The Biochemical and Cellular Basis for Nutraceutical Strategies to Attenuate Neurodegeneration in Parkinson’s Disease


Abstract
:1. Introduction
1.1. Pathology
1.2. Treatment
1.3. Previous Studies on Therapeutic Agents to Slow Progression of PD
2. Review
2.1. Energy Failure—Loss of OXPHOS, Rise in Anaerobic Glycolysis & Lactate, ATP Depletion
2.2. Loss of DA Regulation and Trafficking—VMAT2
2.3. Dopamine Oxidation
2.3.1. Enzymatic Oxidation of DA, the Neuromelanin Pathway & DA-Quinones
2.3.2. Non Enzymatic Oxidation of DA, 6-OHDA, Release of Iron & Oxidative Stress
2.3.3. Enzymatic DA Oxidation by MAO-DA Aldehydes & H2O2
2.4. Excitotoxicity
2.5. Inflammation
3. Nutraceuticals
3.1. Energy—Biochemistry and Metabolism
3.1.1. Pyruvic Acid
3.1.2. Niacin
3.1.3. Magnesium
3.1.4. B Vitamins and Regulation of Physiological Homocysteine
3.1.5. B Complex Vitamins, Riboflavin and Mitochondrial Disorders
3.1.6. Creatine, Chromium
3.2. Plant Polyphenols—Attenuation of DA Oxidation
3.2.1. Tyrosinase Inhibitors
3.2.2. COX Inhibitors
3.2.3. Lipoxygenase Inhibitors
3.2.4. Phospholipase A2 Inhibitors
3.2.5. Xanthine Oxidase Inhibitors
3.2.6. Xanthine Oxidase and Superoxide Scavengers
3.3. Histidine, Quercetin and Zinc
3.4. N Acetyl Cysteine
3.5. Hydrogen Peroxide Scavengers
3.6. Iron Chelators
3.7. Heme Oxygenase Inhibitors
3.8. Zinc and Selenium
3.9. Anti-Inflammatory Nutraceuticals
3.10. Toxic Protein Aggregates
3.10.1. Nutraceuticals—Reduction of aggregated α-SYNUCLEIN (PARK1)
3.10.2. (Parkin) E3 ubiquitin ligase and Proteosomal Dysfunction
3.10.3. Nutraceuticals, Autophagy and mTOR signaling
4. Conclusion
Acknowledgment
Abbreviations
| 6-OHDA | 6 Hydroxydopamine |
| AADC | Aromatic amino acid decarboxylase |
| AMPA | α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate |
| AP-1 | Activator protein 1 |
| ATP | Adenosine triphosphate |
| BBB | Blood brain barrier |
| CBF | Cerebral blood flow |
| CMRG | Cerebral metabolic rate of glucose |
| CMRG | Cerebral metabolic rate of glucose |
| COMT | Catechol-O-methyltransferase |
| COX | Cyclooxygenase |
| CSF | Cerebral spinal fluid |
| DA | Dopamine |
| DAergic | Dopaminergic |
| DAT | Dopamine transporter |
| DOPAC | 3,4-dihydroxyphenylacetic acid |
| DTBZ | [11C]-dihydrotetrabenazine |
| ERK | Extracellular signal-regulated kinases |
| FAD | Flavin adenine dinucleotide |
| FD | [18F]-fluoro-l dopa |
| FDG | [18F]-Fluoro-deoxyglucose |
| FMN | Flavin mononucleotide |
| GABA | Γ-Aminobutyric acid |
| GSHPx | Glutathione peroxidase |
| GSH | Reduced Glutathione |
| H2O2 | Hydrogen Peroxide |
| HO-1 | Heme oxygenase 1 |
| HVA | Homovanillic acid |
| IKK | I kappaB kinase |
| IL | Interleukin |
| INOS | Inducible NOS |
| mRNA | Messenger ribonucleic acid |
| mTOR | Mammalian target of rapamycin |
| NA | Nucleus accumbens, |
| NAC | N acetyl L cysteine |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH | Nicotinamide adenine dinucleotide reduced |
| NADPH | Nicotinamide adenine dinucleotide phosphate reduced |
| NF-κB | Nuclear factor-kappa B |
| NM | Neuromelanin |
| NMDA | N-methyl-D-aspartate |
| NNMT | Nicotinamide N-methyl transferase |
| NOS | Nitric oxide synthase |
| NT | Neurotransmitter |
| O2− | Superoxide |
| OXPHOS | Oxidative Phosphorylation |
