
Alzheimer’s Disease: Mechanism and Approach to Cell Therapy

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Gene Mutations Related to Early-Onset and Late-Onset AD
3. Amyloid Plaques and Immunotherapy
4. Tau Pathology and Immunotherapy
5. Metabolic Changes in Senescence and AD
5.1. Protein Metabolism in AD
5.2. Cholesterol Metabolism (Lipid Rafts and PrPC) in AD
5.3. Glucose Metabolism in AD
5.4. Oxidative Stress and Metabolism
5.5. Insulin Metabolism and AD
6. Glia and AD
6.1. Microglia
6.2. Astrocytes
7. Models of AD and Senescence
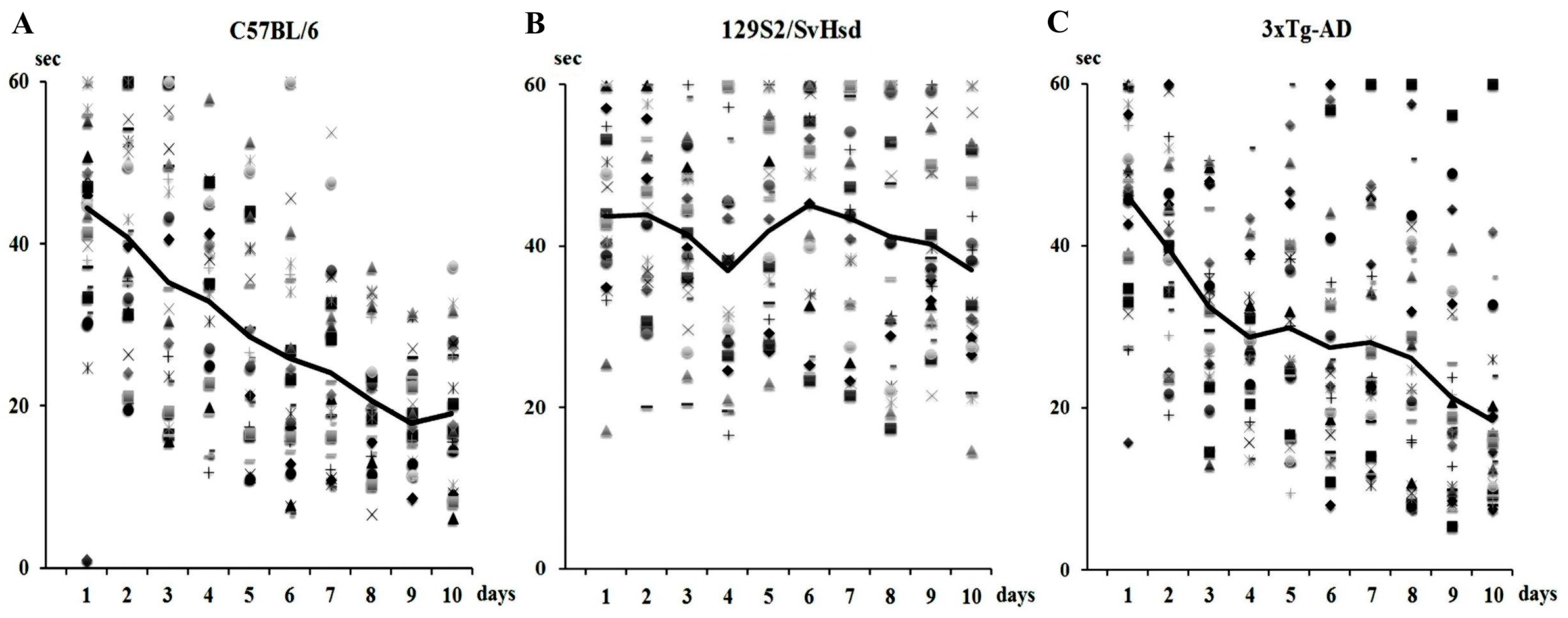
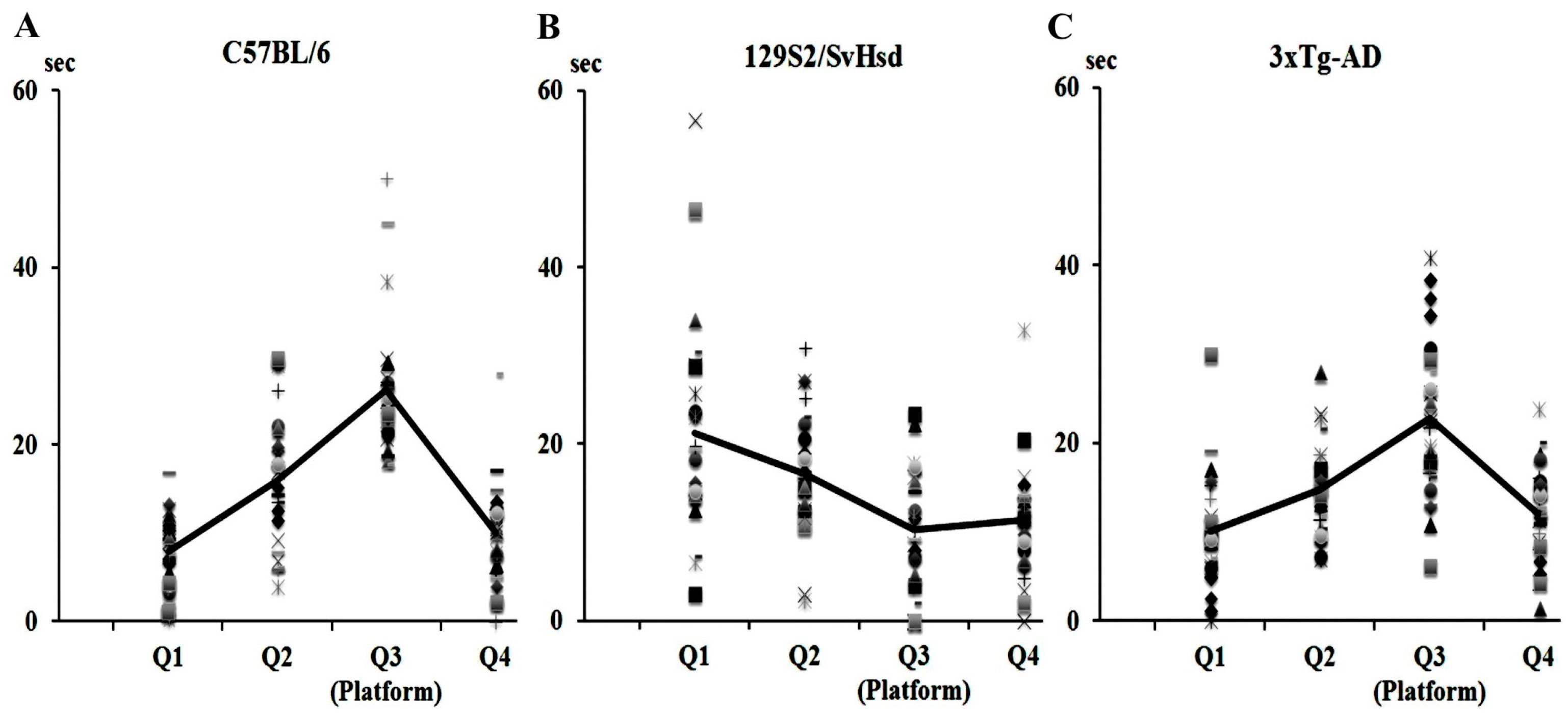
7.1. Animal Models of AD


7.2. Animal Model of Senescence
8. Stem Cells for Treating and Modeling AD
8.1. Mesenchymal Stem Cells
8.2. Neural Stem Cells and Neurogenesis
8.3. Genetically Modified Cells
8.4. iPS Cells as AD Models
9. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Brien, C. Auguste D. and Alzheimer’s disease. Science 1996, 273, 28. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Almeida, C.G.; Takahashi, R.H. Intraneuronal Aβ accumulation and origin of plaques in Alzheimer’s disease. Neurobiol. Aging 2005, 26, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Oskar Fischer and the study of dementia. Brain 2009, 132, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Maurer, K.; Volk, S.; Gerbaldo, H. Augaste D and Alzheimer’s disease. Lancet 1997, 349, 1546–1549. [Google Scholar] [CrossRef]
- Graeber, M.B.; Kosel, S.; Egensperger, R.; Banati, R.B.; Muller, U.; Bise, K.; Hoff, P.; Moller, H.J.; Fujisawa, K.; Mehraein, P. Rediscovery of the case described by Alois Alzheimer in 1911: Historical, histological and molecular genetic analysis. Neurogenetics 1997, 1, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Kosel, S.; Grasbon-Frodl, E.; Moller, H.J.; Mehraein, P. Histopathology and APOE genotype of the first Alzheimer disease patient, Auguste D. Neurogenetics 1998, 1, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Muller, U.; Winter, P.; Graeber, M.B. A presenilin 1 mutation in the first case of Alzheimer’s disease. Lancet Neurol. 2013, 12, 129–130. [Google Scholar] [CrossRef]
- Rupp, C.; Beyreuther, K.; Maurer, K.; Kins, S. A presenilin 1 mutation in the first case of Alzheimer’s disease: Revised. Alzheimers Dement. 2014, 10, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Imtiaz, B.; Tolppanen, A.M.; Kivipelto, M.; Soininen, H. Future direction in Alzheimer’s disease from risk factors to prevention. Biochem. Pharmacol. 2014, 88, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Cacquevel, M.; Aeschbach, L.; Houacine, J.; Fraering, P.C. Alzheimer’s disease-linked mutations in presenilin-1 result in a drastic loss of activity in purified γ-secretase complexes. PLoS ONE 2012, 7, e35133. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Aisen, P.S.; de Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosonal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 2011, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Westaway, D.; Xia, W.; Carlson, G.; Diehl, T.; Levesque, G.; Johnson-Wood, K.; Lee, M.; Seubert, P.; Davis, A.; et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat. Med. 1997, 3, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Selfridge, J.E.; Lezi, E.; Lu, J.; Swerdlow, R.H. Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol. Dis. 2013, 51, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Nathan, B.P.; Pitas, R.E. Apolipoprotein E. Structure, function, and possible roles in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1996, 777, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef] [PubMed]
- Beffert, U.; Stolt, P.C.; Herz, J. Functions of lipoprotein receptors in neurons. J. Lipid Res. 2004, 45, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Cedazo-Minguez, A. Apolipoprotein E and Alzheimer’s disease: Molecular mechanisms and therapeutic opportunities. J. Cell. Mol. Med. 2007, 11, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Roses, A.D. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med. 1996, 47, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. The genetics of Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2012, 107, 79–100. [Google Scholar] [PubMed]
- Slooter, A.J.; Cruts, M.; Kalmijn, S.; Hofman, A.; Breteler, M.M.; van Broeckhoven, C.; van Duijn, C.M. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: The Rotterdam study. Arch. Neurol. 1988, 55, 964–968. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; Destefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, S.; Wood, N.W. Genome-wide association studies: The key to unlocking neurodegeneration? Nat. Neurosci. 2010, 13, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Haooingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Teavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, J.; Hansmannel, F.; Gistelinck, M.; Mounier, A.; van Cauwenberghe, C.; Kolen, K.V.; Geller, F.; Sottejeau, Y.; Harold, D.; Dourlen, P.; et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating Tau pathology. Mol. Psychiatry 2013, 18, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Antúnez, C.; Boada, M.; González-Pérez, A.; Gayán, J.; Ramírez-Lorca, R.; Marín, J.; Hernández, I.; Moreno-Rey, C.; Morón, F.J.; López-Arrieta, J.; et al. The membrane-spanning 4-domains, subfamily A (MS4A) gene cluster contains a common variant associated with Alzheimer’s disease. Genome Med. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid β. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Jeng, A.T.; Nowotny, P.; Cady, J.; Cruchaga, C.; Goate, A.M. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS ONE 2012, 7, e50976. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrsquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Rogaeva, E.; Meng, Y.; Lee, J.H.; Gu, Y.; Kawarai, T.; Zou, F.; Katayama, T.; Baldwin, C.T.; Cheng, R.; Hasegawa, H.; et al. The neuronal sortilin-related SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 2007, 39, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, A.; Koike, A.; Jun, G.; Wang, L.S.; Takahashi, S.; Matsubara, E.; Kawarabayashi, T.; Shoji, M.; Tomita, N.; Arai, H.; et al. SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS ONE 2013, 8, e58618. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.; Holmans, P.A.; Hamshere, M.L.; Harold, D.; Moskvina, V.; Ivanov, D.; Pocklington, A.; Abraham, R.; Hollingworth, P.; Sims, R.; et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS ONE 2010, 5, e13950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B. Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.A.; Cardoso, I.; Goncalves, I. Key enzymes and proteins in amyloid-β production and clearance. In Alzheimer’s Disease Pathogenesis—Core Concepts, Shifting Paradigms and Therapeutic Targets; de la Monte, S., Ed.; InTech: Shanghai, China, 2011; pp. 53–86. [Google Scholar]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Hardy, J.A. Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Herrmann, F.R.; Bussiere, T.; Bouras, C.; Kovari, E.; Perl, D.P.; Mossison, J.H.; Gold, G.; Hof, P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Naslund, J.; Haroutunian, V.; Mohs, R.; Davis, K.L.; Davies, P.; Greengard, P.; Buxbaum, J.D. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA 2000, 283, 1571–1577. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Master, C.L. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Schankar, G.M.; Kukskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Bitan, G.; Fradinger, E.A.; Spring, S.M.; Teplow, D.B. Neurotoxic protein oligomers—What you see is not always what you get. Amyloid 2005, 12, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; de Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid β oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–560. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q. Alkon, D.L. Role of insulin and insulin receptor in learning and memory. Mol. Cell. Endocrinol. 2001, 177, 125–134. [Google Scholar] [CrossRef]
