. Increased ascorbate radical formation and ascorbate depletion in plasma from women with preeclampsia: Implications for oxidative stress. Free Radic. Biol. Med. 1997, 23, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Mutlu-Türkoglu, U.; Ademoglu, E.; Ibrahimoglu, L.; Aykaç-Toker, G.; Uysal, M. Imbalance between lipid peroxidation and antioxidant status in preeclampsia. Gynecol. Obstet. Investig. 1998, 46, 37–40. [Google Scholar] [CrossRef]
- Song, J.H.; Simons, C.; Cao, L.; Shin, S.H.; Hong, M.; Chung, I.M. Rapid uptake of oxidized ascorbate induces loss of cellular glutathione and oxidative stress in liver slices. Exp. Mol. Med. 2003, 35, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hunt, J.A.; Groves, J.T. Manganese porphyrins as redox-coupled peroxynitrite reductases. J. Am. Chem. Soc. 1998, 120, 6053–6061. [Google Scholar] [CrossRef]
- Jendryczko, A.; Drozdz, M. Plasma retinol, β-carotene and vitamin E levels in relation to the future risk of preeclampsia. Zent. Gynakol. 1989, 111, 1121–1123. [Google Scholar]
- Rosta, K.; Molvarec, A.; Enzsöly, A.; Nagy, B.; Rónai, Z.; Fekete, A.; Sasvári-Székely, M.; Rigó, J., Jr.; Vér, A. Association of extracellular superoxide dismutase (SOD3) Ala40Thr gene polymorphism with preeclampsia complicated by severe fetal growth restriction. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 142, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A.; Squadrito, G.L. The chemistry of peroxynitrite: A product from the reaction of nitric oxide with superoxide. Am. J. Physiol. 1995, 268, L699–L722. [Google Scholar] [PubMed]
- Wang, Y.; Walsh, S.W. Increased superoxide generation is associated with decreased superoxide dismutase activity and mRNA expression in placental trophoblast cells in preeclampsia. Placenta 2001, 22, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Ademuyiwa, O.; Odusoga, O.L.; Adebawo, O.O.; Ugbaja, R. Endogenous antioxidant defences in plasma and erythrocytes of pregnant women during different trimesters of pregnancy. Acta Obstet. Gynecol. Scand. 2007, 86, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Góth, L. Catalase deficiency and type 2 diabetes. Diabetes Care 2008, 31, e93. [Google Scholar] [CrossRef] [PubMed]
- Dordević, N.Z.; Babić, G.M.; Marković, S.D.; Ognjanović, B.I.; Stajn, A.S.; Zikić, R.V.; Saicić, Z.S. Oxidative stress and changes in antioxidative defense system in erythrocytes of preeclampsia in women. Reprod. Toxicol. 2008, 25, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Walsh, S.W. Antioxidant activities and mRNA expression of superoxide dismutase, catalase, and glutathione peroxidase in normal and preeclamptic placentas. J. Soc. Gynecol. Investig. 1996, 3, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.K.; Siddiqui, M.U.; Islam, N.; Moin, S. Erythrocyte markers of oxidative stress in higher age-group preeclamptic and normal pregnant mothers. Hypertens. Pregnancy 2010, 29, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, F.; Renner, A.; Rath, W.; Kuhn, W.; Wieland, E. Lipid hydroperoxides and free radical scavenging enzyme activities in preeclampsia and HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome: No evidence for circulating primary products of lipid peroxidation. Am. J. Obstet. Gynecol. 2001, 185, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.W.; Wang, Y. Deficient glutathione peroxidase activity in preeclampsia is associated with increased placental production of thromboxane and lipid peroxides. Am. J. Obstet. Gynecol. 1993, 169, 1456–1461. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Csiszar, A.; Kaminski, P.M.; Wolin, M.S.; Koller, A. Chronic high pressure-induced arterial oxidative stress: Involvement of protein kinase C-dependent NAD(P)H oxidase and local renin-angiotensin system. Am. J. Pathol. 2004, 165, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Cianchetti, S.; del Fiorentino, A.; Colognato, R.; Stefano, R.; Franzoni, F.; Pedrinelli, R. Anti-inflammatory and anti-oxidant properties of telmisartan in cultured human umbilical vein endothelial cells. Atherosclerosis 2007, 198, 22–28. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsubara, K.; Higaki, T.; Matsubara, Y.; Nawa, A. Nitric Oxide and Reactive Oxygen Species in the Pathogenesis of Preeclampsia. Int. J. Mol. Sci. 2015, 16, 4600-4614. https://doi.org/10.3390/ijms16034600
Matsubara K, Higaki T, Matsubara Y, Nawa A. Nitric Oxide and Reactive Oxygen Species in the Pathogenesis of Preeclampsia. International Journal of Molecular Sciences. 2015; 16(3):4600-4614. https://doi.org/10.3390/ijms16034600
Chicago/Turabian StyleMatsubara, Keiichi, Takashi Higaki, Yuko Matsubara, and Akihiro Nawa. 2015. "Nitric Oxide and Reactive Oxygen Species in the Pathogenesis of Preeclampsia" International Journal of Molecular Sciences 16, no. 3: 4600-4614. https://doi.org/10.3390/ijms16034600