Looking into a Conceptual Framework of ROS–miRNA–Atrial Fibrillation

Abstract
:1. Introduction
2. Atrial Fibrillation (AF) and Atrial Remodeling
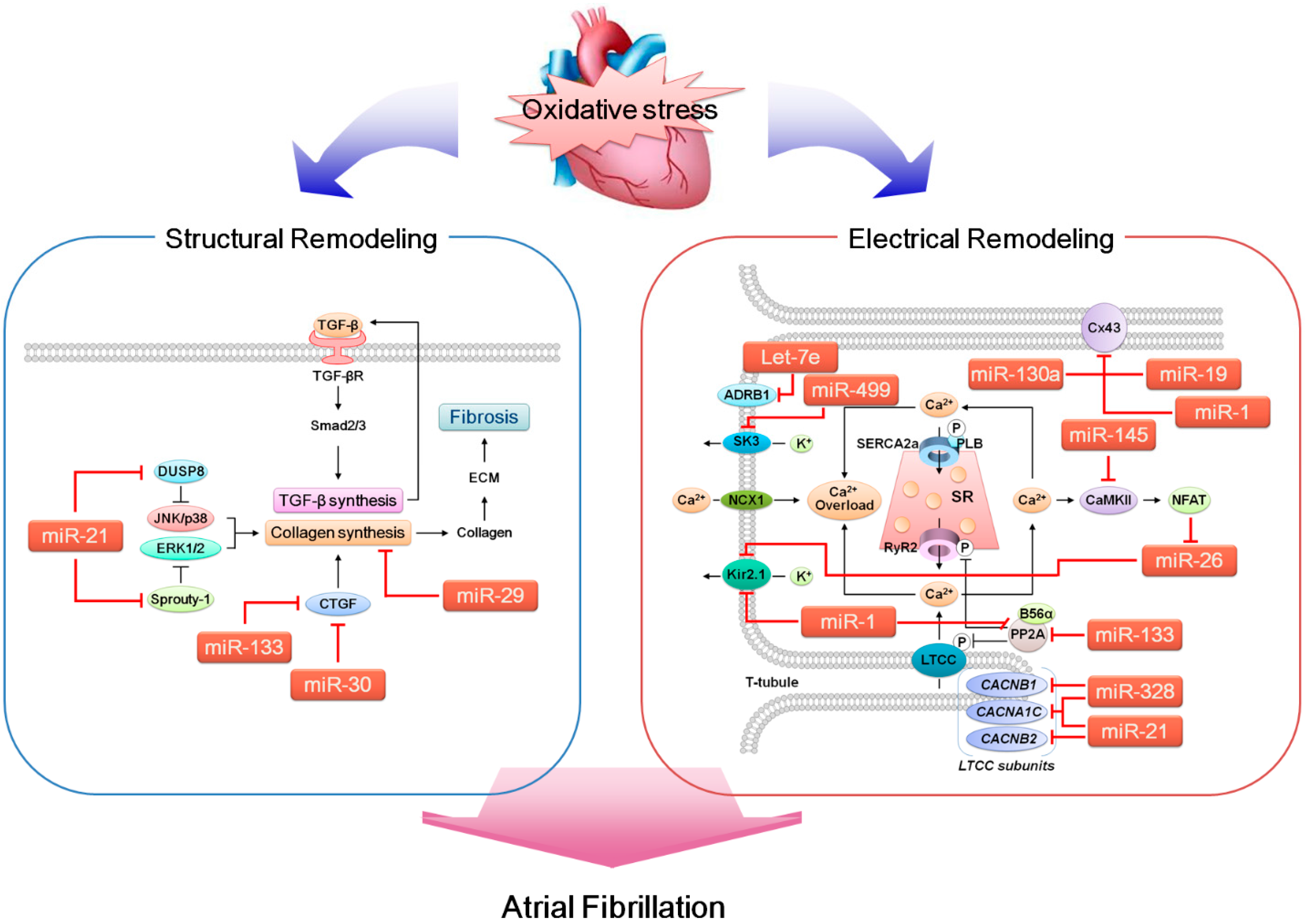
2.1. Electrical Remodeling of the Heart
2.1.1. Ca2+ Regulation and Key Molecules Involved
2.1.2. K+ Regulation and Key Molecules Involved
2.1.3. Gap Junction Ion Channels
2.2. Structural Remodeling of the Heart
3. MicroRNAs (miRNAs)
4. Reactive Oxygen Species (ROS)
5. ROS, miRNA and AF

{kind=link}
| miRNA | Changes in CVDs (Ref. if miR Expression Was Confirmed in Other Study) | Targeted Protein (mRNA) and Subsequent Results | Ref. | |
|---|---|---|---|---|
| miR-1 | Decreased in AF | Potassium channel, inwardly rectifying, Kir2.1 (KCNJ2) | Increased Ik1 | [94] |
| Decreased in hypertrophy | Connexin 43 (GJA1) | Cx43 displacement due to hyper-phosphorylation | [95] | |
| Increased in HF ([96]) | Protein phosphatase 2A (PPP2CA) | Excessive RyR2 phosphorylation by CaMKII | [97] | |
| miR-19a | Increased in AF ([98]) | Phosphatase and tensin homolog (PTEN) | Increased hypertrophy | [99] |
| Connexin 43 (GJA1) | Disturbed electrical coupling | |||
| miR-21 | Increased in MI Increased in AF | Sprouty1 (SPRY1) | Increased fibrosis | [100] |
| Calcium channel, voltage-dependent, l type, alpha 1C subunit (CACNA1C) | Shortened APD | [101] | ||
| Calcium channel, voltage-dependent, beta 2 subunit (CACNB2) | Decreased ICaL | |||
| mir-26 | Decreased in AF | Potassium channel, inwardly rectifying, Kir2.1 (KCNJ2) | Increased Ik1 | [102] |
| miR-29b | Decreased in CHF | Collagen, type I, alpha 1 (COL1A1) | Increased fibrosis | [103] |
| Collagen, type III, alpha 1 (COL3A1) | ||||
| Fibrillin (FBN) | ||||
| miR-30 | Reduced in AF ([104]) | Connective tissue growth factor (CTGF) | Increased fibrosis | [105] |
| mir-130a | Increased in Atherosclerosis ([106]) | Connexin 43 (GJA1) | Disturbed electrical coupling | [107] |
| miR-133 | Reduced in AF ([104]) | Connective tissue growth factor (CTGF) | Increased fibrosis | [105] |
| Increased in HF ([96]) | Protein phosphatase 2A (PPP2CA) | Excessive RyR2 phosphorylation by CaMKII | [97] | |
