Pine Bark and Green Tea Concentrated Extracts: Antioxidant Activity and Comprehensive Characterization of Bioactive Compounds by HPLC–ESI-QTOF-MS


Abstract
:1. Introduction

2. Results and Discussion
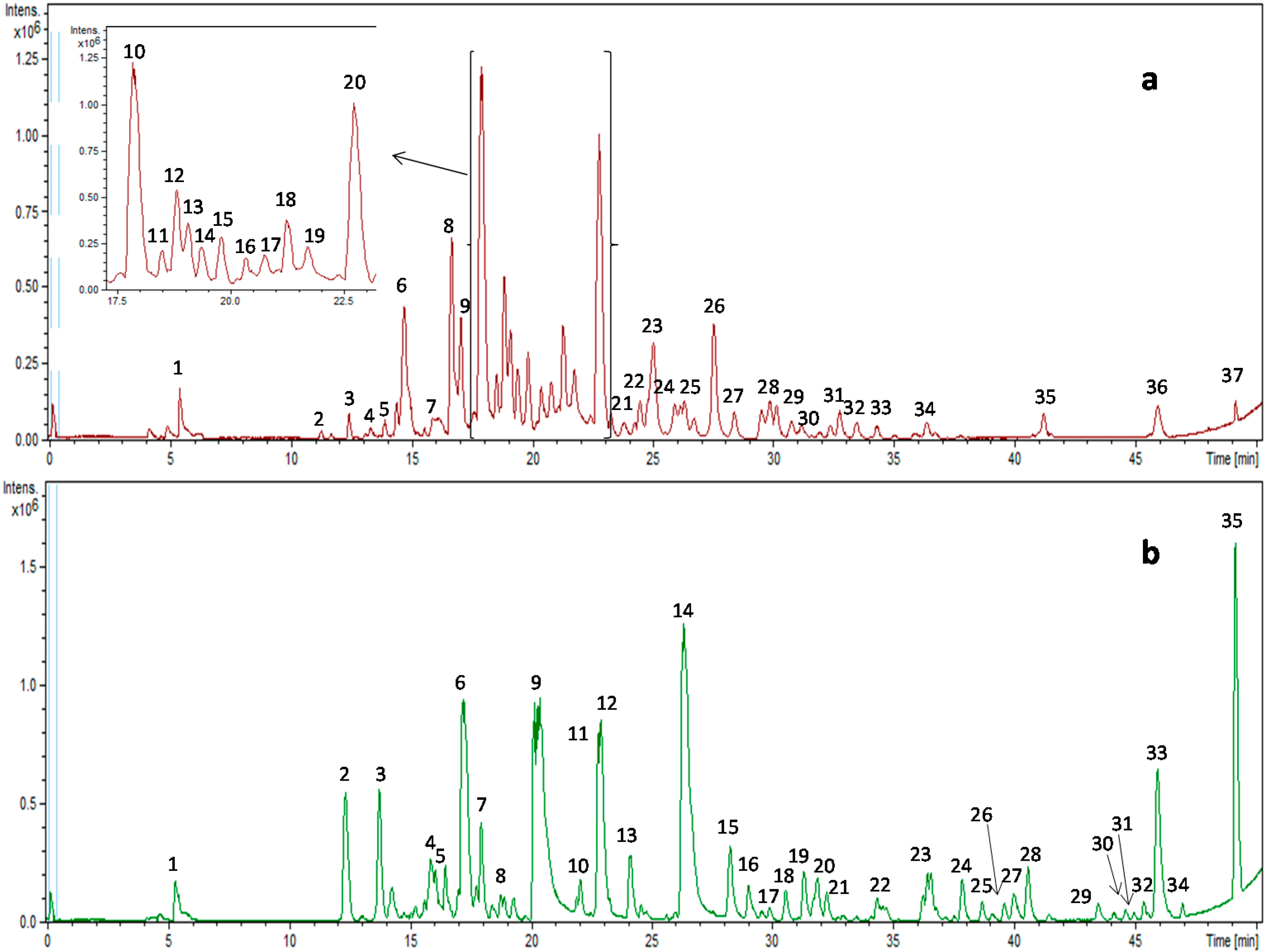
2.1. Characterization of Polar Compounds by High-Performance Liquid Chromatography Coupled to Electrospray Ionization Mass Spectrometry (HPLC–ESI-QTOF-MS)
2.1.1. Pine Bark Extract
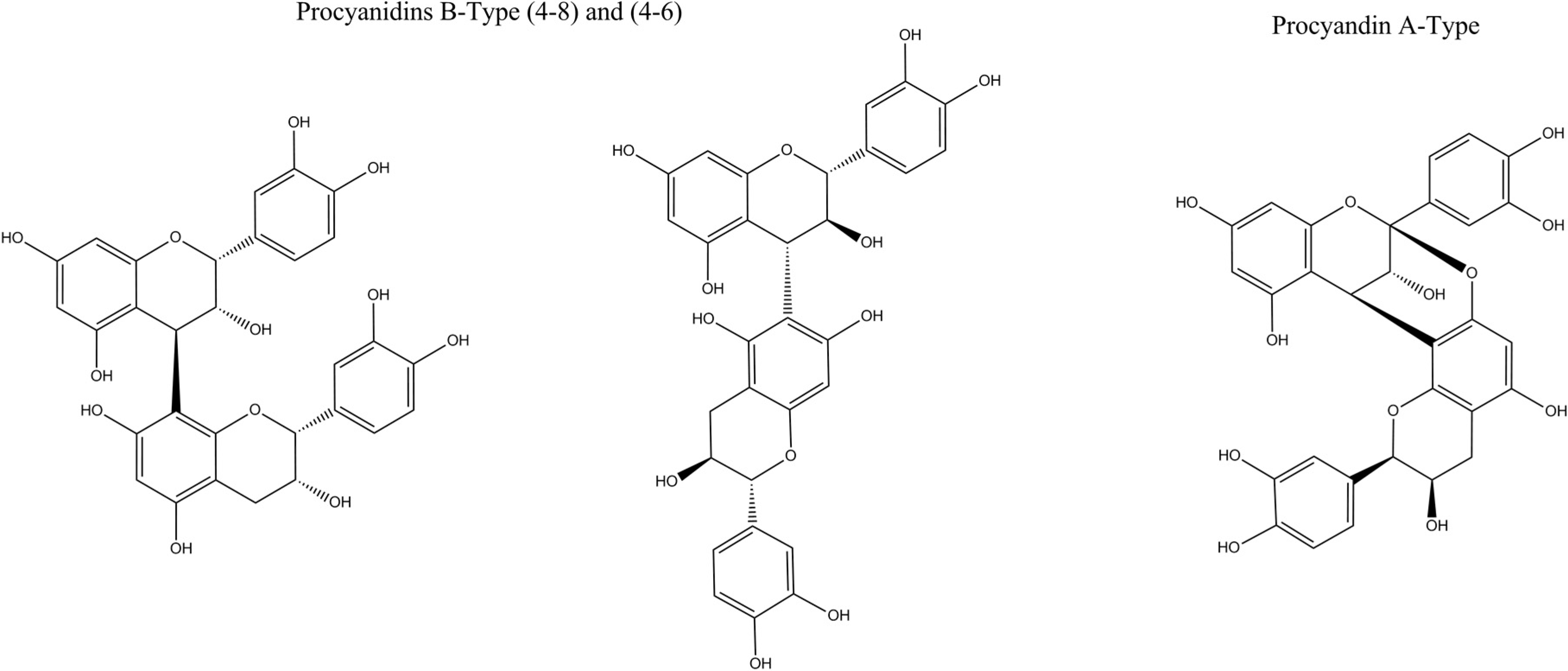
Flavan-3-ol and Its Derivatives


{kind=link}
{kind=link}
{kind=link}
| Peak | Proposed Compound | RT (min) | [M–H]− Measured | [M–H]− Calculated | Error (ppm) | mSigma | Fragmentation Pattern | Molecular Formula | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Sucrose | 5.4 | 3,411,098 | 3,411,089 | 2.6 | 9 | non fragmented | C12H22O11 | – |
| 2 | Procyanidin C | 11.3 | 8,651,981 | 8,651,985 | 0.5 | 30 | 577, 289 | C45H38O18 | [1,13,15,24,25,26,27] |
| 3 | Gardenoside | 12.4 | 4,031,257 | 4,031,246 | 2.7 | 6.2 | non fragmented | C17H24O11 | – |
| 4 | Procyanidin A (isomer 1) | 13.3 | 5,751,191 | 5,751,195 | 0.6 | 22 | 289 | C30H24O12 | – |
| 5 | Procyanidin A (isomer 2) | 13.9 | 5,751,189 | 5,751,195 | 1 | 34.8 | 289 | C30H24O12 | – |
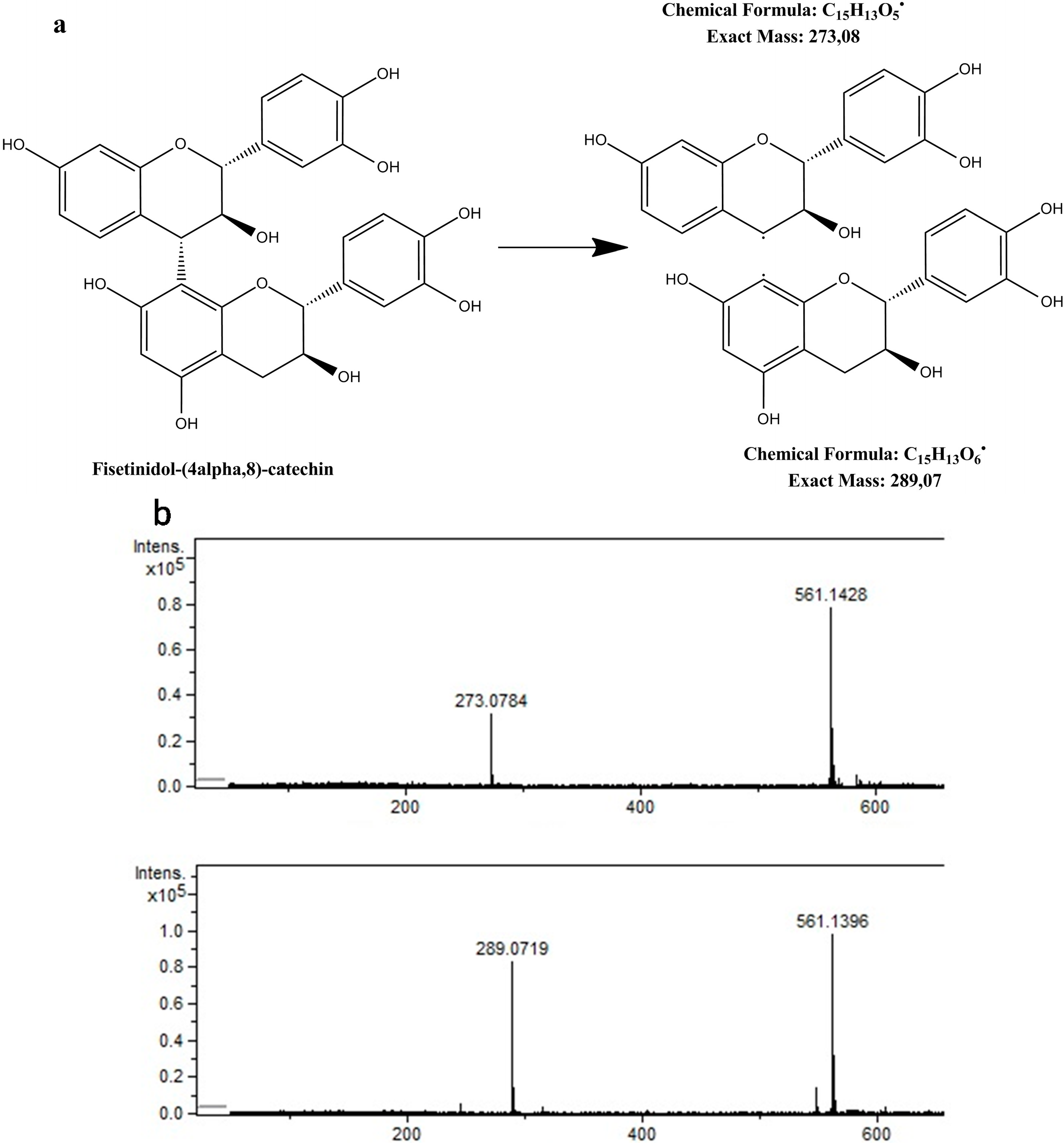
| 6 | Procyanidin B (isomer 1) | 14.7 | 5,771,366 | 5,771,351 | 2.6 | 5.3 | 425 | C30H26O12 | [1,13,15,24,25,26,27] |
| 7 | Procyanidin B (isomer 2) | 15.9 | 5,771,347 | 5,771,351 | 0.7 | 13.2 | 425, 289 | C30H26O12 | [1,13,15,24,25,26,27] |
| 8 | Chalcan-flavan-3-ol dimer (isomer 1) | 16.7 | 5,791,532 | 5,791,508 | 4.2 | 5.2 | 561 | C30H28O12 | – |
| 9 | Procyanidin trimer A-type (isomer 1) | 17 | 8,631,842 | 8,631,829 | 0.4 | 27.4 | 289, 285 | C45H36O18 | – |
| 10 | (−)-epicatechin | 17.9 | 2,890,727 | 2,890,718 | 3.4 | 4.9 | 245 | C15H14O6 | [1,13,15,24,25,26,27] |
| 11 | Chalcan-flavan-3-ol dimer (isomer 2) | 18.5 | 5,791,512 | 5,791,508 | 0.7 | 4.3 | 289 | C30H28O12 | – |
| 12 | Chalcan-flavan-3-ol dimer (isomer 3) | 18.8 | 579,152 | 5,791,508 | 2.1 | 6.2 | 561, 289 | C30H28O12 | – |
| 13 | Chalcan-flavan-3-ol dimer (isomer 4) | 19.1 | 5,791,528 | 5,791,508 | 3.5 | 6.5 | 561 | C30H28O12 | – |
| 14 | Procyanidin trimer A-type (isomer 2) | 19.4 | 8,631,869 | 8,631,829 | 4.6 | 10.3 | 289 | C45H36O18 | – |
| 15 | Chalcan-flavan-3-ol dimer (isomer 5) | 19.8 | 5,791,516 | 5,791,508 | 1.3 | 5 | 561, 289 | C30H28O12 | – |
| 16 | Chalcan-flavan-3-ol dimer (isomer 6) | 20.4 | 579,152 | 5,791,508 | 2 | 4 | 561 | C30H28O12 | – |
| 17 | (Epi)fisetinidol-(epi)catechin (isomer 1) | 20.8 | 5,611,422 | 5,611,402 | 3.4 | 5.9 | 273 | C30H26O11 | – |
| 18 | Procyanidin A (isomer 3) | 21.2 | 5,751,195 | 5,751,195 | 0.1 | 18.9 | 289 | C30H24O12 | – |
| 19 | (Epi)fisetinidol-(epi)catechin (isomer 2) | 21.7 | 5,611,428 | 5,611,402 | 4.7 | 6.5 | 289, 273 | C30H26O11 | – |
| 20 | (+)-catechin | 22.7 | 2,890,729 | 2,890,718 | 3.8 | 7.8 | 245 | C15H14O6 | [1,13,15,24,25,26,27] |
| 21 | (Epi)fisetinidol-(epi)catechin (isomer 3) | 23.8 | 5,611,406 | 5,611,402 | 0.6 | 38.5 | 289 | C30H26O11 | – |
