
Lipoprotein-Associated Oxidative Stress: A New Twist to the Postprandial Hypothesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Oxidative Stress and Reactive Oxygen Species (ROS)
2. Oxidized Low-Density Lipoproteins (LDL) and Atherosclerosis
3. Biomarkers of Oxidative Modification
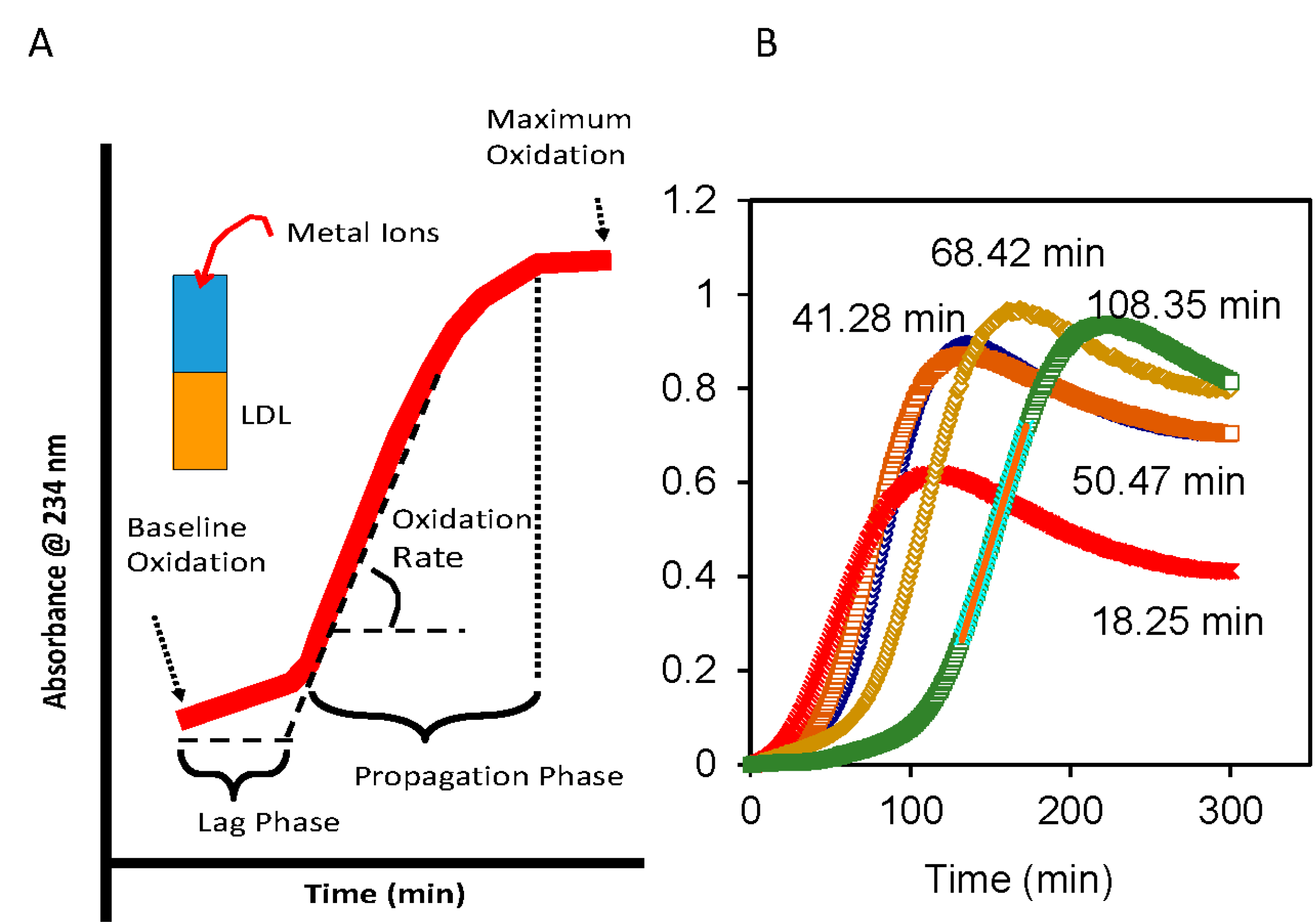
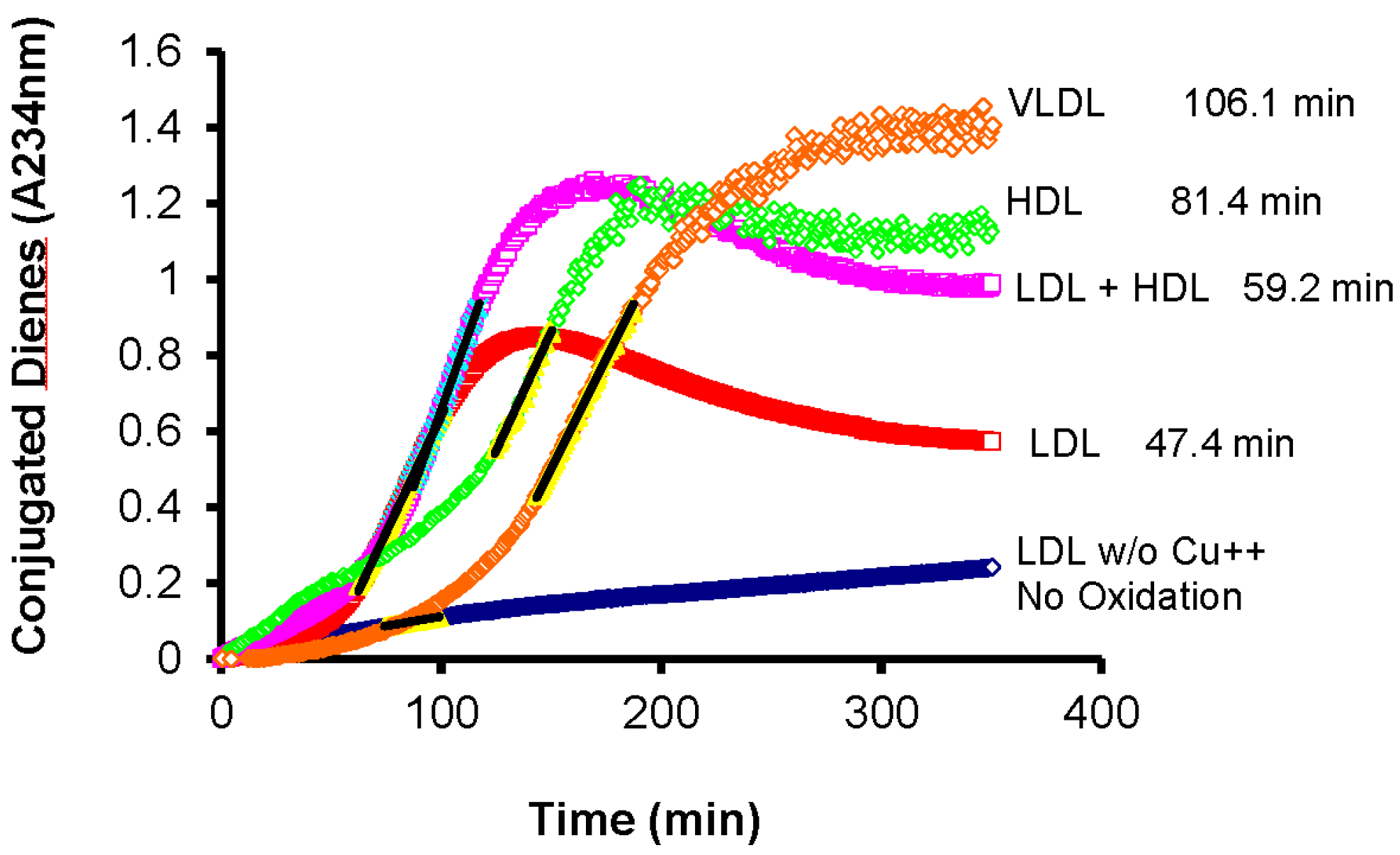
4. Oxidative Susceptibility of Plasma Lipoproteins


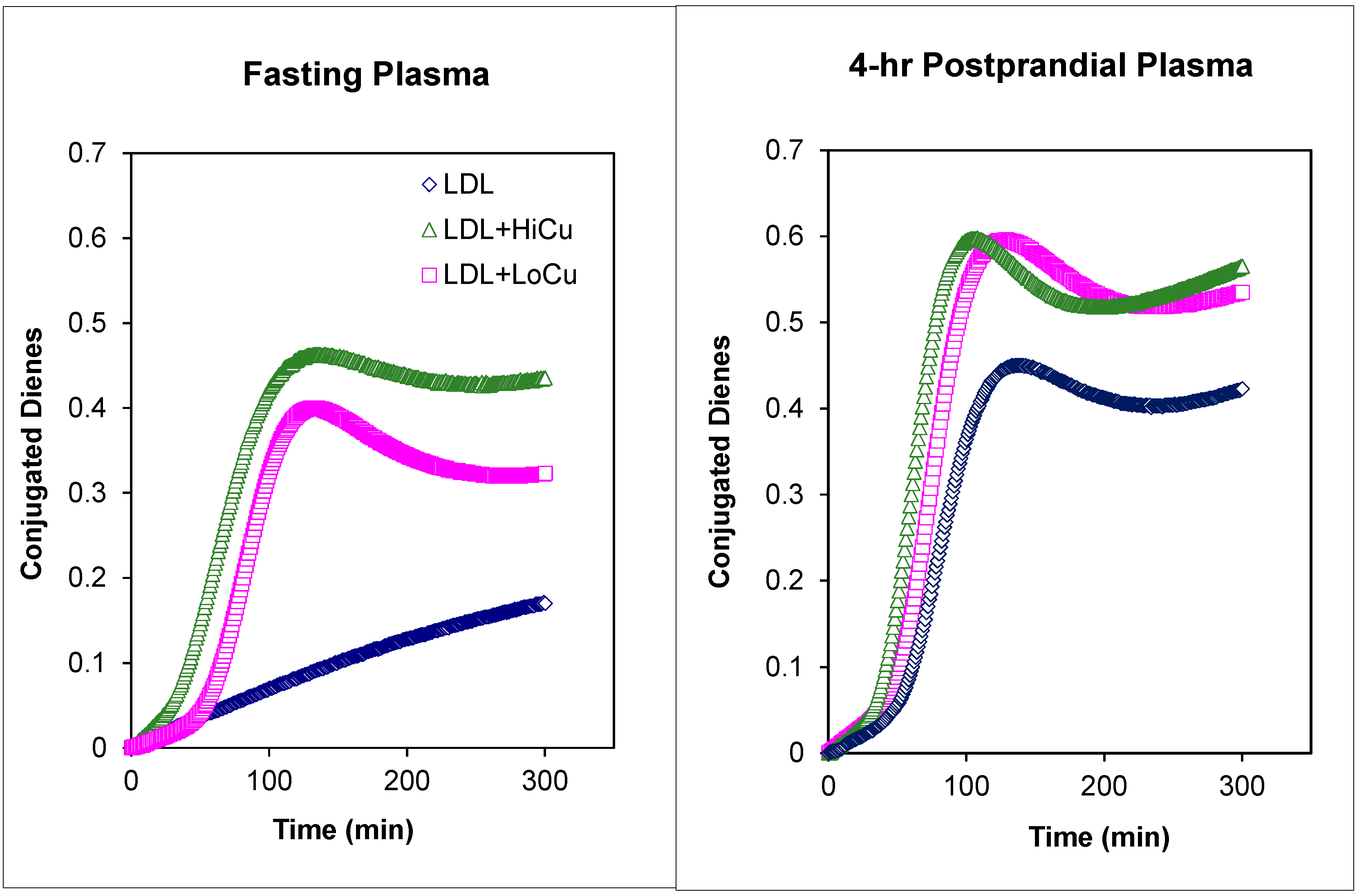
5. Plasma Lipoproteins and the Arterial Wall: Fasting versus Postprandial



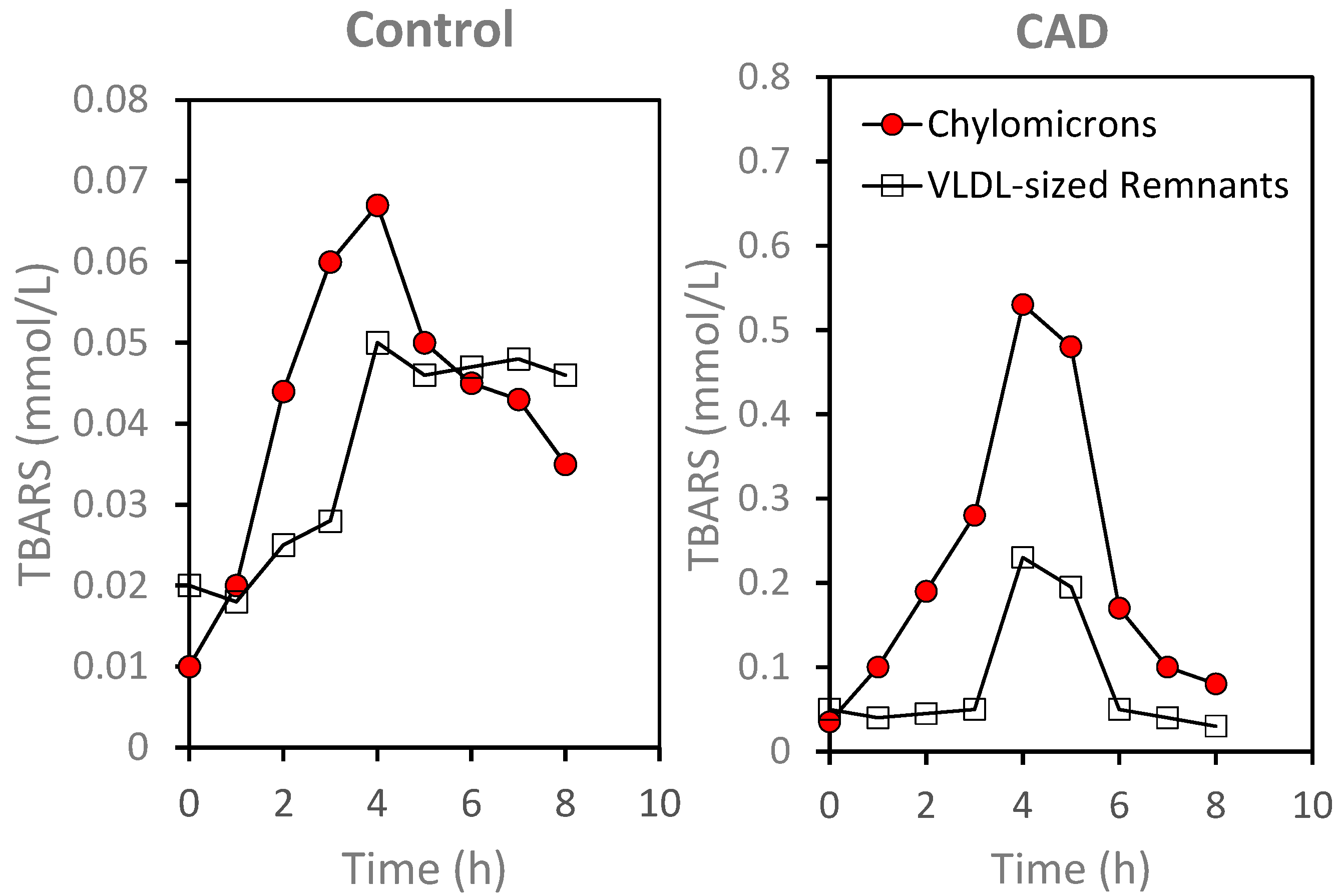
6. Oxidative Stress: Protective Response Leading to Disease
7. Discussion
Acknowledgments
Conflicts of Interest
References
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Farkas-Epperson, M.; Le, N.-A. Lipoproteins as biosensors of endothelial oxidative stress. Clin. Lipidol. 2012, 7, 49–63. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Van Oostrom, A.J.H.H.M.; van Wijk, J.P.H.; Castro-Cabezas, M. Lipemia, inflammation and atherosclerosis: Novel opportunities in the understanding and treatment of atherosclerosis. Drugs 2004, 64, 19–24. [Google Scholar]
- Goldstein, J.L.; Ho, Y.K.; Basu, S.K.; Brown, M.S. Binding sites on macrophages that mediates uptake and degradation of acetylated LDL, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. USA 1979, 76, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Fogelman, A.M.; Schechter, I.; Seager, J.; Hokom, M.; Child, J.S.; Edwards, P.A. Malondialdehyde alteration of low density lipoproteins leads to cholesteryl ester accumulation in human monocyte-macrophages. Proc. Natl. Acad. Sci. USA 1980, 77, 2214–2218. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Hama, S.H.; Ready, S.T.; Ng, C.J.; van Lenten, B.J.; Laks, H.; Fogelman, A.M. Oxidized lipids as mediators of coronary heart disease. Curr. Opin. Lipidol. 2002, 13, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Palinski, W.; Rosenfeld, M.E.; Yla-Herttuala, S.; Gurtner, G.C.; Socher, S.S.; Butler, S.W.; Parthasarathy, S.; Carew, T.E.; Steinberg, D.; Witztum, J.L. LDL undergoes oxidative modification in vivo. Proc. Natl. Acad. Sci. USA 1989, 86, 1372–1376. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, P.; Bon, G.B.; Cazzolato, G. Presence of a modified LDL in humans. Arteriosclerosis 1988, 8, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Yla-Herttuala, S.; Palinski, W.; Rosenfeld, M.E.; Parthasarathy, S.; Carew, T.E.; Butler, S.; Witztum, J.L.; Steinberg, D. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J. Clin. Investig. 1989, 84, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Holvoet, P.; Vanhaecke, J.; Janssens, S.; van de Werf, F.; Collen, D. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation 1998, 98, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Hulthe, J.; Fagerberg, B. Circulating oxidized LDL is associated with subclinical atherosclerosis development and inflammatory cytokines (AIR study). Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1162–1167. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, Y.; Oka, Y.; Katagiri, H. Circulating oxidized LDL: A biomarker and a pathogenic factor. Curr. Opin. Lipidol. 2009, 20, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Itabe, H. Oxidized low-density lipoproteins: What is understood and what remains to be clarified. Biol. Pharm. Bull. 2003, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond cholesterol: Modifications of low-density lipoprotein that increase its atherogenecity. N. Engl. J. Med. 1989, 320, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Witztum, J.L.; Steinberg, D. Role of oxidized low density lipoprotein in atherogenesis. J. Clin. Investig. 1991, 88, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Witztum, J.L. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2311–2316. [Google Scholar] [CrossRef] [PubMed]