| miR-145 | Decreased in AF ([108]) | Ca2+/calmodulin-dependent protein kinase 2 delta (CAMK2D) | Excessive RyR2 phosphorylation by CaMKII | [109] |
| miR-328 | Increased in AF | Calcium channel, voltage-dependent, l-Type, alpha 1C subunit (CACNA1C) | Shortened APD | [108] |
| Increased in hypertrophy ([ 110]) | Calcium channel, voltage-dependent, beta 1 subunit (CACNB1) | Decreased ICaL | [111] | |
| miR-499 | Increased in AF | Small-conductance calcium-activated potassium channel 3 (KCNN3) | Altered conduction | [112] |
| Increased in hypertrophy ([113]) | Increased AF | |||
| Let-7e | Decreased in Acute MI | Beta 1 adrenergic receptor (ADRB1) | Increased arrhythmia score | [114] |
5.1. miRNAs that Likely Contribute to Arrhythmogenesis
5.1.1. miR-1
5.1.2. miR-19a (miR-17-92 Cluster)
5.1.3. miR-21
5.1.4. miR-26
5.1.5. miR-29b
5.1.6. miR-30
5.1.7. miR-130a
5.1.8. miR-133
5.1.9. miR-145
5.1.10. miR-328
5.1.11. miR-499
5.1.12. Let-7e
5.2. AF-Related miRNAs and ROS
| miRNA | Modulation by ROS | Stimulation | Experimental System | Ref. |
|---|---|---|---|---|
| miR-1 | Increased | H2O2 (50–400 µM) | H9c2 | [129] |
| Increased | H2O2 (100 µM, 6 h) | NRCM | [130] | |
| Decreased | H2O2 (100 µM, 6 h) | NRVM | [131] | |
| miR-19a | Decreased | H2O2 (200 µM, 6 h) | VSMC | [132] |
| miR-21 | Increased | H2O2 (10–100 µM, 6 h) | NRCM | [133] |
| Increased | H2O2 (25–200 µM, 6 h) | VSMC | [132] | |
| Increased | H2O2 (100 µM, 6 h) | NRVM | [131] | |
| miR-26 | Increased | H2O2 (100 µM, 6 h) | NRCM | [130] |
| Decreased | H2O2 (200 µM, 6 h) | VSMC | [132] | |
| miR-29b | Decreased | H2O2 (200 µM, 6 h) | VSMC | [132] |
| miR-30 | Decreased | H2O2 (100 µM, 6 h) | NRCM | [134] |
| miR-130a | Decreased | H2O2 (200 µM, 6 h) | VSMC | [132] |
| miR-133 | Decreased | H2O2 (100 µM, 24 h) | NRCM | [135] |
| Decreased | H2O2 (100 µM, 6 h) | NRVM | [131] | |
| miR-145 | Decreased | H2O2 (50 µM, 0.5–8 h) | NRVM | [136] |
| miR-328 | Decreased | H2O2 (200 µM, 6 h) | VSMC | [132] |
| miR-499 | Increased | H2O2 (50–200 µM, 6 h) | NRVM | [131] |
| Let-7e | Decreased | H2O2 (50 µM, 1 h) | HCT116 colon cancer cells | [137] |
| Increased | H2O2 (200 µM, 6 h) | VSMC | [132] |