- Barry, A.E.; Klyubin, I.; McDonald, J.M.; Mably, A.J.; Farrell, M.A.; Scott, M.; Walsh, D.M.; Rowan, M.J. Alzheimer’s disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 2011, 31, 7259–7263. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimers Dis. 2013, 33, S67–S78. [Google Scholar] [PubMed]
- Pei, J.J.; Hugon, J. mTOR-dependent signalling in Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 2525–2532. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.K.; Verdile, G.; Bates, K.A.; Martins, R.N. Immunization in Alzheimer’s disease: Naive hope or realistic clinical potential? Mol. Psychiatry 2009, 14, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.; Arnold, S.E.; van Dyck, C.H.; Aisen, P.S.; Snider, B.J.; Porsteinsson, A.P.; Friedrich, S.; Dean, R.A.; Gonzales, C.; Sethuraman, G.; et al. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 2012, 8, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Goure, W.F.; Krafft, G.A.; Jerecic, J.; Hefti, F. Targeting the proper amyloid-β neuronal toxins: A path forward for Alzheimer’s disease immunotherapeutics. Alzheimers Res. Ther. 2014, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Alzheimer’s Disease Cooperative Study Steering Committee, Solanezumab Study Group. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Siemers, E.R.; Sundell, K.L.; Carlson, C.; Case, M.; Sethuraman, G.; Liu-Seifert, H.; Dowsett, S.A.; Pontecorvo, M.J.; Dean, R.A.; Demattos, R. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Goni, F. Immunotherapy for Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, M.C.; Brashear, H.R.; Logovinsky, V.; Ryan, J.M.; Feldman, H.H.; Siemers, E.R.; Abushakra, S.; Hartley, D.M.; Petersen, R.C.; Khachaturian, A.S.; et al. Can we prevent Alzheimer’s disease? Secondary “prevention” trials in Alzheimer’s disease. Alzheimers Dement. 2013, 9, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of Tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [PubMed]
- Caceres, A.; Kosik, K.S. Inhibition of neurite polarity by Tau antisense oligonucleotides in primary cerebellar neurons. Nature 1990, 343, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-specific functional impairments in distinct Tau isoforms of hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.L.; Cleveland, D.W. Going new places using an old MAP: Tau, microtubules and human neurodegenerative disease. Curr. Opin. Cell Biol. 2001, 13, 41–48. [Google Scholar] [CrossRef]
- Gomez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 1997, 41, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Munoz, M.J.; Kiritoshi, T.; Neugebauer, V.; Jackson, G.R.; Kayed, R. Alzheimer brain-derived Tau oligomers propagate pathology from endogenous Tau. Sci. Rep. 2012, 2, 700. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Roder, H.; Hanna, A.; Carlson, A.; Rangachari, V.; Yue, M.; Wszolek, Z.; Ashe, K.; Knight, J.; Dickson, D.; et al. Accumulation of pathological Tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 2007, 27, 3650–3662. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; Delacourte, A.; Buee, L. Tau protein as a differential biomarker of tauopathies. Biochim. Biophys. Acta 2005, 1739, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, K.C.; Lynch, T.; Pavlou, E.; Higgins, M.; Nygaard, T.G. Localization of disinhibition–dementia–parkinsonism–amyotrophy complex to 17q2-22. Am. J. Hum. Genet. 1994, 55, 1159–1165. [Google Scholar] [PubMed]
- Goedert, M. Tau gene mutations and their effects. Mov. Disord. 2005, 12, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Duff, K. Linking Aβ and Tau in late-onset Alzheimer’s disease: A dual pathway hypothesis. Neuron 2008, 60, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Perry, G. The complexities of the pathology—pathogenesis relationship in Alzheimer disease. Biochem. Pharmacol. 2014, 88, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-term effects of Aβ42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008, 372, 216–223. [Google Scholar] [CrossRef]
- Rosenmann, H.; Grigoriadis, N.; Karussis, D.; Boimel, M.; Touloumi, O.; Ovadia, H.; Abramsky, O. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal Tau protein. Arch. Neurol. 2006, 63, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Boutajangout, A.; Ingadottir, J.; Davies, P.; Sigurdsson, E.M. Passive immunization targeting pathological phospho-Tau protein in a mouse model reduces functional decline and clears Tau aggregates from the brain. J. Neurochem. 2011, 118, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Wu, S.; Murray, T.K.; Kinley, R.; Cella, C.V.; Sims, H.; Buckner, N.; Hanmer, J.; Davies, P.; O’Neill, M.J.; et al. Passive immunization with anti-Tau antibodies in two transgenic models: Reduction of Tau pathology and delay of disease progression. J. Biol. Chem. 2011, 286, 34457–34467. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Farr, S.A. The role of amyloid-β in the regulation of memory. Biochem. Pharmacol. 2014, 88, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Muller, U.C.; Zheng, H. Physiological functions of APP family proteins. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.D.; Suchowerska, A.K.; van der Hoven, J.; de Silva, D.M.; Wu, C.W.; van Eersel, J.; Ittner, A.; Ittner, L.M. Lessons from Tau-deficient mice. Int. J. Alzheimers Dis. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Dillin, A. Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta 2013, 1833, 3507–3517. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K.; Ojala, J. ER stress in Alzheimer’s disease: A novel neuronal trigger for inflammation and Alzheimer’s pathology. J. Neuroinflamm. 2009, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α-mediated stimulation of inositol 1,4,5-tirphoate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Takeda, K.; Ichijo, H. The ASK1-MAP kinase signaling in ER stress and neurodegenerative diseases. Curr. Mol. Med. 2006, 6, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, I.; Garwood, C.; Hanger, D.P.; Anderton, B.H.; Noble, W. Kinase activities increase during the development of tauopathy in htau mice. J. Neurochem. 2007, 103, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Ploia, C.; Antoniou, X.; Sclip, A.; Grande, V.; Cardinetti, D.; Colombo, A.; Canu, N.; Benussi, L.; Ghidoni, R.; Forloni, G.; et al. JNK plays a key role in Tau hyperphosphorylation in Alzheimer’s disease models. J. Alzheimers Dis. 2011, 26, 315–329. [Google Scholar] [PubMed]
- Cuanalo-Contreras, K.; Mukherjee, A.; Soto, C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Rossner, S.; Sastre, M.; Bourne, K.; Lichtenthaler, S.F. Transcriptional and translational regulation of BACE1 expression—Implications for Alzheimer’s disease. Prog. Neurobiol. 2006, 79, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Resende, R.; Moreira, P.I.; Proenca, T.; Deshpande, A.; Busciglio, J.; Pereira, C.; Oliveira, C.R. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic. Biol. Med. 2008, 44, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Morawe, T.; Hiebel, C.; Kern, A.; Behl, C. Protein homeostasis, aging and Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef] [PubMed]
- Kakimura, J.; Kitamura, Y.; Takata, K.; Umeki, M.; Suzuki, S.; Shibagaki, K.; Taniguchi, T.; Nomura, Y.; Gebicke-Haerter, P.J.; Smith, M.A.; et al. Microglial activation and amyloid-β clearance induced by exogenous heat-shock proteins. FASEB J. 2002, 16, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Koren, J., III; Jinwal, U.K.; Lee, D.C.; Jones, J.R.; Shults, C.L.; Johnson, A.G.; Anderson, L.J.; Dickey, C.A. Chaperone signalling complexes in Alzheimer’s disease. J. Cell. Mol. Med. 2009, 13, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmus, M.M.; de Waal, R.M.; Verbeek, M.M. Heat shock proteins and amateur chaperones in amyloid-β accumulation and clearance in Alzheimer’s disease. Mol. Neurobiol. 2007, 35, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; O’Leary, J.C., III; Borysov, S.I.; Jones, J.R.; Li, Q.; Koren, J., III; Abisambra, J.F.; Vestal, G.D.; Lawson, L.Y.; Johnson, A.G.; et al. Hsc70 rapidly engages Tau after microtubule destabilization. J. Biol. Chem. 2010, 285, 16798–16805. [Google Scholar] [CrossRef] [PubMed]