- Chisolm, G.M.; Steinburg, D. The oxidative modification hypothesis of atherogenesis: An overview. Free Radic. Biol. Med. 2000, 28, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Sigurdardottir, V.; Fagerberg, B.; Hulthe, J. Circulating oxidized low-density lipoprotein (LDL) is associated with risk factors of the metabolic syndrome and LDL size in clinically healthy 58-year-old men (AIR study). J. Intern. Med. 2002, 252, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Mokuno, H.; Matsunaga, E.; Miyazaki, T.; Sumiyoshi, K.; Miyauchi, K.; Daida, H. Circulating oxidized LDL is an independent predictor for cardiac event in patients with coronary artery disease. Atherosclerosis 2004, 174, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Memon, R.; Staprans, I.; Noor, M. Infection and inflammation induce LDL oxidation in vivo. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1536–1540. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.A.; Mensah, G.A.; Alexander, R.W.; Anderson, J.L.; Canno, R.O., III; Criqui, M.; Fadl, Y.Y.; Fortmann, S.P.; Hong, Y.; Myer, G.L.; et al. Markers of inflammation and cardiovascular disease: Application to clinical and public health practice: A statement for healthcare professionals from the centers for disease control and prevention and the american heart association. Circulation 2003, 107, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Szmitko, P.E.; Wang, C.H.; Weisel, R.D.; Jeffries, G.A.; Anderson, T.J.; Verma, S. Biomarkers of vascular disease linking inflammation to endothelial activation: Part II. Circulation 2003, 108, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Brennan, M.L.; Shen, Z.; MacPherson, J.C.; Schmitt, D.; Molenda, C.E.; Hazen, S.L. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J. Biol. Chem. 2002, 277, 46116–46122. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Glomset, J.A. Atherosclerosis and the arterial smooth muscle cell: Proliferation of smooth muscle cell is a key event in the genesis of the lesions of atherosclerosis. Science 1973, 180, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Proctor, S.; Vine, D.; Mamo, J. Arterial retention of apolipoprotein B(48)- and B(100)-containing lipoproteins in atherogenesis. Curr. Opin. Lipidol. 2002, 13, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Frei, B. Endogenous antioxidant defenses in human blood plasma. In Oxidative Stress: Oxidants and Antioxidants; Sies, H., Ed.; Academic Press: London, UK, 1991; pp. 213–243. [Google Scholar]
- Cushing, S.D.; Berliner, J.A.; Valente, A.J.; Navab, M.; Parhami, F.; Gerrity, R.; Schwartz, C.J.; Fogelman, A.M. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. USA 1990, 87, 5134–5138. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.T.; Parthasarathy, S.; Fong, L.G.; Steinberg, D. Oxidatively modified low density lipoproteins: A potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc. Natl. Acad. Sci. USA 1987, 84, 2995–2998. [Google Scholar] [CrossRef] [PubMed]