5.3. Perspective: Can ROS-Dependent Dysregulation of miRNA Really Cause AF in Humans?
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Alpert, J.S. Atrial fibrillation: A growth industry in the 21st century. Eur. Heart J. 2000, 21, 1207–1208. [Google Scholar] [PubMed]
- Menezes, A.R.; Lavie, C.J.; DiNicolantonio, J.J.; O’Keefe, J.; Morin, D.P.; Khatib, S.; Milani, R.V. Atrial fibrillation in the 21st century: A current understanding of risk factors and primary prevention strategies. Mayo Clin. Proc. 2013, 88, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Iaccarino, G.; de Luca, N.; Trimarco, B.; Condorelli, G. Atrial fibrillation and microRNAs. Front. Physiol. 2014, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Burstein, B.; Dobrev, D. Atrial remodeling and atrial fibrillation: Mechanisms and implications. Circ. Arrhythm. Electrophysiol. 2008, 1, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Zhang, J.; Zhang, Y.; Chen, H.; Liu, D.; Ping, P.; Weiss, J.N.; Cai, H. Oxidative stress in atrial fibrillation: An emerging role of NADPH oxidase. J. Mol. Cell. Cardiol. 2013, 62, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Greco, S.; Gaetano, C.; Martelli, F. Oxidative stress and microRNAs in vascular diseases. Int. J. Mol. Sci. 2013, 14, 17319–17346. [Google Scholar] [PubMed]
- Nattel, S.; Harada, M. Atrial remodeling and atrial fibrillation: Recent advances and translational perspectives. J. Am. Coll. Cardiol. 2014, 63, 2335–2345. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Voigt, N.; Dobrev, D. New directions in antiarrhythmic drug therapy for atrial fibrillation. Future Cardiol. 2013, 9, 71–88. [Google Scholar] [CrossRef]
- Wakili, R.; Voigt, N.; Kaab, S.; Dobrev, D.; Nattel, S. Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Investig. 2011, 121, 2955–2968. [Google Scholar] [CrossRef] [PubMed]
- Brundel, B.J.; van Gelder, I.C.; Henning, R.H.; Tuinenburg, A.E.; Wietses, M.; Grandjean, J.G.; Wilde, A.A.; van Gilst, W.H.; Crijns, H.J. Alterations in potassium channel gene expression in atria of patients with persistent and paroxysmal atrial fibrillation: Differential regulation of protein and mRNA levels for K+ channels. J. Am. Coll. Cardiol. 2001, 37, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Bosch, R.F.; Zeng, X.; Grammer, J.B.; Popovic, K.; Mewis, C.; Kuhlkamp, V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc. Res. 1999, 44, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Burstein, B.; Nattel, S. Atrial fibrosis: Mechanisms and clinical relevance in atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.H.; Wakili, R.; Qi, X.Y.; Chartier, D.; Boknik, P.; Kaab, S.; Ravens, U.; Coutu, P.; Dobrev, D.; Nattel, S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ. Arrhythm. Electrophysiol. 2008, 1, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.S.; Tan, A.Y. Autonomic nerve activity and atrial fibrillation. Heart Rhythm 2007, 4, S61–S64. [Google Scholar] [CrossRef] [PubMed]
- Dinanian, S.; Boixel, C.; Juin, C.; Hulot, J.S.; Coulombe, A.; Rucker-Martin, C.; Bonnet, N.; le Grand, B.; Slama, M.; Mercadier, J.J.; et al. Downregulation of the calcium current in human right atrial myocytes from patients in sinus rhythm but with a high risk of atrial fibrillation. Eur. Heart J. 2008, 29, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, R.; Ronca-Testoni, S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: Modulation by endogenous effectors, drugs and disease states. Pharmacol. Rev. 1997, 49, 1–51. [Google Scholar] [PubMed]
- Shan, J.; Xie, W.; Betzenhauser, M.; Reiken, S.; Chen, B.X.; Wronska, A.; Marks, A.R. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2012, 111, 708–717. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.H.; Asahi, M.; Tupling, A.R. The regulation of SERCA-type pumps by phospholamban and sarcolipin. Ann. N. Y. Acad. Sci. 2003, 986, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Voigt, N.; Li, N.; Wang, Q.; Wang, W.; Trafford, A.W.; Abu-Taha, I.; Sun, Q.; Wieland, T.; Ravens, U.; Nattel, S.; et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+–Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012, 125, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.H.; Herting, J.; Tirilomis, T.; Renner, A.; Neef, S.; Toischer, K.; Ellenberger, D.; Forster, A.; Schmitto, J.D.; Gummert, J.; et al. Ca2+/calmodulin-dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation 2013, 128, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Hudasek, K.; Brown, S.T.; Fearon, I.M. H2O2 regulates recombinant Ca2+ channel alpha1C subunits but does not mediate their sensitivity to acute hypoxia. Biochem. Biophys. Res. Commun. 2004, 318, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.E.; Sulakhe, P.V. Sarcoplasmic reticulum Ca2+-pump dysfunction in rat cardiomyocytes briefly exposed to hydroxyl radicals. Free Radic. Biol. Med. 1997, 22, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Goldhaber, J.I. Free radicals enhance Na+/Ca2+ exchange in ventricular myocytes. Am. J. Physiol. 1996, 271, H823–H833. [Google Scholar] [PubMed]
- Anzai, K.; Ogawa, K.; Ozawa, T.; Yamamoto, H. Oxidative modification of ion channel activity of ryanodine receptor. Antioxid. Redox Signal. 2000, 2, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Dhamoon, A.S.; Jalife, J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm 2005, 2, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed]