- Sahara, N.; Murayama, M.; Mizoroki, T.; Urushitani, M.; Imai, Y.; Takahashi, R.; Murata, S.; Tanaka, K.; Takashima, A. In vivo evidence of CHIP up-regulation attenuating Tau aggregation. J. Neurochem. 2005, 94, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Koren, J.; Zhang, Y.J.; Xu, Y.F.; Jinwal, U.K.; Birnbaum, M.J.; Monks, B.; Sun, M.; Cheng, J.Q.; Patterson, C.; et al. Akt and CHIP coregulate Tau degradation through coordinated interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 3622–3627. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Dotti, C.G.; Ledesma, M.D. Brain cholesterol in normal and pathological aging. Biochim. Biophys. Acta 2010, 1801, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Eckel, R.H. What are lipoproteins doing in the brain? Trends Endocrinol. Metab. 2014, 25, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.G.; Schroeder, F.; Igbavboa, U.; Avdulov, N.A.; Chochina, S.V. Brain membrane cholesterol domains, aging and amyloid β-peptides. Neurobiol. Aging 2002, 23, 685–694. [Google Scholar] [CrossRef]
- Ariga, T.; Wakade, C.; Yu, R.K. The pathological roles of ganglioside metabolism in Alzheimer’s disease: Effects of gangliosides on neurogenesis. Int. J. Alzheimers Dis. 2011, 2011, 193618. [Google Scholar] [CrossRef] [PubMed]
- Kakio, A.; Nishimoto, S.I.; Yanagisawa, K.; Kazutsumi, Y.; Matsuzaki, K. Cholesterol-dependent formation on GM1 ganglioside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J. Biol. Chem. 2001, 276, 24985–24990. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N.; Garmy, N. Cholesterol accelerates the binding of Alzheimer’s β-amyloid peptide to ganglioside GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front. Physiol. 2013, 4, 120. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Yanagisawa, K. Sphingomyelin accumulation provides a favorable milieu for GM1 ganglioside-induced assembly of amyloid β-protein. Neurosci. Lett. 2010, 481, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Hanzal-Bayer, M.F.; Hancock, J.F. Lipid rats and membrane traffic. FEBS Lett. 2007, 581, 2098–2104. [Google Scholar] [CrossRef] [PubMed]
- Cordy, J.M.; Hooper, N.M.; Turner, A.J. The involvement of lipid rafts in Alzheimer’s disease. Mol. Membr. Biol. 2006, 23, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef] [PubMed]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.A.; Nalivaeva, N.N.; Turner, A.J. Lipid rafts and Alzheimer’s disease: Protein–lipid interactions and perturbation of signaling. Front. Physiol. 2012, 3, 189. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Kuchenbecker, J.; Rothhaar, T.L.; Grosgen, S.; Hundsdorfer, B.; Burg, V.K.; Friess, P.; Muller, U.; Grimm, H.S.; Riemenschneider, M.; et al. Plasmalogen synthesis is regulated via alkyl-dihydorxyacetonephosphate-synthase by amyloid precursor protein processing and is affected in Alzheimer’s disease. J. Neurochem. 2011, 116, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Rothhaar, T.L.; Grosgen, S.; Haupenthal, V.J.; Burg, V.K.; Hundsdorfer, B.; Mett, J.; Riemenschneider, M.; Grimm, H.S.; Hartmann, T.; Grimm, M.O. Plasmalogens inhibit APP processing by directly affecting γ-secretase activity in Alzheimer’s disease. Sci. World J. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, K.; Vogt, D.L.; Liang, M.; Shen, Y.; Lamb, B.T.; Pimplikar, S.W. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18367–18372. [Google Scholar] [PubMed]
- Fabelo, N.; Marin, V.; Marin, R.; Moreno, D.; Ferrer, I.; Diaz, M. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol. Aging 2014, 35, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Geekiyanage, H.; Upadhye, A.; Chan, C. Inhibition of serine palmitoyltransferase reduces Aβ and Tau hyperphosphorylation in a murine model: A safe therapeutic strategy for Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2037–2051. [Google Scholar] [PubMed]
- Riemann, D.; Hansen, G.H.; Niels-Christiansen, L.L.; Thorsen, E.; Immerdal, L.; Santos, A.N.; Kehlen, A.; Langner, J.; Danielsen, E.M. Caveolae/lipid rafts in fibroblast-like synoviocytes: Ectopeptidase-rich membrane microdomains. Biochem. J. 2001, 354, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated Tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Bulloj, A.; Leal, M.C.; Surace, E.I.; Zhang, X.; Xu, H.; Ledesma, M.D.; Castano, E.M.; Morelli, L. Detergent resistant membrane-associated IDE in brain tissue and cultured cells: Relevance to Aβ and insulin degradation. Mol. Neurodegener. 2008, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Parkin, E.T.; Watt, N.T.; Hussain, I.; Eckman, E.A.; Eckman, C.B.; Manson, J.C.; Baybutt, H.N.; Turner, A.J.; Hooper, N.M. Cellular prion protein regulates β-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2007, 104, 11062–11067. [Google Scholar] [CrossRef] [PubMed]
- Prybylowski, K.; Chang, K.; Sans, N.; Kan, L.; Vicini, S.; Wenthold, R.J. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 2005, 47, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Strittmatter, S.M. Amyloid-β induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion 2013, 7, 37–41. [Google Scholar] [CrossRef] [PubMed]
- De Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of Tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Ronicke, R.; Mikhaylova, M.; Ronicke, S.; Meinhardt, J.; Schroder, U.H.; Fandrich, M.; Reiser, G.; Kreutz, M.R.; Reymann, K.G. Early neuronal dysfunction by amyloid β oligomers depends on activation of NR2B-cnotaining NMDA receptors. Neurobiol. Aging 2011, 32, 2219–2228. [Google Scholar] [CrossRef] [PubMed]
- Williamson, R.; Usardi, A.; Hanger, D.P.; Anderton, B.H. Membrane-bound β-amyloid oligomers are recruited into lipid rafts by a fyn-dependent mechanism. FASEB J. 2008, 22, 1552–1559. [Google Scholar] [CrossRef] [PubMed]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Lauren, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Calella, A.M.; Farinelli, M.; Nuvolone, M.; Mirante, O.; Moos, R.; Falsig, J.; Mansuy, I.M.; Aguzzi, A. Prion protein and Aβ-related synaptic toxicity impairment. EMBO Mol. Med. 2010, 2, 306–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, W.; Lee, H.P.; Zou, W.Q.; Wang, X.; Perry, G.; Zhu, X.; Smith, M.A.; Petersen, R.B.; Lee, H.G. Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum. Mol. Genet. 2012, 21, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, A.J.; Panico, S.; Freir, D.B.; Wright, D.; Terry, C.; Risse, E.; Herron, C.E.; O’Malley, T.; Wadsworth, J.D.; Farrow, M.A.; et al. Amyloid-β nanotubes are associated with prion protein-dependent synaptotoxicity. Nat. Commun. 2013, 4, 2416. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.; Lee, G.; Sjoberg, M.; Maccioni, R.B. Tau phosphorylation by Cdk5 and Fyn in response to amyloid peptide Abera (25–35): Involvement of lipid rafts. J. Alzheimers Dis. 2009, 16, 149–156. [Google Scholar] [PubMed]