| PARP-1 | Poly [ADP-ribose] polymerase 1 |
| PD | Parkinson’s disease |
| PDE | Phosphodiesterase |
| PDPC | Plant derived polyphenolic compounds |
| PET | Positron emission tomography |
| PGE2 | Prostaglandin E2 |
| PGH2 | Prostaglandin H2 synthase |
| PI3K | Phosphoinositide 3 kinase |
| PINK-1 | PTEN-induced putative kinase 1 |
| PK | Pyruvate kinase |
| PLA2 | Phospholipase A2 |
| PPO | Polyphenol Oxidase |
| PY | Pyruvate |
| ROS | Reactive Oxygen Species |
| Se | Selenium |
| SLP | Substrate level phosphorylation |
| JNK | c-jun N-terminal kinase |
| LC | Locus coeruleus |
| LDH | Lactate Dehydrogenase |
| l-DOPA | l-3,4-dihydroxyphenylalanine |
| LOX | Lipoxygenase |
| MAO | Monoamine oxidase |
| MAPK | Mitogen-activated protein kinase |
| MP | 11C-methylphenidate |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| SN | Substantia nigra |
| SNc | Substantia nigra pars compacta |
| SNCA | α-synuclein |
| SOD | Superoxide dismutase |
| SPECT | Single photon emission computed tomography |
| TH | Tyrosine hydroxylase |
| TNF | Tumor necrosis factor |
| UPS | Ubiquitin-proteasome system |
| VMAT2 | Type-2-vesicular monoamine transporter |
| Zn | Zinc |
References
- Burch, D; Sheerin, F. Parkinson’s disease. Lancet 2005, 365, 622–627. [Google Scholar]
- Lin, TK; Liou, CW; Chen, SD; Chuang, YC; Tiao, MM; Wang, PW; Chen, JB; Chuang, JH. Mitochondrial dysfunction and biogenesis in the pathogenesis of Parkinson’s disease. Chang Gung Med. J 2009, 32, 589–599. [Google Scholar]
- Dagda, RK; Chu, CT. Mitochondrial quality control: insights on how Parkinson’s disease related genes PINK1, parkin, and Omi/HtrA2 interact to maintain mitochondrial homeostasis. J. Bioenerg. Biomembr 2009, 41, 473–479. [Google Scholar]
- Nishioka, K; Vilariño-Güell, C; Cobb, SA; Kachergus, JM; Ross, OA; Hentati, E; Hentati, F; Farrer, MJ. Genetic variation of the mitochondrial complex I subunit NDUFV2 and Parkinson’s disease. Parkinsonism Relat. Disord 2010, 10, 686–687. [Google Scholar]
- Levy, OA; Malagelada, C; Greene, LA. Cell death pathways in Parkinson’s disease: Proximal triggers, distal effectors, and final steps. Apoptosis 2009, 14, 478–500. [Google Scholar]
- Nagatsu, T. Parkinson’s disease: changes in apoptosis-related factors suggesting possible gene therapy. J. Neural. Transm 2002, 109, 731–745. [Google Scholar]
- Tofaris, GK; Spillantini, MG. Alpha-synuclein dysfunction in Lewy body diseases. Mov. Disord 2005, 20, S37–S44. [Google Scholar]
- Bharath, S; Hsu, M; Kaur, D; Rajagopalan, S; Andersen, JK. Glutathione, iron and Parkinson’s disease. Biochem. Pharmacol 2002, 64, 1037–1048. [Google Scholar]
- Johnson, MD; Yu, LR; Conrads, TP; Kinoshita, Y; Uo, T; McBee, JK; Veenstra, TD; Morrison, RS. The proteomics of neurodegeneration. Am. J Pharmacogenomics 2005, 5, 259–270. [Google Scholar]
- Hald, A; Lotharius, J. Oxidative stress and inflammation in Parkinson’s disease: is there a causal link? Exp. Neurol 2005, 193, 279–290. [Google Scholar]
- Sato, S; Mizuno, Y; Hattori, N. Urinary 8-hydroxydeoxyguanosine levels as a biomarker for progression of Parkinson disease. Neurology 2005, 64, 1081–1083. [Google Scholar]