| 22 | (Epi)fisetinidol-(epi)catechin (isomer 4) | 24.4 | 5,611,409 | 5,611,402 | 1.1 | 9.6 | 273 | C30H26O11 | – |
| 23 | Procyanidin A (isomer 4) | 25 | 5,751,207 | 5,751,195 | 2 | 8 | 423 | C30H24O12 | – |
| 24 | (Epi)fisetinidol-(epi)catechin (isomer 5) | 25.9 | 5,611,413 | 5,611,402 | 2 | 2.7 | non fragmented | C30H26O11 | – |
| 25 | Procyanidin A (isomer 5) | 26.3 | 5,751,188 | 5,751,195 | 1.3 | 12.7 | 289 | C30H24O12 | – |
| 26 | Procyanidin A (isomer 6) | 27.5 | 5,751,221 | 5,751,195 | 4.6 | 21.8 | 289 | C30H24O12 | – |
| 27 | (Epi)fisetinidol-(epi)catechin (isomer 6) | 28.3 | 5,611,402 | 5,611,402 | 0 | 5 | non fragmented | C30H26O11 | – |
| 28 | (Epi)fisetinidol-(epi)catechin (isomer 7) | 29.8 | 5,611,416 | 5,611,402 | 2.3 | 3 | 289, 273 | C30H26O11 | – |
| 29 | Procyanidin A (isomer 7) | 30.7 | 57,512 | 5,751,195 | 0.8 | 14.3 | 289 | C30H24O12 | – |
| 30 | (Epi)fisetinidol-(epi)catechin (isomer 8) | 31.1 | 56,114 | 5,611,402 | 0.5 | 11.8 | 245 | C30H26O11 | – |
| 31 | Procyanidin A (isomer 8) | 32.7 | 5,751,205 | 5,751,195 | 1.7 | 10.7 | 285 | C30H24O12 | – |
| 32 | (Epi)fisetinidol-(epi)catechin (isomer 9) | 33.4 | 5,611,418 | 5,611,402 | 2.8 | 7.9 | 289 | C30H26O11 | – |
| 33 | Quercetin rhamnosylrutinoside | 34,2 | 7,552,041 | 755,204 | 0.2 | 11.9 | 301 | C33H40O20 | [28] |
| 34 | Rutin | 36.3 | 6,091,476 | 6,091,461 | 0.7 | 14.4 | 301 | C27H30O16 | [28] |
| 35 | Isorhamnetin rutinoside | 41.1 | 6,231,614 | 6,231,618 | 0.6 | 10.5 | 315 | C28H32O16 | [25] |
| 36 | Quercetin | 45.8 | 3,010,357 | 3,010,354 | 0.9 | 7.4 | non fragmented | C15H10O7 | [28,29] |
| 37 | Kaempferol | 49 | 285,041 | 2,850,405 | 1.7 | 11.2 | non fragmented | C15H10O6 | [30,31] |
Monomeric Forms
B and A-Type Oligomeric Forms
Flavonols
Other Compounds

2.1.2. Green Tea Extract
| Peak | Proposed Compound | RT (min) | [M−H]− Measured | [M−H]− Calculated | Error (ppm) | mSigma | Fragmentation Pattern | Molecular Formula | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Quinic acid | 5.3 | 1,910,562 | 1,910,561 | 0.4 | 7 | 127 | C7H12O6 | [40,41] |
| 2 | Gallic acid | 12.3 | 1,690,143 | 1,690,142 | 0.4 | 4.3 | 125 | C7H6O5 | [17,42] |
| 3 | (Epi)gallocatechin (isomer 1) | 13.7 | 3,050,663 | 3,050,667 | 1.1 | 6.6 | 169 | C15H14O7 | [17,40,43,44] |
| 4 | (Epi)gallocatechin-(epi)gallocatechin gallate | 15.8 | 7,451,397 | 745,141 | 1.8 | 9.2 | 457, 169 | C37H30O17 | [40] |
| 5 | (Epi)gallocatechin gallate glucoside | 16.5 | 6,191,308 | 6,191,305 | 0.5 | 4.7 | 457, 305 | C28H28O16 | – |
| 6 | (Epi)gallocatechin (isomer 2) | 17.2 | 3,050,677 | 3,050,667 | 3.4 | 3 | 261, 219, 179, 165 | C15H14O7 | [17,40,43,44] |
| 7 | (−)-epicatechin | 17.9 | 2,890,725 | 2,890,718 | 2.4 | 2 | 245 | C15H14O6 | [17,42,44] |
| 8 | Procyanidin B gallate (isomer 1) | 18.9 | 7,291,447 | 7,291,461 | 2 | 15.8 | 577, 169 | C37H30O16 | [43,45] |
| 9 | (Epi)gallocatechin gallate (isomer 1) | 20.3 | 4,570,789 | 4,570,776 | 2.7 | 2.7 | 169 | C22H18O11 | [17,40] |
| 10 | (Epi)gallocatechin digallate | 22 | 6,090,911 | 6,090,886 | 4.1 | 5 | 457, 305, 169 | C29H22O15 | [40,46] |
| 11 | (+)-catechin | 22.8 | 2,890,728 | 2,890,718 | 3.5 | 1.3 | 245 | C15H14O6 | [17,42,44] |