- Atienza, F.; Almendral, J.; Moreno, J.; Vaidyanathan, R.; Talkachou, A.; Kalifa, J.; Arenal, A.; Villacastin, J.P.; Torrecilla, E.G.; Sanchez, A.; et al. Activation of inward rectifier potassium channels accelerates atrial fibrillation in humans: Evidence for a reentrant mechanism. Circulation 2006, 114, 2434–2442. [Google Scholar] [CrossRef]
- Dobrev, D.; Wettwer, E.; Kortner, A.; Knaut, M.; Schuler, S.; Ravens, U. Human inward rectifier potassium channels in chronic and postoperative atrial fibrillation. Cardiovas. Res. 2002, 54, 397–404. [Google Scholar] [CrossRef]
- Anumonwo, J.M.; Lopatin, A.N. Cardiac strong inward rectifier potassium channels. J. Mol. Cell. Cardiol. 2010, 48, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Kovoor, P.; Wickman, K.; Maguire, C.T.; Pu, W.; Gehrmann, J.; Berul, C.I.; Clapham, D.E. Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 2001, 37, 2136–2143. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.J.; Ehrlich, J.R.; Chartier, D.; Qi, X.Y.; Xiao, L.; Nattel, S. Kir3-based inward rectifier potassium current: Potential role in atrial tachycardia remodeling effects on atrial repolarization and arrhythmias. Circulation 2006, 113, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.J.; Kim, Y.H.; Yashima, M.; Athill, C.A.; Ting, C.T.; Karagueuzian, H.S.; Chen, P.S. Progressive action potential duration shortening and the conversion from atrial flutter to atrial fibrillation in the isolated canine right atrium. J. Am. Coll. Cardiol. 2001, 38, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H.; Wakili, R.; Heo, J.H.; Chartier, D.; Kim, H.S.; Kim, S.J.; Lee, J.W.; Qi, X.Y.; Nattel, S.; Cha, T.J. Role of constitutively active acetylcholine-mediated potassium current in atrial contractile dysfunction caused by atrial tachycardia remodelling. Europace 2010, 12, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Li, T.; Lei, M.; Tan, X.Q.; Yang, Y.; Liu, T.P.; Pei, J.; Zeng, X.R. Increased small conductance calcium-activated potassium channel (SK2 channel) current in atrial myocytes of patients with persistent atrial fibrillation. Zhonghua Xin Xue Guan Bing Za Zhi 2011, 39, 147–151. (In Chinese) [Google Scholar] [PubMed]
- Qi, X.Y.; Diness, J.G.; Brundel, B.J.; Zhou, X.B.; Naud, P.; Wu, C.T.; Huang, H.; Harada, M.; Aflaki, M.; Dobrev, D.; et al. Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation 2014, 129, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Alekseev, A.E.; Liu, X.K.; Park, S.; Zingman, L.V.; Bienengraeber, M.; Sattiraju, S.; Ballew, J.D.; Jahangir, A.; Terzic, A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet. 2006, 15, 2185–2191. [Google Scholar] [CrossRef] [PubMed]
- Bilodeau, M.T.; Trotter, B.W. Kv1.5 blockers for the treatment of atrial fibrillation: Approaches to optimization of potency and selectivity and translation to in vivo pharmacology. Curr. Top. Med. Chem. 2009, 9, 436–451. [Google Scholar] [CrossRef]
- Tamargo, J.; Caballero, R.; Gomez, R.; Delpon, E. I(Kur)/Kv1.5 channel blockers for the treatment of atrial fibrillation. Expert Opin. Investig. Drugs 2009, 18, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Caouette, D.; Dongmo, C.; Berube, J.; Fournier, D.; Daleau, P. Hydrogen peroxide modulates the Kv1.5 channel expressed in a mammalian cell line. Naunyn-Schmied. Arch. Pharmacol. 2003, 368, 479–486. [Google Scholar] [CrossRef]
- Kolbe, K.; Schonherr, R.; Gessner, G.; Sahoo, N.; Hoshi, T.; Heinemann, S.H. Cysteine 723 in the C-linker segment confers oxidative inhibition of hERG1 potassium channels. J. Physiol. 2010, 588, 2999–3009. [Google Scholar] [PubMed]
- Dhein, S. Pharmacology of gap junctions in the cardiovascular system. Cardiovasc. Res. 2004, 62, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Saffitz, J.E. Connexins, conduction, and atrial fibrillation. N. Engl. J. Med. 2006, 354, 2712–2714. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Iwasaki, Y.K.; Nattel, S. Connexins and atrial fibrillation: Filling in the gaps. Circulation 2012, 125, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Casaclang-Verzosa, G.; Gersh, B.J.; Tsang, T.S. Structural and functional remodeling of the left atrium: Clinical and therapeutic implications for atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Corradi, D.; Callegari, S.; Maestri, R.; Benussi, S.; Alfieri, O. Structural remodeling in atrial fibrillation. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Moe, G.W.; Nili, N.; Rezaei, E.; Eskandarian, M.; Butany, J.; Strauss, B.H. The cardiac atria are chambers of active remodeling and dynamic collagen turnover during evolving heart failure. J. Am. Coll. Cardiol. 2004, 43, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Burstein, B.; Comtois, P.; Michael, G.; Nishida, K.; Villeneuve, L.; Yeh, Y.H.; Nattel, S. Changes in connexin expression and the atrial fibrillation substrate in congestive heart failure. Circ. Res. 2009, 105, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Brilla, C.G.; Campbell, S.E.; Guarda, E.; Zhou, G.; Sriram, K. Myocardial fibrosis: Role of angiotensin II and aldosterone. Basic Res. Cardiol. 1993, 88, 107–124. [Google Scholar] [PubMed]