- Shechter, I.; Fogelman, A.M.; Haberland, M.E.; Seager, J.; Hokom, M.; Edwards, P.A. The metabolism of native and MDA-altered LDL by human monocyte-macrophages. J. Lipid Res. 1981, 22, 63–71. [Google Scholar] [PubMed]
- Hessler, J.R.; Robertson, A.L., Jr.; Chisolm, G.M., III. LDL-induced cytotoxicity and its inhibition by HDL in human vascular smooth muscle and endothelial cells in culture. Atherosclerosis 1979, 32, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, M.K.; McNally, A.K.; Chisolm, G.M. Lipoxygenase-mediated transformation of human LDL to an oxidized and cytotoxic complex. J. Lipid Res. 1991, 32, 63–70. [Google Scholar] [PubMed]
- Mabile, L.; Salvayre, R.; Bonnafe, M.J.; Negre-Salvayre, A. Oxidizability and subsequent cytotoxicity of chylomicrons to monocytic U937 and endothelial cells are dependent on dietary fatty acid composition. Free Radic. Biol. Med. 1995, 19, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.; Berlett, B.S. Reactive oxygen-mediated protein oxidation in ageing and disease. Chem. Res. Toxicol. 1997, 10, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Dedon, P.C.; Tannenbaum, S.R. Reactive nitrogen species in the chemical biology of inflammation. Arch. Biochem. Biophys. 2004, 433, 12–22. [Google Scholar] [CrossRef]
- Tarpey, M.M.; Wink, D.A.; Grisham, M.B. Methods for detection of reactive metabolites of oxygen and nitrogen: In vitro and in vivo considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R431–R444. [Google Scholar] [CrossRef] [PubMed]
- Cracowski, J.L.; Durand, T.; Bessard, G. Isoprostanes as a biomarker of lipid peroxidation in humans: Physiology, pharmacology and clinical implications. Trends Pharmacol. Sci. 2002, 23, 230–236. [Google Scholar] [CrossRef]
- Draper, H.H.; Csallany, A.S.; Hadley, M. Urinary aldehydes as indicators of lipid peroxidation in vivo. Free Radic. Biol. Med. 2000, 29, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.T.; Fu, S.; Stocker, R.; Davies, M.J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 1997, 324, 1–18. [Google Scholar] [PubMed]
- Requena, J.R.; Levine, R.L.; Stadtman, E.R. Recent advances in the analysis of oxidized proteins. Amino Acids 2003, 25, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malondialdehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, P.; Barnes, P.J.; Roberts, L.J., II. Isoprostanes: Markers and mediators of oxidative stress. FASEB J. 2004, 18, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Slatter, D.A.; Bolton, C.H.; Bailey, A.J. The importance of lipid-derived malondialdehyde in diabetes mellitus. Diabetologia 2000, 43, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Heinecke, J.W. Oxidized amino acids: Culprits in human atherosclerosis and indicators of oxidative stress. Free Radic. Biol. Med. 2002, 32, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Bellamo, G.; Robino, G.; Barrera, G.; Dianzani, M.U. 4-Hydroxynonenal as a biological signal: Molecular basis and pathophysiological implications. Antioxid. Redox Signal. 1999, 1, 255–284. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; O’Brien, K.D.; McDonald, T.O.; Fu, X.; Oram, J.F.; Uchida, K.; Heinecke, J.W. Acrolein modifies apolipoprotein A–I in the human artery wall. Ann. N. Y. Acad. Sci. 2005, 1043, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Rossi, R.; Milzani, A.; Colombo, R.; Dalle-Donne, I. S-Glutathionylation: From redox regulation of protein functions to human diseases. J. Cell. Mol. Med. 2004, 8, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Gaut, J.; Yeh, G.; Tran, H.; Byun, J.; Henderson, J.P.; Richter, G.M.; Brennan, M.