- Sui, Z.; Kovacs, A.D.; Maggirwar, S.B. Recruitment of active glycogen synthase kinase-3 into neuronal lipid rafts. Biochem. Biophys. Res. Commun. 2006, 345, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, M.; Dudek, H.; Kwon, Y.T.; Ramos, Y.F.; Tsai, L.H. The Cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996, 10, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Ishiguro, K.; Hisanaga, S. Physiological and pathological phosphorylation of Tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Skuntz, S.; Pant, H.C. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res. 2012, 43, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Mazanetz, M.P.; Fischer, P.M. Untangling Tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 2007, 6, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Thangavel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of Tau by fyn: Implications of Alzheimer’s disease. J. Neurosci. 2004, 24, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Yen, S.H.; Lee, G. Disease-related modifications in Tau affect the interaction between Fyn and Tau. J. Biol. Chem. 2005, 280, 35119–35125. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of Tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Usardi, A.; Pooler, A.M.; Seereeram, A.M.; Reynolds, C.H.; Derkinderen, P.; Anderton, B.; Hanger, D.P.; Noble, W.; Williamson, R. Tyrosine phosphorylation of Tau regulates its interactions with Fyn SH2 domains, but not SH3 domains, altering the cellular localization of tau. FEBS J. 2011, 278, 2927–2937. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Shi, J.; Tranimukai, H.; Gu, J.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Reduced O-GlcNAcylation links lower brain glucose metabolism and Tau pathology in Alzheimer’s disease. Brain 2009, 132, 1820–1832. [Google Scholar] [CrossRef] [PubMed]
- Velliquette, R.A.; O’Connor, T.; Vassar, R. Energy inhibition elevates β-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: Possible early events in Alzheimer’s disease pathogenesis. J. Neurosci. 2005, 25, 10874–10883. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [PubMed]
- Ankarcrona, M.; Mangialasche, F.; Winblad, B. Rethinking Alzheimer’s disease therapy: Are mitochondria the key? J. Alzheimers Dis. 2010, 20, S579–S590. [Google Scholar] [PubMed]
- Jacobsen, K.T.; Iverfeldt, K. O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-β precursor protein (APP). Biochem. Biophys. Res. Commun. 2011, 404, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Paterson, A.J.; Zhang, F.; McAndrew, J.; Fukuchi, K.; Wyss, J.M.; Peng, L.; Hu, Y.; Kudlow, J.E. Accumulation of protein O-GlcNAc modification inhibits proteasomes in the brain and coincides with neuronal apoptosis in brain areas with high O-GlcNAc metabolism. J. Neurochem. 2004, 89, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Brister, M.A.; Pandey, A.K.; Bielska, A.A.; Zondlo, N.J. OGlcNAcylation and phosphorylation have opposing structural effects in Tau: Phosphothreonine induces particular conformational order. J. Am. Chem. Soc. 2014, 136, 3803–3816. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Iqbal, K.; Grundke-Igbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of Tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [PubMed]
- Tallent, M.K.; Varghis, N.; Skorobogatko, Y.; Hernandez-Cuebas, L.; Whelan, K.; Vocadlo, D.J.; Vosseller, K. In vivo modulation of O-GlcNAc levels regulates hippocampal synaptic plasticity through interplay with phosphorylation. J. Biol. Chem. 2009, 284, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adh. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Pocernich, C.B.; Butterfield, D.A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Biophys. Acta 2012, 1822, 625–630. [Google Scholar] [PubMed]
- Liu, R.; Choi, J. Age-associated decline in γ-glutamylcysteine synthetase gene expression in rats. Free Radic. Biol. Med. 2000, 28, 566–574. [Google Scholar] [CrossRef]
- Saharan, S.; Mandal, P.K. The emerging role of glutathione in Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 519–529. [Google Scholar] [PubMed]
- Lee, M.; Cho, T.; Jantaratnotai, N.; Wang, Y.T.; McGeer, E.; McGeer, P.L. Depletion of GSH in glial cells induces neurotoxicity: Relevance to aging and degenerative neurological diseases. FASEB J. 2010, 24, 2533–2545. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Chen, Y.; Liu, H.; Zhang, K.; Zhang, T.; Lin, A.; Jing, N. Hydrogen peroxide promotes Aβ production through JNK-dependent activation of γ-secretase. J. Biol. Chem. 2008, 283, 17721–17730. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, M.; Tangpong, J.; Keller, J.N.; Murphy, M.P.; Markesbery, W.R.; Kiningham, K.K., St.; Clair, D.K. β-amyloid mediated nitration of manganese superoxide dismutase: Implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am. J. Pathol. 2006, 168, 1608–1617. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Atwood, C.S.; Scarpa, R.C.; Huang, X.; Moir, R.D.; Jones, W.D.; Fairlie, D.P.; Tanzi, R.E.; Bush, A.I. Characterization of copper interactions with Alzheimer amyloid β peptides: Identification of an attomolar-affinity copper binding site on amyloid β1-42. J. Neurochem. 2000, 75, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Cuajungco, M.P.; Atwood, C.S.; Hartshorn, M.A.; Tyndall, J.D.; Hanson, G.R.; Stokes, K.C.; Leopold, M.; Multhaup, G.; Goldstein, L.E.; et al. Cu(II) potentiation of Alzheimer Aβ neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 1999, 274, 37111–37116. [Google Scholar] [CrossRef] [PubMed]
- Al-Hilaly, Y.K.; Williams, T.L.; Stewart-Parker, M.; Ford, L.; Skaria, E.; Cole, M.; Bucher, W.G.; Morris, K.L.; Sada, A.A.; Thorpe, J.R.; et al. A central role for dityrosine crosslinking of amyloid-β in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 83. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J. Neurosci. 1998, 18, 8126–8132. [Google Scholar] [PubMed]
- Cuajungco, M.P.; Goldstein, L.E.; Nunomura, A.; Smith, M.A.; Lim, J.T.; Atwood, C.S.; Huang, X.; Farrag, Y.W.; Perry, G.; Bush, A.I. Evidence that the β-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of Aβ by zinc. J. Biol. Chem. 2000, 275, 19439–19442. [Google Scholar] [CrossRef] [PubMed]
- Craddock, T.J.; Tuszynski, J.A.; Chopra, D.; Casey, N.; Goldstein, L.E.; Hameroff, S.R.; Tanzi, R.E. The zinc dyshoemostasis hypothesis of Alzheimer’s disease. PLoS ONE 2012, 7, e33552. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Munch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Valente, T.; Gella, A.; Fernandez-Busquets, X.; Unzeta, M.; Durany, N. Immunohistochemical analysis of human brain suggests pathological synergism of Alzheimer’s disease and diabetes mellitus. Neurobiol. Dis. 2010, 37, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Son, S.M.; Jin, S.M.; Hong, H.S.; Shin, D.H.; Kim, S.J.; Huh, K.; Mook-Jung, I. RAGE regulates BACE1 and Aβ generation via NFAT1 activation in Alzheimer’s disease animal model. FASEB J. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Bierhaus, A.; Nawroth, P.P.; Stern, D.M. RAGE and Alzheimer’s disease: A progression factor for amyloid-β-induced cellular perturbation? J. Alzheimers Dis. 2009, 16, 833–843. [Google Scholar] [PubMed]
- Martel, C.L.; Mackic, J.B.; McComb, J.G.; Ghiso, J.; Zlokovic, B.V. Blood–brain barrier uptake of the 40 and 42 amino acid sequences of circulating Alzheimer’s amyloid β in guinea pigs. Neurosci. Lett. 1996, 206, 157–160. [Google Scholar] [CrossRef]
- Deane, R.; du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumlation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Manolopoulos, K.N.; Klotz, L.O.; Korsten, P.; Bornstein, S.R.; Barthel, A. Linking Alzheimer’s disease to insulin resistance: The FoxO response to oxidative stress. Mol. Psychiatry 2010, 15, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Hardas, S.S.; Sultana, R.; Clark, A.M.; Beckett, T.L.; Szweda, L.I.; Murphy, M.P.; Butterfield, D.A. Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol. 2013, 1, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Siegel, S.J.; Bieschke, J.; Powers, E.T.; Kelly, J.W. The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry 2007, 46, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R.; Lovell, M.A. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- Schulingkamp, R.J.; Pagano, T.C.; Hung, D.; Raffa, R.B. Insulin receptors and insulin action in the brain: Review and clinical implications. Neurosci. Biobehav. Rev. 2000, 24, 855–872. [Google Scholar] [CrossRef]