- Pennathur, S; Jackson-Lewis, V; Przedborski, S; Heinecke, JW. Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and o,o′-dityrosine in brain tissue of 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine-treated mice, a model of oxidative stress in Parkinson’s disease. J. Biol. Chem 1999, 274, 34621–34628. [Google Scholar]
- Takeuchi, H; Mizuno, T; Zhang, G; Wang, J; Kawanokuchi, J; Kuno, R; Suzumura, A. Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J. Biol. Chem 2005, 280, 10444–10454. [Google Scholar]
- Cheng, HC; Ulane, CM; Burke, RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol 2010, 67, 715–725. [Google Scholar]
- Fahn, S; Sulzer, D. Neurodegeneration and neuroprotection in Parkinson disease. NeuroRx 2004, 1, 139–154. [Google Scholar]
- Bertram, L; Tanzi, RE. The genetic epidemiology of neurodegenerative disease. J. Clin. Invest 2005, 115, 1449–1457. [Google Scholar]
- Hyun, DH; Lee, M; Halliwell, B; Jenner, P. Effect of overexpression of wild-type or mutant parkin on the cellular response induced by toxic insults. J. Neurosci. Res 2005, 82, 232–244. [Google Scholar]
- Mortiboys, H; Johansen, KK; Aasly, JO; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar]
- Henchcliffe, C; Beal, MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol 2008, 4, 600–609. [Google Scholar]
- Arvanitakis, Z; Wilson, RS; Schneider, JA; Bienias, JL; Evans, DA; Bennett, DA. Diabetes mellitus and progression of rigidity and gait disturbance in older persons. Neurology 2004, 63, 996–1001. [Google Scholar]
- Klein, RC; de Jong, BM; de Vries, JJ; Leenders, KL. Direct comparison between regional cerebral metabolism in progressive supranuclear palsy and Parkinson’s disease. Mov. Disord 2005, 20, 1021–1030. [Google Scholar]
- Lesage, S; Brice, A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet 2009, 18, R48–R59. [Google Scholar]
- Tanner, CM; Ross, GW; Jewell, SA; Hauser, RA; Jankovic, J; Factor, SA; Bressman, S; Deligtisch, A; Marras, C; Lyons, KE; Bhudhikanok, GS; Roucoux, DF; Meng, C; Abbott, RD; Langston, JW. Occupation and risk of parkinsonism: a multicenter case-control study. Arch. Neurol 2009, 66, 1106–1113. [Google Scholar]
- Nguyen, N; Pradel, V; Micallef, J; Montastruc, JL; Blin, O. Drug-induced Parkinson syndromes. Therapie 2004, 59, 105–112. [Google Scholar]
- Sanyal, J; Chakraborty, DP; Sarkar, B; Banerjee, TK; Mukherjee, SC; Ray, BC; Rao, VR. Environmental and familial risk factors of Parkinsons disease: case-control study. Can. J. Neurol. Sci 2010, 37, 637–642. [Google Scholar]
- Allam, MF; Del Castillo, AS; Navajas, RF. Parkinson’s disease risk factors: genetic, environmental, or both? Neurol. Res 2005, 27, 206–208. [Google Scholar]
- Logroscino, G. The role of early life environmental risk factors in Parkinson disease: what is the evidence? Environ. Health Perspect 2005, 113, 1234–1238. [Google Scholar]
- Pavese, N; Brooks, DJ. Imaging neurodegeneration in Parkinson’s disease. Biochim. Biophys Acta 2009, 1792, 722–729. [Google Scholar]
- Thobois, S; Guillouet, S; Broussolle, E. Contributions of PET and SPECT to the understanding of the pathophysiology of Parkinson’s disease. Neurophysiol. Clin 2001, 31, 321–340. [Google Scholar]
- Au, WL; Adams, JR; Troiano, AR; Stoessl, AJ. Parkinson’s disease: in vivo assessment of disease progression using positron emission tomography. Brain Res. Mol. Brain Res 2005, 134, 24–33. [Google Scholar]