-L.; Lusis, A.J.; Belaaouaj, A.; Hotchkiss, R.S.; et al. Neutrophils employ the myeloperoxidase system to generate antimicrobial brominating and chlorinating oxidants during sepsis. Proc. Natl. Acad. Sci. USA 2001, 98, 11961–11966. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.L.; Wu, W.; Fu, X.; Shen, Z.; Song, W.; Frost, H.; Vadseth, C.; Narine, L.; Lenkiewicz, E.; Borchers, M.T.; et al. A tale of two controversies: Defining both the role of peroxidases in nitrtyrosine formation in vivo using eosinophil peroxidase nad myeloperoxidase-deficient mice, and the nature of peroxidase-generated reactive nitrogen species. J. Biol. Chem. 2002, 277, 17415–17422. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Nukuna, B.; Brennan, M.; Brennan, M.L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A–I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Pennathur, S.; Bergt, C.; Shao, B.; Byun, J.; Kassim, S.Y.; Singh, P.; Green, P.S.; McDonald, T.O.; Brunzell, J.; Chait, A.; et al. Human atherosclerotic intima and blood of patients with established coronary artery disease contain high density lipoprotein damaged by reactive nitrogen species. J. Biol. Chem. 2004, 279, 42977–42983. [Google Scholar] [CrossRef] [PubMed]
- Bergt, C.; Pennathur, S.; Fu, X.; Byun, J.; O’Brien, K.; McDonald, T.O.; Singh, P.; Anantharamaiah, G.M.; Chait, A.; Brunzell, J.; et al. The myeloperoxidase product hypochlorous acid oxidizes hdl in the human artery wall and impairs ABCA1 cholesterol transport. Proc. Natl. Acad. Sci. USA 2004, 101, 13032–13037. [Google Scholar] [CrossRef] [PubMed]
- Palinski, W.; Horkko, S.; Miller, E.; Steinbrecher, U.P.; Powell, H.C.; Curtiss, L.K.; Witztum, J.L. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J. Clin. Investig. 1996, 98, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Palinski, W.; Ord, V.A.; Plump, A.S.; Breslow, J.L.; Steinberg, D.; Witztum, J.L. ApoE-deficient mice are a model of lipoprotein oxidation in atherogenesis. Demonstration of oxidation-specific epitopes in lesions and high titers of autoantibodies to malondialdehyde-lysine in serum. Arterioscler. Thromb. 1994, 14, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Striegl, G.; Puhl, H.; Rotheneder, M. Continuous monitoring of in vitro oxidation of human LDL. Free Radic. Biol. Med. 1989, 6, 67–75. [Google Scholar] [CrossRef]
- Le, N.-A.; Farkas-Epperson, M.; Sweeney, M.; Wilson, P.; Brown, W. Effect of ABT-335 (fenofibric acid) on meal-induced oxidative stress in patients with metabolic syndrome. 2013, 231, 268–273. [Google Scholar]
- McEneny, J.; O’Kane, M.J.; Moles, K.W.; McMaster, C.; McMaster, D.; Mercer, C.; Trimble, E.R.; Young, I.S. Very low density lipoprotein subfractions in type II diabetes mellitus: Alterations in composition and susceptibility to oxidation. Diabetologia 2000, 43, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, A.; McKinstry, L.A.; Lewis, J.K.; Lum, J.; Louie, A.; Schellenberg, G.D.; Hatsukami, T.S.; Chait, A.; Jarvik, G.P. Ex vivo measures of LDL oxidative susceptibility predict carotid artery disease. Atherosclerosis 2005, 179, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Ryglewicz, D.; Rodo, M.; Roszczynko, M.; Baranska-Gieruszczak, M.; Szirkowiec, W.; Swiderska, M.; Wehr, H. Dynamics of ldl oxidation in ischemic stroke patients. Acta Neurol. Scand. 2002, 105, 185–188. [Google Scholar] [CrossRef] [PubMed]