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Induction of cardiac fibrosis by transforming growth factor-beta(1). Mol. Genet. Metab. 2000, 71, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Ponten, A.; Folestad, E.B.; Pietras, K.; Eriksson, U. Platelet-derived growth factor D induces cardiac fibrosis and proliferation of vascular smooth muscle cells in heart-specific transgenic mice. Circ. Res. 2005, 97, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Boixel, C.; Fontaine, V.; Rucker-Martin, C.; Milliez, P.; Louedec, L.; Michel, J.B.; Jacob, M.P.; Hatem, S.N. Fibrosis of the left atria during progression of heart failure is associated with increased matrix metalloproteinases in the rat. J. Am. Coll. Cardiol. 2003, 42, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Burstein, B.; Qi, X.Y.; Yeh, Y.H.; Calderone, A.; Nattel, S. Atrial cardiomyocyte tachycardia alters cardiac fibroblast function: A novel consideration in atrial remodeling. Cardiovasc. Res. 2007, 76, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Sevignani, C.; Calin, G.A.; Siracusa, L.D.; Croce, C.M. Mammalian microRNAs: A small world for fine-tuning gene expression. Mamm. Genome 2006, 17, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Ardekani, A.M.; Naeini, M.M. The role of microRNAs in human diseases. Avicenna J. Med. Biotechnol. 2010, 2, 161–179. [Google Scholar] [PubMed]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Latronico, M.V.; Cavarretta, E. MicroRNAs in cardiovascular diseases: Current knowledge and the road ahead. J. Am. Coll. Cardiol. 2014, 63, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Weiz, L.; Burwinkel, B. Extracellular miRNAs: The mystery of their origin and function. Trends Biochem. Sci. 2012, 37, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Koppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci. Signal. 2009, 2, ra81. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Nagaeva, O.; Kargl, D.; Baranov, V.; Mincheva-Nilsson, L. Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PLoS One 2011, 6, e16899. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M. TLRs as miRNA receptors. Cancer Res. 2012, 72, 6333–6337. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Rippo, M.R.; Prattichizzo, F.; Babini, L.; Graciotti, L.; Recchioni, R.; Procopio, A.D. Toll like receptor signaling in “inflammaging”: microRNA as new players. Immun. Ageing 2013, 10, 11. [Google Scholar] [PubMed]
- Ohshima, K.; Inoue, K.; Fujiwara, A.; Hatakeyama, K.; Kanto, K.; Watanabe, Y.; Muramatsu, K.; Fukuda, Y.; Ogura, S.; Yamaguchi, K.; et al. Let-7 microRNA family is selectively secreted into the extracellular environment via exosomes in a metastatic gastric cancer cell line. PLoS One 2010, 5, e13247. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Pashkow, F.J. Introduction. Oxidative stress and heart disease. Am. J. Cardiol. 2008, 101, 1D–2D. [Google Scholar] [CrossRef] [PubMed]
- Ogura, S.; Shimosawa, T. Oxidative stress and organ damages. Curr. Hypertens. Rep. 2014, 16, 452. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [PubMed]
- Hong, S.Y.; Roze, L.V.; Linz, J.E. Oxidative stress-related transcription factors in the regulation of secondary metabolism. Toxins 2013, 5, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Forstermann, U. Oxidative stress in vascular disease and its pharmacological prevention. Trends Pharmacol. Sci. 2013, 34, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Ceconi, C.; Boraso, A.; Cargnoni, A.; Ferrari, R. Oxidative stress in cardiovascular disease: Myth or fact? Arch. Biochem. Biophys. 2003, 420, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Otani, H. Oxidative stress as pathogenesis of cardiovascular risk associated with metabolic syndrome. Antioxid. Redox Signal. 2011, 15, 1911–1926. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, M.; Gonano, L.A.; Back, T.G.; Chen, S.R.; Vila Petroff, M. Role of CaMKII and ROS in rapid pacing-induced apoptosis. J. Mol. Cell. Cardiol. 2013, 63, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Donoso, P.; Sanchez, G.; Bull, R.; Hidalgo, C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front. Biosci. 2011, 16, 553–567. [Google Scholar] [CrossRef]
- Amberg, G.C.; Earley, S.; Glapa, S.A. Local regulation of arterial l-type calcium channels by reactive oxygen species. Circ. Res. 2010, 107, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Sovari, A.A.; Dudley, S.C., Jr. Reactive oxygen species-targeted therapeutic interventions for atrial fibrillation. Front. Physiol. 2012, 3, 311. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.R.; Chen, K.; Keaney, J.F., Jr. Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J. Biol. Chem. 2002, 277, 6017–6024. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sun, X.; Wedgwood, S.; Black, S.M. Hydrogen peroxide decreases endothelial nitric oxide synthase promoter activity through the inhibition of AP-1 activity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L370–L377. [Google Scholar] [CrossRef]
- Kumar, S.; Sun, X.; Wiseman, D.A.; Tian, J.; Umapathy, N.S.; Verin, A.D.; Black, S.M. Hydrogen peroxide decreases endothelial nitric oxide synthase promoter activity through the inhibition of Sp1 activity. DNA Cell Biol. 2009, 28, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Jourd’heuil, D.; Jourd’heuil, F.L.; Kutchukian, P.S.; Musah, R.A.; Wink, D.A.; Grisham, M.B. Reaction of superoxide and nitric oxide with peroxynitrite. Implications for peroxynitrite-mediated oxidation reactions in vivo. J. Biol. Chem. 2001, 276, 28799–28805. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Wauquier, F.; Eid, A.A.; Roman, L.J.; Ghosh-Choudhury, G.; Khazim, K.; Block, K.; Gorin, Y. Nox4 NADPH oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin II: Role of mitochondrial reactive oxygen species. J. Biol. Chem. 2013, 288, 28668–28686. [Google Scholar] [CrossRef] [PubMed]