| 12 | (Epi)gallocatechin gallate (isomer 2) | 22.9 | 4,570,798 | 4,570,776 | 4.8 | 1.4 | 289, 169 | C22H18O11 | [17,40] |
| 13 | (Epi)gallocatechin methyl gallate | 24.1 | 4,710,938 | 4,710,933 | 1.2 | 3.3 | 305, 183 | C23H20O11 | [41,45,46] |
| 14 | (Epi)catechin gallate (isomer 1) | 26.3 | 4,410,843 | 4,410,827 | 3.7 | 1.8 | 169 | C22H18O10 | [17,40] |
| 15 | (Epi)catechin gallate (isomer 2) | 28.2 | 4,410,844 | 4,410,827 | 3.8 | 4.7 | 289, 169 | C22H18O10 | [17] |
| 16 | Procyanidin B gallate (isomer 2) | 29 | 7,291,464 | 7,291,461 | 0.4 | 79.7 | 441, 289, 169 | C37H30O16 | [43,45] |
| 17 | Eriodictyol | 29.9 | 2,870,565 | 2,870,561 | 1.3 | 9.4 | non fragmented | C15H12O6 | [17] |
| 18 | (Epi)catechin methyl gallate | 30.5 | 4,550,987 | 4,550,984 | 0.6 | 15.8 | 289, 183 | C23H20O10 | [47] |
| 19 | Epiafzelechin gallate | 31.3 | 425,088 | 4,250,878 | 1.1 | 8.4 | 169 | C22H18O9 | [48] |
| 20 | Myricetin glucoside | 31.8 | 4,790,815 | 4,790,831 | 3.2 | 5.8 | 317 | C21H20O13 | [40,44] |
| 21 | Genistein glucoside (isomer 1) | 32.2 | 4,310,985 | 4,310,984 | 0.3 | 4.6 | 269 | C21H20O10 | – |
| 22 | Genistein glucoside (isomer 2) | 34.3 | 4,310,981 | 4,310,984 | 0.6 | 9.9 | 269 | C21H20O10 | – |
| 23 | Rutin | 36.4 | 6,091,486 | 6,091,461 | 4.1 | 7.3 | 463 | C27H30O16 | [40,45] |
| 24 | Naringenin | 37.8 | 271,062 | 2,710,612 | 3.1 | 2.7 | non fragmented | C15H12O5 | [17,49] |
| 25 | Kaempferol glucosylrutinoside | 38.6 | 7,552,056 | 755,204 | 2.1 | 12.5 | 447, 285 | C33H40O20 | [44,45] |
| 26 | Kaempferol-glucoside | 39.5 | 4,470,937 | 4,470,993 | 1 | 9.8 | 285 | C21H20O11 | [44,45] |
| 27 | Myricetin | 39.9 | 3,170,308 | 3,170,303 | 1.6 | 12.3 | non fragmented | C15H10O8 | [40,49] |
| 28 | Kaempferol rutinoside | 40.5 | 593,151 | 5,931,512 | 0.3 | 3.6 | 447 | C27H30O15 | [45] |
| 29 | Morin | 43.4 | 3,010,355 | 3,010,354 | 0.4 | 4.7 | non fragmented | C15H10O7 | [50] |
| 30 | Theaflavin gallate | 44.5 | 7,151,309 | 7,151,305 | 0.6 | 16.3 | 563, 545 | C36H28O16 | [17,40,43] |
| 31 | Theaflavin digallate | 44.9 | 8,671,387 | 8,671,414 | 3.1 | 25.1 | 715, 563, 545 | C43H32O20 | [17,40,43] |
| 32 | Theaflavin | 45.3 | 5,631,187 | 5,631,195 | 0.5 | 7.3 | 545 | C29H24O12 | [17,40,43] |
| 33 | Quercetin | 45.8 | 3,010,362 | 3,010,354 | 2.6 | 2.9 | non fragmented | C15H10O7 | [45,49] |
| 34 | Kaempferol-coumaryl-glucoside | 46.9 | 5,931,293 | 5,931,301 | 1.3 | 17.4 | 447 | C29H24O12 | [45] |
| 35 | Kaempferol | 49 | 2,850,418 | 2,850,405 | 4.7 | 1.5 | non fragmented | C15H10O6 | [45,49] |
Flavan-3-ol and Its Derivatives
Flavonols
Flavanones
Isoflavones
Other Compounds
2.2. Total Phenolic and Flavan-3-ol Contents and in Vitro Antioxidant Activities of Pine Bark and Green Tea Extracts
| Assays | Pine Bark | Green Tea |
|---|---|---|
| Folin–Ciocalteu a | 847.62 ± 39.74 [54,56] | 835.23 ± 50.31 [55] |
| Vanillin assay b | 883.33 ± 76.38 | 906.25 ± 150.26 |
| TEAC c | 5.72 ± 0.78 | 9.66 ± 1.27 [57] |
| FRAP d | 4.83 ± 0.15 | 8.4 ± 0.4 |
| ORAC c | 8.4 ± 0.4 [56] | 7.58 ± 0.57 [57] |
3. Experimental Section
3.1. Chemicals
3.2. Sample Preparation