- Dore, S.; Kar, S.; Quirion, R. Insulin-like growth factor I protects and rescues hippocampal neurons against β-amyloid- and human amylin-induced toxicity. Proc. Natl. Acad. Sci. USA 1997, 94, 4772–4777. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.M.; Torres-Aleman, I. The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.; Rydzewski, B.; Baker, S.P.; Raizada, M.K. The cellular and physiological actions of insulin in the central nervous system. Neurochem. Int. 1993, 22, 1–10. [Google Scholar] [CrossRef]
- Sesti, G.; Federici, M.; Hribal, M.L.; Lauro, D.; Sbraccia, P.; Lauro, R. Defects of the isnulin receptor substrate (IRS) system in human metabolic disorders. FASEB J. 2001, 15, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. Insulin signaling in health and disease. Science 2003, 302, 1710–1711. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Brazil, D.P.; Burks, D.J.; Kushner, J.A.; Ye, J.; Flint, C.L.; Farhang-Fallah, J.; Dikkes, P.; Warot, X.M.; Rio, C.; et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes Tau phosphorylation. J. Neurosci. 2003, 23, 7084–7092. [Google Scholar] [PubMed]
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Kustermann, E.; et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.M.; Tseng, V.; Wang, J.; Wang, D.; Matyakhina, L.; Bondy, C.A. Tau is hyperphosphorylated in the insulin-like growth factor-I null brain. Endocrinology 2005, 146, 5086–5091. [Google Scholar] [CrossRef] [PubMed]
- Killick, R.; Scales, G.; Leroy, K.; Causevic, M.; Hooper, C.; Irvine, E.E.; Choudhury, A.I.; Drinkwater, L.; Kerr, F.; Al-Qassab, H.; et al. Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochem. Biophys. Res. Commun. 2009, 386, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neil, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Cohen, P. Mechanism of activation and function of protein kinase B. Curr. Opin. Genet. Dev. 1998, 8, 55–62. [Google Scholar] [CrossRef]
- Frolich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, B.S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Turk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, V.; Werner, E.D.; Giraud, J.; Lee, Y.H.; Shoelson, S.E.; White, M.F. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 2002, 277, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Paz, K.; Hemi, R.; LeRoith, D.; Karasik, A.; Elhanany, E.; Kanety, H.; Zick, Y. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 29911–29918. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement. 2014, 10, S26–S32. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; di Domenico, F.; Barone, E. Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochim. Biophys. Acta 2014, 1842, 1693–1706. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.T.; Chen, M.; Dufour, F.; Alkon, D.L.; Zhao, W.Q. Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn. Mem. 2005, 12, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Cole, G.M.; Frautschy, S.A. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer’s disease. Exp. Gerontol. 2007, 42, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Lacor, P.N.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric Aβ. J. Biol. Chem. 2009, 284, 18742–18753. [Google Scholar] [CrossRef] [PubMed]
- Vekrellis, K.; Ye, Z.; Qiu, W.Q.; Walsh, D.; Hartley, D.; Chesneau, V.; Rosner, M.R.; Selkoe, D.J. Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 2000, 20, 1657–1665. [Google Scholar] [PubMed]
- Zhao, L.; Teter, B.; Morihara, T.; Lim, G.P.; Ambegaokar, S.S.; Ubeda, O.J.; Frautschy, S.A.; Cole, G.M. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: Implications for Alzheimer’s disease intervention. J. Neurosci. 2004, 24, 11120–11126. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H., II; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-β in memory-impaired older adults. J. Alzheimers Dis. 2008, 13, 323–331. [Google Scholar] [PubMed]
- Cook, D.G.; Leverenz, J.B.; McMillan, P.J.; Kulstad, J.J.; Ericksen, S.; Roth, R.A.; Schellenberg, G.D.; Jin, L.W.; Kovacina, K.S.; Craft, S. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with the apolipoprotein E-epsiron4 allele. Am. J. Pathol. 2003, 162, 313–319. [Google Scholar] [CrossRef]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Derecki, N.C.; Katzmarski, N.; Kipnis, J.; Meyer-Luehmann, M. Microglia as a critical player in both developmental and late-life CNS pathogenesis. Acta Neuropathol. 2014, 128, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Ekdahl, C.T.; Claasen, J.H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Ron, N.; Butovsky, O.; Landa, G.; Greenberg, N.; Cohen, H.; Kipnis, J.; Schwartz, M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 2006, 9, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S. Interleukin-1 mediates pathological effects of microglia on Tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [PubMed]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [PubMed]
- Hatori, H.; Nagai, A.; Heisel, R.; Ryu, J.K.; Kim, S.U. Fractalkine and fractalkine receptors in human neurons and glial cells. J. Neurosci. Res. 2002, 69, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Re, D.B.; Przedborski, S. Fractalkine: Moving from chemotaxis to neuroprotection. Nat. Neurosci. 2006, 9, 859–861. [Google Scholar] [PubMed]
- Lyons, A.; Lynch, A.M.; Downer, E.J.; Hanley, R.; O’Sullivan, J.B.; Smith, A.; Lynch, M.A. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attenuates microglial activation in vivo and in vitro. J. Neurochem. 2009, 110, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Deiva, K.; Geeraerts, T.; Salim, H.; Leclerc, P.; Hery, C.; Hugel, B.; Freyssinet, J.M.; Tardieu, M. Fractalkine reduces N-methyl-d-aspartate-induced calcium flux and apoptosis in human neurons through extracellular signal-regulated kinase activation. Eur. J. Neurosci. 2004, 20, 3222–3232. [Google Scholar] [CrossRef] [PubMed]
- Limatola, C.; Lauro, C.; Catalano, M.; Ciotti, M.T.; Bertollini, C.; di Angelantonio, S.; Ragozzino, D.; Eusebi, F. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J. Neuroimmunol. 2005, 166, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Lim, H.K.; Lee, J.Y.; Kim, D.J.; Park, S.; Lee, C.; Lee, C.U. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2008, 436, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Sun, B.; Zhou, Y.; Kauppinen, T.M.; Halabisky, B.; Wes, P.; Ransohoff, R.M.; Gan, L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 32713–32722. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; Downer, E.J.; Crotty, S.; Nolan, Y.M.; Mills, K.H.; Lynch, M.A. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: A role for IL-4. J. Neurosci. 2007, 27, 8309–8313. [Google Scholar] [CrossRef] [PubMed]
- Hernangomez, M.; Carrillo-Salinas, F.J.; Mecha, M.; Correa, F.; Mestre, L.; Loria, F.; Feliu, A.; Docagne, F.; Guaza, C. Brain innate immunity in the regulation of neuroinflammation: Therapeutic strategies by modulating CD200-CD200R interaction involve the cannabinoid system. Curr. Pharm. Des. 2014, 20, 4707–4722. [Google Scholar] [CrossRef] [PubMed]
- Cox, F.F.; Carney, D.; Miller, A.M.; Lynch, M.A. CD200 fusion protein decreases microglial activation in the hippocampus of aged rats. Brain Behav. Immun. 2012, 26, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Barrientos, R.M.; Biedenkapp, J.C.; Rudy, J.W.; Watkins, L.R.; Maier, S.F. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol. Aging 2006, 27, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Dalsing-Hernandez, J.E.; Campbell, N.A.; Lue, L.F. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: A potential mechanism leading to chronic inflammation. Exp. Neurol. 2009, 215, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Costello, D.A.; Lyons, A.; Denieffe, S.; Browne, T.C.; Cox, F.F.; Lynch, M.A. Long term potentiation is impaired in membrane glycoprotein CD200-deficient mice: A role for Toll-like receptor activation. J. Biol. Chem. 2011, 286, 34722–34732. [Google Scholar] [CrossRef] [PubMed]