- Silberman, G.A.; Fan, T.H.; Liu, H.; Jiao, Z.; Xiao, H.D.; Lovelock, J.D.; Boulden, B.M.; Widder, J.; Fredd, S.; Bernstein, K.E.; et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 2010, 121, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Dudley, S.C., Jr.; Hoch, N.E.; McCann, L.A.; Honeycutt, C.; Diamandopoulos, L.; Fukai, T.; Harrison, D.G.; Dikalov, S.I.; Langberg, J. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: Role of the NADPH and xanthine oxidases. Circulation 2005, 112, 1266–1273. [Google Scholar] [CrossRef]
- Rubart, M.; Zipes, D.P. NO hope for patients with atrial fibrillation. Circulation 2002, 106, 2764–2766. [Google Scholar] [CrossRef] [PubMed]
- Morita, N.; Sovari, A.A.; Xie, Y.; Fishbein, M.C.; Mandel, W.J.; Garfinkel, A.; Lin, S.F.; Chen, P.S.; Xie, L.H.; Chen, F.; et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1594–H1605. [Google Scholar] [CrossRef] [PubMed]
- Neuman, R.B.; Bloom, H.L.; Shukrullah, I.; Darrow, L.A.; Kleinbaum, D.; Jones, D.P.; Dudley, S.C., Jr. Oxidative stress markers are associated with persistent atrial fibrillation. Clin. Chem. 2007, 53, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Lim, D.S.; Lee, J.H.; Shim, W.J.; Ro, Y.M.; Park, G.H.; Becker, K.G.; Cho-Chung, Y.S.; Kim, M.K. Gene expression profiling of oxidative stress on atrial fibrillation in humans. Exp. Mol. Med. 2003, 35, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Sovari, A.A.; Dudley, S.C. Antioxidant therapy for atrial fibrillation: Lost in translation? Heart 2012, 98, 1615–1616. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H. MicroRNA regulation of cardiac conduction and arrhythmias. Transl. Res. 2013, 161, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Girmatsion, Z.; Biliczki, P.; Bonauer, A.; Wimmer-Greinecker, G.; Scherer, M.; Moritz, A.; Bukowska, A.; Goette, A.; Nattel, S.; Hohnloser, S.H.; et al. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm 2009, 6, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Curcio, A.; Torella, D.; Iaconetti, C.; Pasceri, E.; Sabatino, J.; Sorrentino, S.; Giampa, S.; Micieli, M.; Polimeni, A.; Henning, B.J.; et al. MicroRNA-1 downregulation increases connexin 43 displacement and induces ventricular tachyarrhythmias in rodent hypertrophic hearts. PLoS One 2013, 8, e70158. [Google Scholar] [CrossRef] [PubMed]
- Belevych, A.E.; Sansom, S.E.; Terentyeva, R.; Ho, H.T.; Nishijima, Y.; Martin, M.M.; Jindal, H.K.; Rochira, J.A.; Kunitomo, Y.; Abdellatif, M.; et al. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS One 2011, 6, e28324. [Google Scholar] [CrossRef]
- Terentyev, D.; Belevych, A.E.; Terentyeva, R.; Martin, M.M.; Malana, G.E.; Kuhn, D.E.; Abdellatif, M.; Feldman, D.S.; Elton, T.S.; Gyorke, S. miR-1 overexpression enhances Ca2+ release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ. Res. 2009, 104, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, C.; Liu, Y.; Wang, S.; Ye, P.; Miao, X.; Xia, J. The expression levels of plasma micoRNAs in atrial fibrillation patients. PLoS One 2012, 7, e44906. [Google Scholar] [CrossRef] [PubMed]
- Danielson, L.S.; Park, D.S.; Rotllan, N.; Chamorro-Jorganes, A.; Guijarro, M.V.; Fernandez-Hernando, C.; Fishman, G.I.; Phoon, C.K.; Hernando, E. Cardiovascular dysregulation of miR-17–92 causes a lethal hypertrophic cardiomyopathy and arrhythmogenesis. FASEB J. 2013, 27, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Cardin, S.; Guasch, E.; Luo, X.; Naud, P.; le Quang, K.; Shi, Y.; Tardif, J.C.; Comtois, P.; Nattel, S. Role for MicroRNA-21 in atrial profibrillatory fibrotic remodeling associated with experimental postinfarction heart failure. Circ. Arrhythm. Electrophysiol. 2012, 5, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Barana, A.; Matamoros, M.; Dolz-Gaiton, P.; Perez-Hernandez, M.; Amoros, I.; Nunez, M.; Sacristan, S.; Pedraz, A.; Pinto, A.; Fernandez-Aviles, F.; et al. Chronic atrial fibrillation increases microRNA-21 in human atrial myocytes decreasing l-type calcium current. Circ. Arrhythm. Electrophysiol. 2014, 7, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Pan, Z.; Shan, H.; Xiao, J.; Sun, X.; Wang, N.; Lin, H.; Xiao, L.; Maguy, A.; Qi, X.Y.; et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J. Clin. Investig. 2013, 123, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Dawson, K.; Wakili, R.; Ordog, B.; Clauss, S.; Chen, Y.; Iwasaki, Y.; Voigt, N.; Qi, X.Y.; Sinner, M.F.; Dobrev, D.; et al. MicroRNA29: A mechanistic contributor and potential biomarker in atrial fibrillation. Circulation 2013, 127. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, S.; Yu, B.; Liu, S. Expression of miR-133 and miR-30 in chronic atrial fibrillation in canines. Mol. Med. Rep. 2012, 5, 1457–1460. [Google Scholar] [PubMed]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; van der Made, I.; Herias, V.; van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 2009, 104, 170–178. [Google Scholar] [CrossRef]