3.3. Instrumentation
3.4. Chromatographic, UV and Spectrophotometric Conditions
3.5. ESI-QTOF-MS Detection
3.6. Total Phenolic and Flavan-3-ol Contents
3.7. Antioxidant Capacity Assays
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Karonen, M.; Loponen, J.; Ossipov, V.; Pihlaja, K. Analysis of procyanidins in pine bark with reversed-phase and normal-phase high-performance liquid chromatography–electrospray ionization mass spectrometry. Anal. Chim. Acta 2004, 522, 105–112. [Google Scholar] [CrossRef]
- Bagchi, D.; Bagchi, M.; Stohs, S.; Das, D. Free radicals and grape seed proanthocyanidin extract: Importance in human health and disease prevention. Toxicology 2000, 148, 187–97. [Google Scholar] [CrossRef]
- Lim, T.K. Edible Medicinal and Non-Medicinal Plants; Springer Netherlands: Dordrecht, The Netherlands, 2014; Volume 6, pp. 450–489. [Google Scholar]
- Lorrain, B.; Ky, I.; Pechamat, L.; Teissedre, P. Evolution of analysis of polyhenols from grapes, wines and extracts. Molecules 2013, 18, 1076–1100. [Google Scholar] [CrossRef]
- Wojdyło, A.; Oszmiański, J.; Czemerys, R. Antioxidant activity and phenolic compounds in 32 selected herbs. Food Chem. 2007, 105, 940–949. [Google Scholar] [CrossRef]
- De la Luz Cádiz-Gurrea, M.; Fernández-Arroyo, S.; Joven, J.; Segura-Carretero, A. Comprehensive characterization by UHPLC–ESI-Q-TOF-MS from an Eryngium bourgatii extract and their antioxidant and anti-inflammatory activities. Food Res. Int. 2013, 50, 197–204. [Google Scholar]
- De la Luz Cádiz-Gurrea, M.; Lozano-Sánchez, J.; Contreras-Gámez, M.; Legeai-Mallet, L.; Fernández-Arroyo, S.; Segura-Carretero, A. Isolation, comprehensive characterization and antioxidant activities of Theobroma cacao extract. J. Funct. Foods 2014, 10, 485–498. [Google Scholar]
- Price, M.; Scoyoc, S. van; Butler, L. A critical evaluation of the vanillin reaction as an assay for tannin in Sorghum grain. J. Agric. Food Chem. 1978, 26, 1214–1218. [Google Scholar] [CrossRef]
- Goldstein, J.; Swain, T. Changes in tannins in ripening fruits. Phytochemistry 1963, 2, 371–383. [Google Scholar]
- Singleton, V.; Rossi, J. Colorimetry of total phenolics with phosphomolybdic-phosphotungstic acid reagents. Am. J. Enol. Vitic. 1965, 16, 144–158. [Google Scholar]
- Porter, L.; Ma, Z.; Chan, B. Cacao procyanidins: Major flavanoids and identification of some minor metabolites. Phytochemistry 1991, 30, 1657–1663. [Google Scholar] [CrossRef]
- Gu, L.; Kelm, M.; Hammerstone, J.F.; Beecher, G.; Cunningham, D.; Vannozzi, S.; Prior, R.L. Fractionation of polymeric procyanidins from lowbush blueberry and quantification of procyanidins in selected foods with an optimized normal-phase HPLC–MS fluorescent detection method. J. Agric. Food Chem. 2002, 50, 4852–4860. [Google Scholar] [CrossRef] [PubMed]
- Packer, L.; Rimbach, G.; Virgili, F. Antioxidant activity and biologic properties of a procyaidin-rich extract from pine (Pinus maritima) bark, Pycnogenol. Free Radic. Biol. Med. 1999, 27, 704–724. [Google Scholar] [CrossRef] [PubMed]