- McEneny, J.; Loughrey, C.M.; McNamee, P.T.; Trimble, E.R.; Young, I.S. Susceptibility of VLDL to oxidation in patients on regular haemodialysis. Atherosclerosis 1997, 129, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Parks, E.J.; German, J.B.; Davis, P.A.; Frankel, E.N.; Kappagoda, C.T.; Rutledge, J.C.; Hyson, D.A.; Schneeman, B.O. Reduced oxidative susceptibility of LDL from patients participating in an intensive atherosclerosis treatment program. Am. J. Clin. Nutr. 1998, 68, 778–785. [Google Scholar] [PubMed]
- Kaikkonen, J.; Porkkala-Sarataho, E.; Tuomainen, T.P.; Nyyssonen, K.; Kosonen, L.; Ristonmaa, U.; Lakka, H.M.; Salonen, R.; Korpela, H.; Salonen, J.T. Exhaustive exercise increases plasma/serum total oxidation resistance in moderately trained men and women, whereas their VLDL + LDL lipoprotein fraction is more susceptible to oxidation. Scand. J. Clin. Lab. Investig. 2002, 62, 599–608. [Google Scholar] [CrossRef]
- Nielsen, N.S.; Marckmann, P.; Hoy, C.E. Effect of meal fat quality on oxidation resistance of postprandial VLDL and LDL particles and plasma triacylglycerol level. Br. J. Nutr. 2000, 84, 855–863. [Google Scholar] [PubMed]
- Parthasarathy, S.; Khoo, J.C.; Miller, E.; Barnett, J.; Witztum, J.L.; Steinberg, D. Low density lipoprotein rich in oleic acid is protected against oxidative modification: Implications for dietary prevention of atherosclerosis. Proc. Natl. Acad. Sci. USA 1990, 87, 3894–3898. [Google Scholar] [CrossRef] [PubMed]
- Reaven, P.; Parthasarathy, S.; Grasse, B.J.; Miller, E.; Almazan, F.; Mattson, F.H.; Khoo, J.C.; Steinberg, D.; Witztum, J.L. Feasibility of using an oleate-rich diet to reduce the susceptibility of low-density lipoprotein to oxidative modification in humans. Am. J. Clin. Nutr. 1991, 54, 701–706. [Google Scholar] [PubMed]
- Reaven, P.; Parthasarathy, S.; Grasse, B.J.; Miller, E.; Steinberg, D.; Witztum, J.L. Effects of oleate-rich and linoleate-rich diets on the susceptibility of LDL to oxidative modification in mildly hypercholesterolemic subjects. J. Clin. Investig. 1993, 91, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Olivecrona, T.; Egelrud, T. Lipoprotein lipase and its interaction with heparin. Horm. Metab. Res. 1974, 4, 23–28. [Google Scholar] [PubMed]
- Fuki, I.V.; Blanchard, N.; Jin, W.; Marchadier, D.H.; Millar, J.S.; Glick, J.M.; Rader, D.J. Endogenously produced endothelial lipase enhances binding and cellular procesin of plasma lipoproteins via heparan sulfate proteoglycan-mediated pathway. J. Biol. Chem. 2003, 278, 34331–34338. [Google Scholar] [CrossRef] [PubMed]
- Quinn, D.; Shirai, K.; Jackson, R.L. Lipoprotein lipase: Mechanism of action and role in lipoprotein metabolism. Prog. Lipid Res. 1983, 22, 35–78. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.W.; Baginsky, M.L. Some functional aspects of apolipoproteins: ApoLp-Ala inhibition of lipoprotein lipase and deinhibition by monoolein. Horm. Metab. Res. 1974, 4, 11–16. [Google Scholar] [PubMed]
- Bengtsson, G.; Olivecrona, T. Lipoprotein lipase. Mechanism of product inhibition. Eur. J. Biochem. 1980, 106, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M.; Herbert, P.N.; Levy, R.I.; Fredrickson, D.S. Further observations on the activation and inhibition of lipoprotein lipase byapolipoproteins. Circ. Res. 1973, 33, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.A.; Catapano, A.L.; Smith, L.C.; Sparrow, J.T.; Gotto, A.M., Jr. Effect of the apolipoprotein C-II/C-III1 ratio on the capacity of purified milk lipoprotein lipase to hydrolyze triglycerides in monolayer vesicles. Atherosclerosis 1996, 127, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Le, N.A.; Cortner, J.A.; Breslow, J.L. Metabolism of intestinal lipoproteins during the postprandial state. In Diabetes; Rifkin, H., Colwell, J.A., Taylor, S.I., Eds.; Elsevier Science Publishers: New York, NY, USA, 1991; pp. 601–608. [Google Scholar]