- Okun, E.; Mattson, M.P.; Arumugam, T.V. Involvement of Fc receptors in disorders of the central nervous system. Neuromol. Med. 2010, 12, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The function of Fcγ receptors in dendritic cells and macrophages. Nat. Rev. Immunol. 2014, 14, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, S.; Larionov, S.; Wang, Y.; Dannenberg, H.; Matozaki, T.; Monsonego, A.; Thal, D.R.; Neumann, H. Signal regulatory protein-β1: A microglial modulator of phagocytosis in Alzheimer’s disease. Am. J. Pathol. 2009, 175, 2528–2539. [Google Scholar] [CrossRef] [PubMed]
- Doens, D.; Fernandez, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Ye, Z.; Kholodenko, D.; Seubert, P.; Selkoe, D.J. Degradation of amyloid β-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J. Biol. Chem. 1997, 272, 6641–6646. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.A.; Turner, A.J. β-amyloid catabolism: Roles for neprilysin (NEP) and other metallopeptidases? J. Neurochem. 2002, 81, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, M.; Miyamoto, K.; Wu, Z.; Nakanishi, H. Possible involvement of cathepsin B released by microglia in methylmercury-induced cerebellar pathological changes in the adult rat. Neurosci. Lett. 2008, 442, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Lucin, K.M.; O’Brien, C.E.; Bieri, G.; Czirr, E.; Mosher, K.I.; Abbey, R.J.; Mastroeni, D.F.; Rogers, J.; Spencer, B.; Masliah, E.; et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 2013, 79, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Vande Walle, L.; Kanneganti, T.D. Deregulated inflammasome signaling in disease. Immunol. Rev. 2011, 243, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht-Talboo, J.; Landreth, G.E. Microglial phagocytosis induced by fibrillar β-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 2005, 25, 8240–8249. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, E.A.; Amiry-Moghaddam, M.; Bergersen, L.H.; Bjaalie, J.G.; Eriksson, J.; Gundersen, V.; Leergaard, T.B.; Morth, J.P.; Storm-Mathisen, J.; Trop, R.; et al. The glia doctorine: Addressing the role of glial cells in healthy brain ageing. Mech. Ageing Dev. 2013, 134, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.; Kim, S.U. Human astrocytes: Secretome profiles of cytokines and chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef] [PubMed]
- Sykova, E. Glial diffusion barriers during aging and pathological states. Prog. Brain Res. 2001, 132, 339–363. [Google Scholar] [PubMed]
- Vargova, L.; Sykova, E. Astrocytes and extracellular matrix in extrasynaptic volume transmission. Philos. Trans. R. Soc. Lond. B 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.A.; Ying, W.; Kauppinen, T.M. Astrocyte influences on ischemic neuronal death. Curr. Mol. Med. 2004, 4, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Wilhelmsson, U.; Bogestal, Y.R.; Pekna, M. The role of astrocytes and complement system in neural plasticity. Int. Rev. Neurobiol. 2007, 82, 95–111. [Google Scholar] [PubMed]
- Apelt, J.; Schliebs, R. Β-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001, 894, 21–30. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Pocernich, C.B. The glutamatergic system and Alzheimer’s disease: Therapeutic implications. CNS Drugs 2003, 17, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Lauderback, C.M.; Hackett, J.M.; Huang, F.F.; Keller, J.N.; Szweda, L.I.; Markesbery, W.R.; Butterfield, D.A. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: The role of Aβ1-42. J. Neurochem. 2001, 78, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Alford, M.; de Teresa, R.; Mallory, M.; Hansen, L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Magnus, T.; Jung, S. Autoantigen specific T cells inhibit glutamate uptake in astrocytes by decreasing expression of astrocytic glutamate transporter GLAST: A mechanism mediated by tumor necrosis factor-α. FASEB J. 2005, 19, 1878–1880. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Wang, Y.X.; Dou, F.F.; Lu, H.Z.; Ma, Z.W.; Lu, P.H.; Xu, X.M. Glutamine synthetase down-regulation reduces astrocyte protection against glutamate excitotoxicity to neurons. Neurochem. Int. 2010, 56, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodriguez, J.J. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: Mechanism of deficient glutamategic transmission? Mol. Neurodegener. 2011, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Kulijewicz-Nawrot, M.; Verkhratsky, A.; Chvatal, A.; Sykova, E.; Rodriguez, J.J. Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer’s disease. J. Anat. 2012, 221, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, T.J.; Spitzer, P.; Klafki, H.W.; Linning, P.; Neff, F.; Knolker, H.J.; Lewczuk, P.; Wiltfang, J.; Kornhuber, J.; Maler, J.M. Astrocytes and microglia but not neurons preferentially generate N-terminally truncated Aβ peptides. Neurobiol. Dis. 2015, 73, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.F.; Turk, J.; et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-β peptide catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef] [PubMed]
- Kanemitsu, H.; Tomiyama, T.; Mori, H. Human neprilysin is capable of degrading amyloid β peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci. Lett. 2003, 350, 113–116. [Google Scholar] [CrossRef]
- Eckman, E.A.; Eckman, C.B. Aβ-degrading enzymes: Modulators of Alzheimer’s disease pathogenesis and targets for therapeutic intervention. Biochem. Soc. Trans. 2005, 33, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Elshourbagy, N.A.; Liao, W.S.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc. Natl. Acad. Sci. USA 1985, 82, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Pitas, R.E.; Boyles, J.K.; Lee, S.H.; Foss, D.; Mahley, R.W. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta 1987, 917, 148–161. [Google Scholar] [CrossRef]
- Nakai, M.; Kawamata, T.; Taniguchi, T.; Maeda, K.; Tanaka, C. Expression of apolipoprotein E mRNA in rat microglia. Neurosci. Lett. 1996, 211, 41–44. [Google Scholar] [CrossRef]
- Hatters, D.M.; Peters-Libeu, C.A.; Weisgraber, K.H. Apolipoprotein E structure: Insights into function. Trends Biochem. Sci. 2006, 31, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Aβ in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and role in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Basak, J.M.; Verghese, P.B.; Yoon, H.; Kim, J.; Hotzman, D.M. Low-density lipoprotein receptor represent an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. J. Biol. Chem. 2012, 287, 13959–13971. [Google Scholar] [CrossRef] [PubMed]
- Myung, N.H.; Zhu, X.; Kruman, I.I.; Castellani, R.J.; Petersen, R.B.; Siedlak, S.L.; Perry, G.; Smith, M.A.; Lee, H.G. Evidence of DNA damage in Alzheimer disease: Phosphorylation of histone H2AX in astrocytes. Age 2008, 30, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; van Eldik, L.; et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Rohn, T.T.; Vyas, V.; Hernandez-Estrada, T.; Nichol, K.E.; Christie, L.A.; Head, E. Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J. Neurosci. 2008, 28, 3051–3059. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Montkowski, A.; Poetting, M.; Mederer, A.; Holsboer, F. Behavioural performance in three substrains of mouse strain 129. Brain Res. 1997, 762, 12–18. [Google Scholar] [CrossRef]
- Balogh, S.A.; McDowell, C.S.; Stavnezer, A.J.; Denenberg, V.H. A behavioral and neuroanatomical assessment of an inbred substrain of 129 mice with behavioral comparisons to C57BL/6J mice. Brain Res. 1999, 836, 38–48. [Google Scholar] [CrossRef]
- Gerlai, R. Gene-targeting studies of mammalian behavior: Is it the mutation or the background genotype? Trends Neurosci. 1996, 19, 177–181. [Google Scholar] [CrossRef]
- Wolfer, D.P.; Muller, U.; Stagliar, M.; Lipp, H.P. Assessing the effects of the 129/Sc genetic background on swimming navigation learning in transgenic mutants: A study using mice with a modified β-amyloid precursor protein gene. Brain Res. 1997, 771, 1–13. [Google Scholar] [CrossRef]