- Ince, I.; Yesil-Celiktas, O.; Karabay-Yavasoglu, N.U.; Elgin, G. Effects of Pinus brutia bark extract and Pycnogenol in a rat model of carrageenan induced inflammation. Phytomedicine 2009, 16, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Sokół-Łętowska, A.; Oszmiański, J.; Wojdyło, A. Antioxidant activity of the phenolic compounds of hawthorn, pine and skullcap. Food Chem. 2007, 103, 853–859. [Google Scholar] [CrossRef]
- Rohdewald, P. A review of the French maritime pine bark extract (Pycnogenol), a herbal medication with a diverse clinical pharmacology. Int. J. Clin. Pharmacol. Ther. 2002, 40, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Horie, H.; Kohata, K. Analysis of tea components by high-performance liquid chromatography and high-performance capillary electrophoresis. J. Chromatogr. 2000, 881, 425–438. [Google Scholar] [CrossRef]
- Graham, H.N. Green tea composition, consumption, and polyphenol chemistry. Prev. Med. 1992, 21, 334–350. [Google Scholar] [CrossRef] [PubMed]
- Mauri, P.; Pietta, P. Electrospray characterization of selected medicinal plant extracts. J. Pharm. Biomed. Anal. 2000, 23, 61–8. [Google Scholar] [CrossRef] [PubMed]
- Gabetta, B.; Fuzzati, N.; Griffini, A.; Lolla, E. Characterization of proanthocyanidins from grape seeds. Fitoterapia 2000, 71, 162–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; White, E. Extraction and characterization of Proanthocyanidins from grape seeds. Open Food Sci. J. 2012, 6, 5–11. [Google Scholar] [CrossRef]
- Revilla, E.; Bourzeix, M.; Alonso, E. Analysis of catechins and proanthocyanidins in grape seeds by HPLC with photodiode array detection. Chromatographia 1991, 31, 465–468. [Google Scholar] [CrossRef]
- Quirantes-Piné, R.; Lozano-Sánchez, J.; Herrero, M.; Ibáñez, E.; Segura-Carretero, A.; Fernández-Gutiérrez, A. HPLC–ESI-QTOF-MS as a powerful analytical tool for characterising phenolic compounds in olive-leaf extracts. Phytochem. Anal. 2013, 24, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Chupin, L.; Motillon, C.; Charrier-El Bouhtoury, F.; Pizzi, A.; Charrier, B. Characterisation of maritime pine (Pinus pinaster) bark tannins extracted under different conditions by spectroscopic methods, FTIR and HPLC. Ind. Crops Prod. 2013, 49, 897–903. [Google Scholar] [CrossRef]
- Apetrei, C.L.; Tuchilus, C.; Aprotosoaie, A.C.; Oprea, A.; Malterud, K.E.; Miron, A. Chemical, antioxidant and antimicrobial investigations of Pinus cembra L. bark and needles. Molecules 2011, 16, 7773–7788. [Google Scholar] [CrossRef] [PubMed]
- Jerez, M.; Touriño, S.; Sineiro, J.; Torres, J.L.; Núñez, M.J. Procyanidins from pine bark: Relationships between structure, composition and antiradical activity. Food Chem. 2007, 104, 518–527. [Google Scholar] [CrossRef]
- Jerez, M.; Selga, A.; Sineiro, J.; Torres, J.L.; Núñez, M.J. A comparison between bark extracts from Pinus pinaster and Pinus radiata: Antioxidant activity and procyanidin composition. Food Chem. 2007, 100, 439–444. [Google Scholar] [CrossRef]
- Willför, S.; Ali, M.; Karonen, M.; Reunanen, M.; Arfan, M.; Harlamow, R. Extractives in bark of different conifer species growing in Pakistan. Holzforschung 2009, 63, 551–558. [Google Scholar] [CrossRef]