- Le, N.A.; Coates, P.M.; Gallagher, P.R.; Cortner, J.A. Kinetics of retinyl esters during postprandial lipemia in man: A compartmental model. Metabolism 1997, 46, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Berman, M.; Hall, M., III; Levy, R.I.; Eisenberg, S.; Bilheimer, D.W.; Phair, R.D.; Goebel, R.H. Metyabolism of apoB and apoC lipoproteins in man: Kinetic studies in normal and hyperlipoproteinemic subjects. J. Lipid Res. 1978, 19, 38–56. [Google Scholar] [PubMed]
- Le, N.A.; Li, X.; Kyung, S.; Brown, W.V. Evidence for the in vivo generation of oxidatively modified epitopes in patients with atherosclerotic endothelium. Metabolism 2000, 49, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Gradek, W.Q.; Harris, M.T.; Yahia, N.; Davis, W.W.; Le, N.-A.; Brown, W.V. Polyunsaturated fatty acids acutely suppress antibodies to malondialdehyde-modified LDL in patients with vascular disease. Am. J. Cardiol. 2004, 93, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Le, N.-A. Oxidized lipids and lipoproteins: Indices of risk or targets for management. Clin. Lipidol. 2009, 4, 41–54. [Google Scholar] [CrossRef]
- Palinski, W.; Miller, E.; Witztum, J.L. Immunization of LDL receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherosclerosis by mechanisms other than induction of high titers of antibodies to oxidative neoepitopes. Proc. Natl. Acad. Sci. USA 1995, 92, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Calara, F.; Regnstrom, J.; Hultgardh-Nilsson, A.; Ameli, S.; Cecek, B.; Shah, P.K. Immunization with homologous oxidized LDL reduces neointimal formation after balloon injury in hypercholesterolemic rabbits. J. Am. Coll. Cardiol. 1997, 30, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, P.J.; Brown, W.V.; Ceriello, A.; Le, N.A.; Goldberg, R.B.; Cooke, J.P.; Robbins, D.C.; Sarwat, S.; Yuan, H.; Jones, C.A.; et al. Meal-induced increases in C-reactive protein, interleukin-6, and tumour necrosis factor α are attenuated by prandial + basal insulin in patients with Type 2 diabetes. Diabet Med. 2011, 28, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Yagi, K. A simple fluorometric assay for lipoperoxide in blood plasma. Biochem. Med. 1976, 15, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Yagi, K. Simple assay for the level of total lipid peroxides in serum or plasma. Methods Mol. Biol. 1998, 108, 101–106. [Google Scholar] [PubMed]
- Davies, K.J.; Qunitanilha, A.T.; Brooks, G.A.; Packer, L. Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Close, G.L.; Ashton, T.; McArdle, A.; Maclaren, D.P.M. The emerging role of free radicals in delayed onset muscle soreness and contraction-induced muscle injury. Comp. Biochem Physiol Part A 2005, 142, 257–266. [Google Scholar] [CrossRef]