- Takeda, T. Senescence-accelerated mouse (SAM) with special references to neurodegeneration models, SAMP8 and SAMP10 mice. Neurochem. Res. 2009, 34, 639–659. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Qiang, J.; Gu, P.; Wang, Y.; Geng, Y.; Wang, M. Age-related autophagy alterations in the brain of senescence accelerated mouse prone 8 (SAMP8) mice. Exp. Gerontol. 2011, 46, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Garcia, O.; Vega-Naredo, I.; Sierra, V.; Caballero, B.; Tomas-Zapico, C.; Camins, A.; Garcia, J.J.; Pallas, M.; Coto-Montes, A. Elevated oxidative stress in the brain of senescence-accelerated mice at 5 months of age. Biogerontology 2006, 7, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Farr, S.A.; Kumar, V.B.; Armbrecht, H.J. The SAMP8 mouse: A model to develop therapeutic interventions for Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Inaba, M.; Guo, K.; Abraham, N.G.; Ikehara, S. Amelioration of cognitive ability in senescence-accelerated mouse prone 8 (SAMP8) by intra-bone marrow-bone marrow transplantation. Neurosci. Lett. 2009, 465, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Rutten, B.P.; Kenis, G.; Peerbooms, O.; Visser, P.J.; Verhey, F.; van Os, J.; Steinbusch, H.W.; van den Hove, D.L. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Prog. Neurobiol. 2010, 90, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, J.K.; Lee, H.; Carter, J.E.; Chang, J.W.; Oh, W.; Yang, Y.S.; Suh, J.G.; Lee, B.H.; Jin, H.K.; et al. Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol. Aging 2012, 33, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Shin, J.Y.; Lee, B.R.; Kim, H.O.; Lee, P.H. Mesenchymal stem cells augment neurogenesis in the subventricular zone and enhance differentiation of neural precursor cells into dopaminergic neurons in the substantia nigra of a parkinsonian model. Cell Transpl. 2012, 21, 1629–1640. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Morales, P.L.; Revilla, A.; Ocana, I.; Gonzalez, C.; Sainz, P.; McGuire, D.; Liste, I. Progress in stem cell therapy for major human neurological disorders. Stem Cell Rev. 2013, 9, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Forostyak, S.; Homola, A.; Turnovcova, K.; Svitil, P.; Jendelova, P.; Sykova, E. Intrathecal delivery of mesenchymal stromal cells protects the structure of altered perineuronal nets in SOD1 rats and amends the course of ALS. Stem Cells 2014, 32, 3163–3172. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Vercelli, A.; Ferrero, I.; Boido, M.; Cantello, R.; Fagioli, F. Transplantation of mesenchymal stem cells in ALS. Prog. Brain Res. 2012, 201, 333–359. [Google Scholar] [PubMed]
- Hayashi, T.; Wakao, S.; Kitada, M.; Ose, T.; Watabe, H.; Kuroda, Y.; Mitsunaga, K.; Matsuse, D.; Shigemoto, T.; Ito, A.; et al. Autologous mesenchymal stem cell-derived dopaminergic neurons function in parkinsonian macaques. J. Clin. Investig. 2013, 123, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Glavaski-Joksimovic, A.; Bohn, M.C. Mesenchymal stem cells and neuroregeneration in Parkinson’s disease. Exp. Neurol. 2013, 247, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.; Eichler, H.; Stoeve, J.; Kluter, H.; Bieback, K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006, 24, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Kassem, M. Abdallah, B.M. Human bone-marrow-derived mesenchymal stem cells: Biological characteristics and potential role in therapy of degenerative diseases. Cell Tissue Res. 2008, 331, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, Y.; Nakajima, H.; Sugiyama, D.; Hirose, I.; Kitamura, T.; Tsuji, K. Human placenta-derived cells have mesenchymal stem/progenitor cell potential. Stem Cells 2004, 22, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Wyse, R.D.; Dunbar, G.L.; Rossignol, J. Use of genetically modified mesenchymal stem cells to treat neurodegenerative diseases. Int. J. Mol. Sci. 2014, 15, 1719–1745. [Google Scholar] [CrossRef] [PubMed]
- Harting, M.T.; Jimenez, F.; Xue, H.; Fischer, U.M.; Baumgartner, J.; Dash, P.K.; Cox, C.S. Intravenous mesenchymal stem cell therapy for traumatic brain injury. J. Neurosurg. 2009, 110, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Danielyan, L.; Schafer, R.; von Ameln-Mayerhofer, A.; Buadze, M.; Geisler, J.; Klopher, T.; Burkhardt, U.; Proksch, B.; Verleysdonk, S.; Ayturan, M.; et al. Intranasal delivery of cells to the brain. Eur. J. Cell Biol. 2009, 88, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.; Samdani, A.F.; Betz, R.R.; Fischer, I.; Neuhuber, B. Grafting of human bone marrow stromal cells into spinal cord injury: A comparison of delivery methods. Spine 2009, 34, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Urdzikova, L.M.; Ruzicka, J.; LaBagnara, M.; Karova, K.; Kubinova, S.; Jirakova, K.; Murali, R.; Sykova, E.; Jhanwar-Uniyal, M.; Jendelova, P. Human mesenchymal stem cells modulate inflammatory cytokines after spinal cord injury in rat. Int. J. Mol. Sci. 2014, 15, 11275–11293. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.M.; Kim, H.S.; Park, K.R.; Shin, J.M.; Kang, A.R.; Lee, K.; Song, S.; Kim, Y.B.; Han, S.B.; Chung, H.M.; et al. Placenta-derived mesenchymal stem cells improve memory dysfunction in an Aβ1-42-induced mouse model of Alzheimer’s disease. Cell Death Dis. 2013, 4, e958. [Google Scholar] [CrossRef] [PubMed]
- Ikehara, S.; Li, M. Stem cell transplantation improves aging-related disease. Front. Cell Dev. Biol. 2014, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jin, H.K.; Bae, J.S. Bone marrow-derived mesenchymal stem cells reduce brain amyloid-β deposition and accelerate the activation of microglia in an acutely induced Alzheimer’s disease mouse model. Neurosci. Lett. 2009, 450, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jin, H.K.; Endo, S.; Schuchman, E.H.; Carter, J.E.; Bae, J.S. Intracerebral transplantation of bone marrow-derived mesenchymal stem cells reduces amyloid-β deposition and rescues memory deficits in Alzheimer’s disease mice by modulation of immune responses. Stem Cells 2010, 28, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Talpalar, A.E.; Ben-Yaakov, K.; Schwartz, M. Activation of microglia by aggregated β-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-γ and IL-4 render them protective. Mol. Cell. Neurosci. 2005, 29, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Koronyo-Hamaoui, M.; Kunis, G.; Ophir, E.; Landa, G.; Cohen, H.; Schwartz, M. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expression insulin-like growth factor 1. Proc. Natl. Acad. Sci. USA 2006, 103, 11784–11789. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, K.; Leelahavanichkul, A.; Yuen, P.S.; Mayer, B.; Parmelee, A.; Doi, K.; Robey, P.G.; Leelahavanichkul, K.; Koller, B.H.; Brown, J.M.; et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009, 15, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Blurton-Jones, M. Can stem cells be used to treat or model Alzheimer disease? Stem Cells 2012, 30, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Xie, N.; Li, W.; Yuan, B.; Shi, Y.; Wang, Y. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2014, 21, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Parr, A.M.; Kulbatski, I.; Tator, C.H. Transplantation of adult rat spinal cord stem/progenitor cells for spinal cord injury. J. Neurotrauma 2007, 24, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.R.; Stoutenger, B.R.; Robinson, A.P.; Spees, J.L.; Prockop, D.J. Human stem/progenitor cells from bone marrow promote neurogenesis of endogenous neural stem cells in the hippocampus of mice. Proc. Natl. Acad. Sci. USA 2005, 102, 18171–18176. [Google Scholar] [CrossRef] [PubMed]
- Kan, I.; Barhum, Y.; Melamed, E.; Offen, D. Mesenchymal stem cells stimulate endogenous neurogenesis in the subventricular zone of adult mice. Stem Cell Rev. 2011, 7, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chang, K.A.; Kim, J.A.; Park, H.G.; Ra, J.C.; Kim, H.S.; Suh, Y.H. The preventive and therapeutic effects of intravenous human adipose-derived stem cells in Alzheimer’s disease mice. PLoS ONE 2012, 7, e45757. [Google Scholar] [CrossRef] [PubMed]