- Braga, M.E.M.; Santos, R.M.S.; Seabra, I.J.; Facanali, R.; Marques, M.O.M.; de Sousa, H.C. Fractioned SFE of antioxidants from maritime pine bark. J. Supercrit. Fluids 2008, 47, 37–48. [Google Scholar] [CrossRef]
- Pan, H.; Lundgren, L. Phenolics from inner bark of Pinus sylvestris. Phytochemistry 1996, 42, 2–6. [Google Scholar] [CrossRef]
- Kıvrak, İ.; Kıvrak, Ş.; Harmandar, M.; Çetintas, Y. Phenolic compounds of pinus brutia ten: chemical investigation and quantitative analysis using an ultra-Performance liquid chromatography tandem mass spectrometry with electrospray ionization source. Rec. Nat. Prod. 2013, 7, 313–319. [Google Scholar]
- Matthews, S.; Mila, I.; Scalbert, A.; Donnelly, D.M.X. Extractable and non-extractable proanthocyanidins in barks. Phytochemistry 1997, 45, 405–410. [Google Scholar] [CrossRef]
- Kusano, R.; Ogawa, S.; Matsuo, Y.; Tanaka, T.; Yazaki, Y.; Kouno, I. α-Amylase and lipase inhibitory activity and structural characterization of acacia bark proanthocyanidins. J. Nat. Prod. 2011, 74, 119–28. [Google Scholar] [CrossRef] [PubMed]
- Antal, D.S.; Schwaiger, S.; Ellmerer-Müller, E.P.; Stuppner, H. Cotinus coggygria wood: Novel flavanone dimer and development of an HPLC/UV/MS method for the simultaneous determination of fourteen phenolic constituents. Planta Med. 2010, 76, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Botha, J.J.; Ferreira, D.; Roux, D.G. Condensed tannins: Direct synthesis, structure, and absolute configuration of four biflavonoids from black wattle bark (“Mimosa”) extract. J. Chem. Soc. Chem. Commun. 1978, 16, 700–702. [Google Scholar] [CrossRef]
- Duan, W.; Ohara, S.; Hashida, K.; Makino, R. Condensed tannins from steamed Acacia mearnsii bark. Holzforschung 2005, 59, 289–294. [Google Scholar] [CrossRef]
- Taniguchi, S.; Kuroda, K.; Doi, K.; Tanabe, M.; Shibata, T.; Yoshida, T.; Hatano, T. Revised structures of gambiriins A1, A2, B1, and B2, chalcane-flavan dimers from gambir (Uncaria gambir extract). Chem. Pharm. Bull. 2007, 55, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, G.; Nishioka, I. Novel biflavonoids, chalcan-flavan dimers from Gambir. Chem. Pharm. Bull. 1980, 28, 3145–3149. [Google Scholar] [CrossRef]
- Köhler, N.; Wray, V.; Winterhalter, P. New approach for the synthesis and isolation of dimeric procyanidins. J. Agric. Food Chem. 2008, 56, 5374–5385. [Google Scholar] [CrossRef] [PubMed]
- Scoparo, C.T.; de Souza, L.M.; Dartora, N.; Sassaki, G.L.; Gorin, P.A J.; Iacomini, M. Analysis of Camellia sinensis green and black teas via ultra high performance liquid chromatography assisted by liquid–liquid partition and two-dimensional liquid chromatography (size exclusion × reversed phase). J. Chromatogr. 2012, 1222, 29–37. [Google Scholar] [CrossRef]
- Bastos, D.; Saldanha, L.; Catharino, R. Phenolic antioxidants identified by ESI-MS from yerba mate (Ilex paraguariensis) and green tea (Camelia sinensis) extracts. Molecules 2007, 12, 423–432. [Google Scholar] [CrossRef]