- Li, T.; Cao, H.; Zhuang, J.; Wan, J.; Guan, M.; Yu, B.; Li, X.; Zhang, W. Identification of miR-130a, miR-27b and miR-210 as serum biomarkers for atherosclerosis obliterans. Clin. Chim. Acta 2011, 412, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Osbourne, A.; Calway, T.; Broman, M.; McSharry, S.; Earley, J.; Kim, G.H. Downregulation of connexin43 by microRNA-130a in cardiomyocytes results in cardiac arrhythmias. J. Mol. Cell. Cardiol. 2014, 74, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhang, H.; Xiao, J.; Wang, Z. Regulation of human cardiac ion channel genes by microRNAs: Theoretical perspective and pathophysiological implications. Cell. Physiol. Biochem. 2010, 25, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.J.; Jang, J.K.; Ham, O.; Song, B.W.; Lee, S.Y.; Lee, C.Y.; Park, J.H.; Lee, J.; Seo, H.H.; Choi, E.; et al. MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIdelta. Biochem. Biophys. Res. Commun. 2013, 435, 720–726. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Gao, X.; Zhang, R.; Zhang, Y.; Liang, H.; Xu, C.; Du, W.; Zhang, Y.; Liu, X.; et al. MicroRNA-328 as a regulator of cardiac hypertrophy. Int. J. Cardiol. 2014, 173, 268–276. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Y.; Wang, N.; Pan, Z.; Gao, X.; Zhang, F.; Zhang, Y.; Shan, H.; Luo, X.; Bai, Y.; et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 2010, 122, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Ling, T.Y.; Wang, X.L.; Chai, Q.; Lau, T.W.; Koestler, C.M.; Park, S.J.; Daly, R.C.; Greason, K.L.; Jen, J.; Wu, L.Q.; et al. Regulation of the SK3 channel by microRNA-499—Potential role in atrial fibrillation. Heart Rhythm 2013, 10, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Matkovich, S.J.; Hu, Y.; Eschenbacher, W.H.; Dorn, L.E.; Dorn, G.W., 2nd. Direct and indirect involvement of microRNA-499 in clinical and experimental cardiomyopathy. Circ. Res. 2012, 111, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, B.; Cui, H.; Du, Y.; Song, Y.; Yang, L.; Zhang, Q.; Sun, F.; Luo, D.; Xu, C.; et al. Let-7e replacement yields potent anti-arrhythmic efficacy via targeting beta 1-adrenergic receptor in rat heart. J. Cell. Mol. Med. 2014, 18, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Cai, J.; Tang, Y.; Zhao, Q. MiR-17–92 cluster is a novel regulatory gene of cardiac ischemic/reperfusion injury. Med. Hypotheses 2013, 81, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Bonauer, A.; Dimmeler, S. The microRNA-17-92 cluster: Still a miRacle? Cell Cycle 2009, 8, 3866–3873. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Penninger, J.M. Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc. Res. 2009, 82, 250–260. [Google Scholar] [CrossRef]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Huebert, R.C.; Li, Q.; Adhikari, N.; Charles, N.J.; Han, X.; Ezzat, M.K.; Grindle, S.; Park, S.; Ormaza, S.; Fermin, D.; et al. Identification and regulation of Sprouty1, a negative inhibitor of the ERK cascade, in the human heart. Physiol. Genomics 2004, 18, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Muda, M.; Theodosiou, A.; Rodrigues, N.; Boschert, U.; Camps, M.; Gillieron, C.; Davies, K.; Ashworth, A.; Arkinstall, S. The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. J. Biol. Chem. 1996, 271, 27205–27208. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.W.; Xu, M.; Huang, H.; Wang, J.; Chen, X. miR-21 Targets DUSP8 to promote collagen synthesis in high glucose treated primary cardiac fibroblasts. Can. J. Cardiol. 2014. [Google Scholar] [CrossRef]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [PubMed]
- Qu, Y.C.; Du, Y.M.; Wu, S.L.; Chen, Q.X.; Wu, H.L.; Zhou, S.F. Activated nuclear factor-kappaB and increased tumor necrosis factor-alpha in atrial tissue of atrial fibrillation. Scand. Cardiovasc. J. 2009, 43, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, T.; Wang, W.; Cutler, M.J.; Wang, Q.; Voigt, N.; Rosenbaum, D.S.; Dobrev, D.; Wehrens, X.H. Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circ. Res. 2012, 110, 465–470. [Google Scholar] [CrossRef] [PubMed]
- McManus, D.D.; Lin, H.; Tanriverdi, K.; Quercio, M.; Yin, X.; Larson, M.G.; Ellinor, P.T.; Levy, D.; Freedman, J.E.; Benjamin, E.J. Relations between circulating microRNAs and atrial fibrillation: Data from the Framingham Offspring Study. Heart Rhythm 2014, 11, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Chandy, K.G.; Fantino, E.; Wittekindt, O.; Kalman, K.; Tong, L.L.; Ho, T.H.; Gutman, G.A.; Crocq, M.A.; Ganguli, R.; Nimgaonkar, V.; et al. Isolation of a novel potassium channel gene hSKCa3 containing a polymorphic CAG repeat: A candidate for schizophrenia and bipolar disorder? Mol. Psychiatry 1998, 3, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Wallukat, G. The beta-adrenergic receptors. Herz 2002, 27, 683–690. [Google Scholar] [CrossRef]
- Kuhlkamp, V.; Bosch, R.; Mewis, C.; Seipel, L. Use of beta-blockers in atrial fibrillation. Am. J. Cardiovasc. Drugs 2002, 2, 37–42. [Google Scholar] [CrossRef]