- Galvan, A; Wichmann, T. Pathophysiology of parkinsonism. Clin. Neurophysiol 2008, 119, 1459–1474. [Google Scholar]
- Pal, PK; Netravathi, M. Management of neurodegenerative disorders: Parkinson’s disease and Alzheimer’s disease. J. Indian Med. Assoc 2005, 103, 168–170. [Google Scholar]
- Samai, M; Sharpe, MA; Gard, PR; Chatterjee, PK. Comparison of the effects of the superoxide dismutase mimetics EUK-134 and tempol on paraquat-induced nephrotoxicity. Free Radic. Biol. Med 2007, 43, 528–534. [Google Scholar]
- Weinreb, O; Amit, T; Bar-Am, O; Youdim, MB. Rasagiline: a novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog. Neurobiol 2010, 92, 330–344. [Google Scholar]
- Lew, MF; Hauser, RA; Hurtig, HI; Ondo, WG; Wojcieszek, J; Goren, T; Fitzer-Attas, CJ. Long-term efficacy of rasagiline in early Parkinson’s disease. Int. J. Neurosci 2010, 120, 404–408. [Google Scholar]
- Weinreb, O; Amit, T; Bar-Am, O; Chillag-Talmor, O; Youdim, MB. Novel neuroprotective mechanism of action of rasagiline is associated with its propargyl moiety: interaction of Bcl-2 family members with PKC pathway. Ann. N. Y. Acad. Sci 2005, 1053, 348–355. [Google Scholar]
- Zheng, H; Gal, S; Weiner, LM; Bar-Am, O; Warshawsky, A; Fridkin, M; Youdim, MB. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: in vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J. Neurochem 2005, 95, 68–78. [Google Scholar]
- Youdim, MB; Fridkin, M; Zheng, H. Bifunctional drug derivatives of MAO-B inhibitor rasagiline and iron chelator VK-28 as a more effective approach to treatment of brain ageing and ageing neurodegenerative diseases. Mech. Ageing Dev 2005, 126, 317–326. [Google Scholar]
- Mandel, SA; Avramovich-Tirosh, Y; Reznichenko, L; Zheng, H; Weinreb, O; Amit, T; Youdim, MB. Multifunctional activities of green tea catechins in neuroprotection. Modulation of cell survival genes, iron-dependent oxidative stress and PKC signaling pathway. Neurosignals 2005, 14, 46–60. [Google Scholar]
- Mandel, S; Maor, G; Youdim, MB. Iron and alpha-synuclein in the substantia nigra of MPTP-treated mice: effect of neuroprotective drugs R-apomorphine and green tea polyphenol (−)-epigallocatechin-3-gallate. J. Mol. Neurosci 2004, 24, 401–416. [Google Scholar]
- Cleren, C; Calingasan, NY; Chen, J; Beal, MF. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J. Neurochem 2005, 94, 995–1004. [Google Scholar]
- Klivenyi, P; Andreassen, OA; Ferrante, RJ; Lancelot, E; Reif, D; Beal, MF. Inhibition of neuronal nitric oxide synthase protects against MPTP toxicity. Neuroreport 2000, 11, 1265–1268. [Google Scholar]
- Watanabe, H; Muramatsu, Y; Kurosaki, R; Michimata, M; Matsubara, M; Imai, Y; Araki, T. Protective effects of neuronal nitric oxide synthase inhibitor in mouse brain against MPTP neurotoxicity: an immunohistological study. Eur. Neuropsychopharmacol 2004, 14, 93–104. [Google Scholar]
- Wang, W; Shi, L; Xie, Y; Ma, C; Li, W; Su, X; Huang, S; Chen, R; Zhu, Z; Mao, Z; Han, Y; Li, M. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci. Res 2004, 48, 195–202. [Google Scholar]
- Teismann, P; Tieu, K; Choi, DK; Wu, DC; Naini, A; Hunot, S; Vila, M; Jackson-Lewis, V; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci USA 2003, 100, 5473–5478. [Google Scholar]