- Zuo, Y.; Chen, H.; Deng, Y. Simultaneous determination of catechins, caffeine and gallic acids in green, Oolong, black and pu-erh teas using HPLC with a photodiode array detector. Talanta 2002, 57, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.; Lee, V.; Tzen, J.; Lee, M. Identification and comparison of phenolic compounds in the preparation of oolong tea manufactured by semifermentation and drying processes. J. Agric. Food Chem. 2007, 55, 7462–7468. [Google Scholar] [CrossRef] [PubMed]
- Van der Hooft, J.J.J.; Akermi, M.; Ünlü, F.Y.; Mihaleva, V.; Gomez Roldan, V.; Bino, R.J.; de Vos, R.C.H.; Vervoort, J. Structural annotation and elucidation of conjugated phenolic compounds in black, green and white tea extracts. J. Agric. Food Chem. 2012, 60, 8841–8850. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, P.; Lin, L.; Harnly, J.; Yu, L.; Li, Z. Tentative identification, quantitation and principal component analysis of green pu-erh, green and white teas using UPLC/DAD/MS. Food Chem. 2011, 126, 1269–1277. [Google Scholar] [CrossRef]
- Wang, D.; Lu, J.; Miao, A.; Xie, Z.; Yang, D. HPLC–DAD-ESI-MS/MS analysis of polyphenols and purine alkaloids in leaves of 22 tea cultivars in China. J. Food Compos. Anal. 2008, 21, 361–369. [Google Scholar] [CrossRef]
- Zeeb, D.; Nelson, B.; Albert, K.; Dalluge, J. Separation and identification of twelve catechins in tea using liquid chromatography/atmospheric pressure chemical ionization-mass spectrometry. Anal. Chem. 2000, 72, 5020–5026. [Google Scholar] [CrossRef] [PubMed]
- Kiehne, A.; Lakenbrink, C.; Engelhardt, U.H. Analysis of proanthocyanidins in tea samples. Z. Leb. Unters. 1997, 205, 153–157. [Google Scholar] [CrossRef]
- Rusak, G.; Komes, D.; Likić, S.; Horžić, D.; Kovač, M. Phenolic content and antioxidative capacity of green and white tea extracts depending on extraction conditions and the solvent used. Food Chem. 2008, 110, 852–858. [Google Scholar] [CrossRef]
- Česla, P.; Fischer, J.; Jandera, P. Separation of phenolic acids and flavone natural antioxidants by two-dimensional method combining liquid chromatography and micellar electrokinetic capillary chromatography. Electrophoresis 2010, 31, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, E.; Sagesaka, Y.M. Simultaneous determination of catechins, caffeine and other phenolic compounds in tea using new HPLC method. J. Food Compos. Anal. 2004, 17, 675–685. [Google Scholar] [CrossRef]
- Higdon, J.; Frei, B. Tea catechins and polyphenols: Health effects, metabolism and antioxidant functions. Crit. Rev. Food Sci. Nutr. 2003, 43, 89–143. [Google Scholar] [CrossRef]
- Fukumoto, L.; Mazza, G. Assessing antioxidant and prooxidant activities of phenolic compounds. J. Agric. Food Chem. 2000, 48, 3597–3604. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.S.; Jang, J.P.; Mun, S.P. Exploitation of polyphenol-rich pine barks for potent antioxidant activity. J. Wood Sci. 2007, 53, 524–528. [Google Scholar] [CrossRef]
- Gramza, A.; Pawlak-Lemanska, K.; Korczak, J.; Wasowicz, E.; Rudzinska, M. Tea extracts as free radical scavengers. Pol. J. Environ. Stud. 2005, 14, 861–867. [Google Scholar]