- Chen, T.; Ding, G.; Jin, Z.; Wagner, M.B.; Yuan, Z. Insulin ameliorates miR-1-induced injury in H9c2 cells under oxidative stress via Akt activation. Mol. Cell. Biochem. 2012, 369, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Choi, E.; Cha, M.J.; Song, B.W.; Ham, O.; Lee, S.Y.; Yoon, C.; Lee, C.Y.; Park, J.H.; Lee, S.H.; et al. Up-regulation of miR-26a promotes apoptosis of hypoxic rat neonatal cardiomyocytes by repressing GSK-3beta protein expression. Biochem. Biophys. Res. Commun. 2012, 423, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jia, Z.; Zhang, C.; Sun, M.; Wang, W.; Chen, P.; Ma, K.; Zhang, Y.; Li, X.; Zhou, C. miR-499 protects cardiomyocytes from H2O2-induced apoptosis via its effects on Pdcd4 and Pacs2. RNA Biol. 2014, 11, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, X.; Cheng, Y.; Yang, J.; Huo, Y.; Zhang, C. Involvement of microRNAs in hydrogen peroxide-mediated gene regulation and cellular injury response in vascular smooth muscle cells. J. Biol. Chem. 2009, 284, 7903–7913. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Liu, X.; Zhang, S.; Lin, Y.; Yang, J.; Zhang, C. MicroRNA-21 protects against the H2O2-induced injury on cardiac myocytes via its target gene PDCD4. J. Mol. Cell. Cardiol. 2009, 47, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Donath, S.; Li, Y.; Qin, D.; Prabhakar, B.S.; Li, P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010, 6, e1000795. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Hu, Y.; Hou, L.; Ju, J.; Li, X.; Du, N.; Guan, X.; Liu, Z.; Zhang, T.; Qin, W.; et al. Beta-Blocker carvedilol protects cardiomyocytes against oxidative stress-induced apoptosis by up-regulating miR-133 expression. J. Mol. Cell. Cardiol. 2014, 75, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yan, G.; Li, Q.; Sun, H.; Hu, Y.; Sun, J.; Xu, B. MicroRNA-145 protects cardiomyocytes against hydrogen peroxide (H2O2)-induced apoptosis through targeting the mitochondria apoptotic pathway. PLoS One 2012, 7, e44907. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.D.; Savage, J.E.; Cao, L.; Soule, B.P.; Ly, D.; DeGraff, W.; Harris, C.C.; Mitchell, J.B.; Simone, N.L. Cellular stress induced alterations in microRNA let-7a and let-7b expression are dependent on p53. PLoS One 2011, 6, e24429. [Google Scholar] [PubMed]
- Michel, J.B.; Li, Z.; Lacolley, P. Smooth muscle cells and vascular diseases. Cardiovasc. Res. 2012, 95, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Agmon, Y.; Khandheria, B.K.; Meissner, I.; Schwartz, G.L.; Petterson, T.M.; O’Fallon, W.M.; Gentile, F.; Spittell, P.C.; Whisnant, J.P.; Wiebers, D.O.; et al. Association of atrial fibrillation and aortic atherosclerosis: A population-based study. Mayo Clin. Proc. 2001, 76, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Willeit, K.; Pechlaner, R.; Egger, G.; Weger, S.; Oberhollenzer, M.; Willeit, J.; Kiechl, S. Carotid atherosclerosis and incident atrial fibrillation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2660–2665. [Google Scholar] [CrossRef] [PubMed]
- Kulagina, N.V.; Michael, A.C. Monitoring hydrogen peroxide in the extracellular space of the brain with amperometric microsensors. Anal. Chem. 2003, 75, 4875–4881. [Google Scholar] [CrossRef] [PubMed]
- Tarpey, M.M.; Wink, D.A.; Grisham, M.B. Methods for detection of reactive metabolites of oxygen and nitrogen: In vitro and in vivo considerations. Am. J. Physiol. 2004, 286, R431–R444. [Google Scholar]
- Ozaydin, M. Atrial fibrillation and inflammation. World J. Cardiol. 2010, 2, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Aldhoon, B.; Kucera, T.; Smorodinova, N.; Martinek, J.; Melenovsky, V.; Kautzner, J. Associations between cardiac fibrosis and permanent atrial fibrillation in advanced heart failure. Physiol. Res. 2013, 62, 247–255. [Google Scholar] [PubMed]
- Li, C.; Wang, F.; Yang, Y.; Fu, F.; Xu, C.; Shi, L.; Li, S.; Xia, Y.; Wu, G.; Cheng, X.; et al. Significant association of SNP rs2106261 in the ZFHX3 gene with atrial fibrillation in a Chinese Han GeneID population. Hum. Genet. 2011, 129, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, J.; Kors, J.A.; Hofman, A.; van Rooij, F.J.; Witteman, J.C. Cigarette smoking and risk of atrial fibrillation: The rotterdam study. Am. Heart J. 2008, 156, 1163–1169. [Google Scholar] [PubMed]
- Djousse, L.; Levy, D.; Benjamin, E.J.; Blease, S.J.; Russ, A.; Larson, M.G.; Massaro, J.M.; D’Agostino, R.B.; Wolf, P.A.; Ellison, R.C. Long-term alcohol consumption and the risk of atrial fibrillation in the framingham study. Am. J. Cardiol. 2004, 93, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Tong, Y.; Zhang, H.M.; Wang, K.; Hu, T.; Shan, G.; Sun, J.; Guo, A.Y. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum. Mutat. 2012, 33, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Li, J.; Chen, F.; Geng, H.; Pan, M. A polymorphism rs11614913 in pre-microRNAs may be associated with atrial fibrillation in Han Chinese population. J. Am. Coll. Cardiol. 2014, 64, GW25-e0274. [Google Scholar] [CrossRef]
- Violi, F.; Loffredo, L. Thromboembolism or atherothromboembolism in atrial fibrillation? Circ. Arrhythm. Electrophysiol. 2012, 5, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Choi, E.; Cha, M.-J.; Hwang, K.-C. Looking into a Conceptual Framework of ROS–miRNA–Atrial Fibrillation. Int. J. Mol. Sci. 2014, 15, 21754-21776. https://doi.org/10.3390/ijms151221754
Lee S, Choi E, Cha M-J, Hwang K-C. Looking into a Conceptual Framework of ROS–miRNA–Atrial Fibrillation. International Journal of Molecular Sciences. 2014; 15(12):21754-21776. https://doi.org/10.3390/ijms151221754
Chicago/Turabian StyleLee, Seahyoung, Eunhyun Choi, Min-Ji Cha, and Ki-Chul Hwang. 2014. "Looking into a Conceptual Framework of ROS–miRNA–Atrial Fibrillation" International Journal of Molecular Sciences 15, no. 12: 21754-21776. https://doi.org/10.3390/ijms151221754