- Tfilin, M.; Sudai, E.; Merenlender, A.; Gispan, I.; Yadid, G.; Turgeman, G. Mesenchymal stem cells increase hippocampal neurogenesis and counteract depressive-like behavior. Mol. Psychiatry 2010, 15, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Lu, C.; Han, Q.; Li, J.; Du, Z.; Liao, L.; Zhao, R.C. Adipose-derived mesenchymal stem cells protect PC12 cells from glutamate excitotoxicity-induced apoptosis by upregulation of XIAP through PI3-K/Akt activation. Toxicology 2011, 279, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Lois, C.; Alvarez-Buylla, A. Long-distance neuronal migration in the adult mammalian brain. Science 1994, 264, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Buylla, A.; Garcia-Verdugo, J.M.; Tramontin, A.D. A unified hypothesis on the lineage of neural stem cells. Nat. Rev. Neurosci. 2001, 2, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Cameron, H.A.; McKay, R.D. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J. Comp. Neurol. 2001, 435, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and functional implications of adult neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Ahlenius, H.; Visan, V.; Kokaia, M.; Lindvall, O.; Kokaia, Z. Neural stem and progenitor cells retain their potential for proliferation and differentiation into functional neurons despite lower number in aged brain. J. Neurosci. 2009, 29, 4408–4419. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.J.; Braun, L.; Jiang, Y.; Hester, M.E.; Zhang, L.; Riolo, M.; Wang, H.; Rao, M.; Altura, R.A.; Kaspar, B.K. Aging brain microenvironment decreases hippocampal neurogenesis through Wnt-mediated survivin signaling. Aging Cell 2012, 11, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Clemenson, G.D.; Gage, F.H. New neurons in an aged brain. Behav. Brain Res. 2012, 227, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Ferron, S.R.; Marques-Torrejon, M.A.; Mira, H.; Flores, I.; Taylor, K.; Blasco, M.A.; Farinas, I. Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J. Neurosci. 2009, 29, 14394–14407. [Google Scholar] [CrossRef] [PubMed]
- Blurton-Jones, M.; Kitazawa, M.; Martinez-Coria, H.; Castello, N.A.; Muller, F.J.; Loring, J.F.; Yamasaki, T.R.; Poon, W.W.; Green, K.N.; LaFerla, F.M. Neural stem cell improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13594–13599. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, K. Possible use of autologous stem cell therapies for Alzheimer’s disease. Curr. Alzheimer Res. 2005, 2, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Waldau, B.; Shetty, A.K. Behavior of neural stem cells in the Alzheimer brain. Cell. Mol. Life Sci. 2008, 65, 2372–2384. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.J.; Siebzehnrubl, F.A.; Kandasamy, M.; Couillard-Despres, S.; Caioni, M.; Poehler, A.M.; Berninger, B.; Sandner, B.; Bogdahn, U.; Goetz, M.; et al. Mesenchymal stem cells promote oligodendroglial differentiation in hippocampal slice cultures. Cell. Physiol. Biochem. 2009, 24, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a toll like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and Toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.E.; Kou, J.; Song, M.; Pattanayak, A.; Jin, J.; Lalonde, R.; Fukuchi, K. MyD88 deficiency ameliorates β-amyloidosis in an animal model of AD. Am. J. Pathol. 2011, 179, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Malm, T.; Koistinaho, J.; Kanninen, K. Utilization of APPswe/PS1dE9 transgenic mice in research of Alzheimer’s disease: Focus on gene therapy and cell-based therapy applications. Int. J. Alzheimers Dis. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Wictorin, K.; Bjorklund, A.; Williams, L.P.; Varon, S.; Gage, F.H. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature 1987, 329, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Banks, W.A.; Kastin, A.J. Permeability of the blood-brain barrier to neurotrophins. Brain Res. 1998, 788, 87–94. [Google Scholar] [CrossRef]
- Williams, L.R. Hypophagia is induced by intracerebroventricular administration of nerve growth factor. Exp. Neurol. 1991, 113, 31–37. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; Sang U, H.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 2005, 11, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S.; Baumann, T.L.; Bakay, R.A.; Ostrove, J.M.; Siffert, J.; Fleisher, A.S.; Herzog, C.D.; Barba, D.; Pay, M.; Salmon, D.P.; et al. A phase 1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimers Dement. 2014, 10, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Connor, B.; Young, D.; Yan, Q.; Faull, R.L.; Synek, B.; Dragunow, M. Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Mol. Brain Res. 1997, 49, 71–81. [Google Scholar] [CrossRef]
- Blurton-Jones, M.; Spencer, B.; Michael, S.; Castello, N.A.; Agazaryan, A.A.; Davis, J.L.; Muller, F.J.; Loring, J.F.; Masliah, E.; LaFerla, F.M. Neural stem cells genetically-modified to express neprilysin reduce pathology in Alzheimer transgenic models. Stem Cell Res. Ther. 2014, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Ait-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; de Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Kosakai, A.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Nabetani, A.; Ishikawa, F.; Arai, Y.; Hirose, N.; et al. Establishment of induced pluripotent stem cells from centenarians for neurodegenerative disease research. PLoS ONE 2012, 7, e41572. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; van Gorp, S.; Nazor, K.L.; Bascolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Isobe, K.; Cheng, Z.; Nishio, N.; Suganya, T.; Tanaka, Y.; Ito, S. iPSCs, aging and age-related diseases. N. Biotechnol. 2014, 31, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. Telomere length, stem cells and aging. Nat. Chem. Biol. 2007, 3, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Strati, K.; Li, H.; Tejera, A.; Schoeftner, S.; Ortega, S.; Serrano, M.; Blasco, M.A. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 2009, 4, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, F.; Okuka, M.; Liu, N.; Ji, G.; Ye, X.; Zuo, B.; Li, M.; Liang, P.; Ge, W.W.; et al. Association of telomere length with authentic pluripotency of ES/iPS cells. Cell Res. 2011, 21, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Miller, V.M. Proteinopathy-induced neuronal senescence: A hypothesis for brain failure in Alzheimer’s and other neurodegenerative diseases. Alzheimers Res. Ther. 2009, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Arendt, T.; Brayne, C. The senescence hypothesis of disease progression in Alzheimer’s disease: An integrated matrix of disease pathways for FAD and SAD. Mol. Neurobiol. 2013, 48, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Amemori, T.; Ermakova, I.V.; Buresova, O.; Zigova, T.; Racekova, E.; Bures, J. Brain transplants enhance rather than reduce the impairment of spatial memory and olfaction in bulbectomized rats. Behav. Neurosci. 1989, 103, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amemori, T.; Jendelova, P.; Ruzicka, J.; Urdzikova, L.M.; Sykova, E. Alzheimer’s Disease: Mechanism and Approach to Cell Therapy. Int. J. Mol. Sci. 2015, 16, 26417-26451. https://doi.org/10.3390/ijms161125961
Amemori T, Jendelova P, Ruzicka J, Urdzikova LM, Sykova E. Alzheimer’s Disease: Mechanism and Approach to Cell Therapy. International Journal of Molecular Sciences. 2015; 16(11):26417-26451. https://doi.org/10.3390/ijms161125961
Chicago/Turabian StyleAmemori, Takashi, Pavla Jendelova, Jiri Ruzicka, Lucia Machova Urdzikova, and Eva Sykova. 2015. "Alzheimer’s Disease: Mechanism and Approach to Cell Therapy" International Journal of Molecular Sciences 16, no. 11: 26417-26451. https://doi.org/10.3390/ijms161125961