- Legault, J.; Girard-Lalancette, K.; Dufour, D.; Pichette, A. Antioxidant potential of bark extracts from boreal forest conifers. Antioxidants 2013, 2, 77–89. [Google Scholar] [CrossRef]
- Seeram, N.P.; Henning, S.M.; Niu, Y.; Lee, R.; Scheuller, H.S.; Heber, D. Catechin and caffeine content of green tea dietary supplements and correlation with antioxidant capacity. J. Agric. Food Chem. 2006, 54, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ou, B.; Prior, R. The chemistry behind antioxidant capacity assays. J. Agric. Food Chem. 2005, 53, 1841–1856. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.; Strain, J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Bruker Daltonics Technical Note 008. In Molecular Formula Determination Under Automation; Bruker Daltonics: Bremen, Germany, 2004.
- Bringmann, G.; Kajahn, I.; Neusüß, C.; Pelzing, M.; Laug, S.; Unger, M.; Holzgrabe, U. Analysis of the glucosinolate pattern of Arabidopsis thaliana seeds by capillary zone electrophoresis coupled to electrospray ionization-mass spectrometry. Electrophoresis 2005, 26, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Wang, S. Antioxidant activity and phenolic compounds in selected herbs. J. Agric. Food Chem. 2001, 49, 5165–5170. [Google Scholar] [CrossRef] [PubMed]
- Makkar, H.; Becker, K. Vanillin–HCl method for condensed tannins: Effect of organic solvents used for extraction of tannins. J. Chem. Ecol. 1993, 19, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Laporta, O.; Pérez-Fons, L.; Mallavia, R. Isolation, characterization and antioxidant capacity assessment of the bioactive compounds derived from Hypoxis rooperi corm extract (African potato). Food Chem. 2007, 101, 1425–1437. [Google Scholar] [CrossRef]
- Morales-Soto, A.; García-Salas, P.; Rodríguez-Pérez, C.; Jiménez-Sánchez, C.; Cádiz-Gurrea, M.D.L.L.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Antioxidant capacity of 44 cultivars of fruits and vegetables grown in Andalusia (Spain). Food Res. Int. 2014, 58, 35–46. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Prior, R. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De la Luz Cádiz-Gurrea, M.; Fernández-Arroyo, S.; Segura-Carretero, A. Pine Bark and Green Tea Concentrated Extracts: Antioxidant Activity and Comprehensive Characterization of Bioactive Compounds by HPLC–ESI-QTOF-MS. Int. J. Mol. Sci. 2014, 15, 20382-20402. https://doi.org/10.3390/ijms151120382
De la Luz Cádiz-Gurrea M, Fernández-Arroyo S, Segura-Carretero A. Pine Bark and Green Tea Concentrated Extracts: Antioxidant Activity and Comprehensive Characterization of Bioactive Compounds by HPLC–ESI-QTOF-MS. International Journal of Molecular Sciences. 2014; 15(11):20382-20402. https://doi.org/10.3390/ijms151120382
Chicago/Turabian StyleDe la Luz Cádiz-Gurrea, María, Salvador Fernández-Arroyo, and Antonio Segura-Carretero. 2014. "Pine Bark and Green Tea Concentrated Extracts: Antioxidant Activity and Comprehensive Characterization of Bioactive Compounds by HPLC–ESI-QTOF-MS" International Journal of Molecular Sciences 15, no. 11: 20382-20402. https://doi.org/10.3390/ijms151120382