- Palomero, J.; Pye, D.; Kabayo, T.; Spiller, D.; Jackson, M. In situ detectioon and measurement of intracellular reactive oxygen species in single isolated mature skeletal muscle fiber by real time fluorescence microscopy. Antioxid Redox Signal 2008, 10, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Lu, D.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Gomez-Cabrera, M.C.; Vina, J. Role of free radicals and antioxidant signaling in skeletal muscle health and pathology. Infect. Disord. Drug Targets 2009, 9, 428–444. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Talbert, E.E.; Adhihetty, P.J. Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J. Physiol. 2011, 589, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.G.; Tresini, M. Oxidative stress and gene regulation. Free Rad. Biol. Med. 2000, 28, 463–499. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Zhang, Y. Antioxidant and anti-inflammatory effects of exercise: Role of redox signaling. Free Radic. Res. 2014, 48, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Morales-Alamo, D.; Calbet, J.A. Free radicals and sprint exercise in humans. Free Radic. Res. 2014, 48, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Chyu, K.Y.; Shah, P. Can we vaccinate against atherosclerosis? J. Cardiovasc. Pharmacol. Ther. 2014, 19, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Salonen, J.T.; Yla-Herttuala, S.; Yamamoto, R.; Butler, S.; Korpola, H.; Salonen, R.; Nyyssonen, K.; Palinski, W.; Witztum, J.L. Autoantibody against LDL and progression of carotid atherosclerosis. Lancet 1992, 339, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Maggi, E.; Finardi, G.; Poli, M.; Bollati, P.; Filipponi, M.; Stefano, P.L.; Paolini, G.; Grossi, A.; Clot, P.; Albano, E. Specificity of autoantibodies against oxLDL predicting myocardial infarction. Carotid Artery Dis. 1993, 4, 1119–1122. [Google Scholar] [CrossRef]
- Thavendiranathan, P.; Bagai, A.; Brookhart, M.; Choudhry, N. Primary prevention of cardiovascualr diseases with statin therapy: A meta-analysis of randomized controlled trials. Arch. Intern. Med. 2006, 166, 2307–2313. [Google Scholar] [CrossRef] [PubMed]
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Kearney, P.M.; Blackwell, L.; Collins, R.; Keech, A.; Simes, J.; Peto, R.; Armitage, J.; Baigent, C. Cholesterol treatmnet trialists’ collaborators: Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials: A meta-analysis. Lancet 2008, 371, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Pfeffer, M.A.; Moye, L.A.; Rouleau, J.L.; Rutherford, J.D.; Cole, T.G.; Brown, L.; Warnica, J.W.; Arnold, J.M.; Wun, C.C.; et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels: Cholesterol and Recurrent Events Trial investigators. N. Engl. J. Med. 1996, 335, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.A.; Danielson, E.; Rifai, N.; Ridker, P.M. Effect of statin therapy on C-reactive protein levels: The pravastatin inflammation/CRP evaluation (PRINCE): A randomized trial and cohort study. JAMA 2001, 286, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Hoogeveen, R.C.; Bang, H.; Coresh, J.; Folsom, A.R.; Heiss, G.; Sha, A.R. Lipoprotein-associated phospholipase A2, high-sensitivity C-reactive protein, and risk for incident coronary heart disease in middle-aged men and women in the Atherosclerosis Risk in Communities (aric) study. Circulation 2004, 109, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Koenig, W.; Khuseyinova, N.; Lowel, H.; Trischler, G.; Meisenger, C. Lipoprotein-associated phospholipase A2 adds to risk prediction of incident coronary events by C-reactive protein in apparently healthy middle-aged men from the general population: Results from the 14-year follow-up of a large cohort from southern Germany. Circulation 2004, 110, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, S.; Zeller, T.; Saarela, O.; Havulinna, A.S.; Kee, F.; Tunstall-Pedoe, H.; Kuulasmaa, K.; Yarnell, J.; Schnabel, R.B.; Wild, P.S.; et al. Contribution of 30 biomarkers to 10-year cardiovascular risk estimation in 2 poplation cohorts. Circulation 2010, 121, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, O.; Mohanty, B.D.; Martin, S.S.; Joshi, P.H.; Blaha, M.J.; Nasir, K.; Blumenthal, R.S.; Budoff, M.J. High-sensitivity C-reactive protein and cardiovascualr disease: A resolute belief or an elusive link. J. Am. Coll. Cardiol. 2013, 62, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Le, N.A. Reducing oxidized lipids to prevent cardiovascular disease. Curr. Treatment. Options Cardiovasc. Med. 2008, 10, 263–272. [Google Scholar] [CrossRef]
- Tsimikas, S.; Aikawa, M.; Miller, F.J., Jr.; Miller, E.R.; Torzewski, M.; Lentz, S.R.; Bergmark, C.; Heistad, D.D.; Libby, P.; Witztum, J.L. Increased plasma oxidized phospholipid:apolipoprotein B-100 ratio with concomitant depletion of oxidized phospholipids from atherosclerotic lesions after dietary lipid-lowering: A potential biomarker of early atherosclerosis regression. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Ky, B.; Burke, A.; Tsimikas, S.; Wolfe, M.L.; Tadesse, M.G.; Szapary, P.O.; Witztum, J.L.; Fitzgerald, G.A.; Rader, D.J. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J. Am. Coll. Cardiol. 2008, 51, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Tuzcu, E.M.; Schoenhagen, P.; Brown, B.G.; Ganz, P.; Vogel, R.A.; Crowe, T.; Howard, G.; Cooper, C.J.; Brodie, B.; et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: A randomized controlled trial. JAMA 2004, 291, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Chae, A.; Miller, E.; Messig, M.; Ntanios, F.; DeMaria, A.N.; Nissen, S.E.; Witztum, J.L.; Tsimikas, S. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: Volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J. Am. Coll. Cardiol. 2008, 52, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Diwadkar, V.A.; Anderson, J.W.; Bridges, S.R.; Gowri, M.S.; Oelgten, P.R. Postprandial low-density lipoproteins in type 2 diabetes are oxidized more extensively than fasting diabetes and control samples. Proc. Soc. Exp. Biol. Med. 1999, 222, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Miki, T.; Sha, S.; Hirata, K.; Ishikawa, Y.; Yokohama, M. Serum levels of TBARS are associated with risk of coronary heart disease. J. Atheroscler. Thromb. 2011, 18, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.F.; Jacobs, R.F.; Jeffers, B.; Ghadanfar, M.M.; Gm Preston, J.B.; Mason, R.P. Serum levels of thiobarbituric acid reactive substances predict cardiovascular events in patients with stable coronary artery disease: A longitudinal analysis of the PREVENT study. J. Am. Coll. Cardiol. 2004, 44, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, N.-A. Lipoprotein-Associated Oxidative Stress: A New Twist to the Postprandial Hypothesis. Int. J. Mol. Sci. 2015, 16, 401-419. https://doi.org/10.3390/ijms16010401
Le N-A. Lipoprotein-Associated Oxidative Stress: A New Twist to the Postprandial Hypothesis. International Journal of Molecular Sciences. 2015; 16(1):401-419. https://doi.org/10.3390/ijms16010401
Chicago/Turabian StyleLe, Ngoc-Anh. 2015. "Lipoprotein-Associated Oxidative Stress: A New Twist to the Postprandial Hypothesis" International Journal of Molecular Sciences 16, no. 1: 401-419. https://doi.org/10.3390/ijms16010401