- Kuan, CY; Burke, RE. Targeting the JNK signaling pathway for stroke and Parkinson’s diseases therapy. Curr. Drug Targets CNS Neurol. Disord 2005, 4, 63–67. [Google Scholar]
- Silva, RM; Kuan, CY; Rakic, P; Burke, RE. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: a new therapeutic target in Parkinson’s disease. Mov. Disord 2005, 20, 653–664. [Google Scholar]
- Testa, CM; Sherer, TB; Greenamyre, JT. Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Brain Res. Mol. Brain Res 2005, 134, 109–118. [Google Scholar]
- Virmani, A; Gaetani, F; Binienda, Z. Effects of metabolic modifiers such as carnitines, coenzyme Q10, and PUFAs against different forms of neurotoxic insults: metabolic inhibitors, MPTP, and methamphetamine. Ann. N. Y. Acad. Sci 2005, 1053, 183–191. [Google Scholar]
- Bhat, V; Weiner, WJ. Parkinson’s disease. Diagnosis and the initiation of therapy. Minerva Med 2005, 96, 145–154. [Google Scholar]
- Shults, CW. Therapeutic role of coenzyme Q(10) in Parkinson’s disease. Pharmacol. Ther 2005, 107, 120–130. [Google Scholar]
- Etminan, M; Gill, SS; Samii, A. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson’s disease: a meta-analysis. Lancet Neurol 2005, 4, 362–365. [Google Scholar]
- Andres, RH; Huber, AW; Schlattner, U; Pérez-Bouza, A; Krebs, SH; Seiler, RW; Wallimann, T; Widmer, HR. Effects of creatine treatment on the survival of dopaminergic neurons in cultured fetal ventral mesencephalic tissue. Neuroscience 2005, 133, 701–713. [Google Scholar]
- Klivenyi, P; Gardian, G; Calingasan, NY; Yang, L; Beal, MF. Additive neuroprotective effects of creatine and a cyclooxygenase 2 inhibitor against dopamine depletion in the 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. J. Mol. Neurosci 2003, 21, 191–198. [Google Scholar]
- Borah, A; Mohanakumar, KP. Melatonin inhibits 6-hydroxydopamine production in the brain to protect against experimental Parkinsonism in rodents. J. Pineal Res 2009, 47, 293–300. [Google Scholar]
- Bi, J; Wang, XB; Chen, L; Hao, S; An, LJ; Jiang, B; Guo, L. Catalpol protects mesencephalic neurons against MPTP induced neurotoxicity via attenuation of mitochondrial dysfunction and MAO-B activity. Toxicol In Vitro 2008, 22, 1883–1889. [Google Scholar]
- Bahat-Stroomza, M; Gilgun-Sherki, Y; Offen, D; Panet, H; Saada, A; Krool-Galron, N; Barzilai, A; Atlas, D; Melamed, E. A novel thiol antioxidant that crosses the blood brain barrier protects dopaminergic neurons in experimental models of Parkinson’s disease. Eur. J. Neurosci 2005, 21, 637–646. [Google Scholar]
- Levy, YS; Gilgun-Sherki, Y; Melamed, E; Offen, D. Therapeutic potential of neurotrophic factors in neurodegenerative diseases. BioDrugs 2005, 19, 97–127. [Google Scholar]
- Slevin, JT; Gerhardt, GA; Smith, CD; Gash, DM; Kryscio, R; Young, B. Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J. Neurosurg 2005, 102, 216–222. [Google Scholar]
- D’Astous, M; Morissette, M; Tanguay, B; Callier, S; Di Paolo, T. Dehydroepiandrosterone (DHEA) such as 17beta-estradiol prevents MPTP-induced dopamine depletion in mice. Synapse 2003, 47, 10–14. [Google Scholar]
- D’Astous, M; Morissette, M; Di Paolo, T. Effect of estrogen receptor agonists treatment in MPTP mice: evidence of neuroprotection by an ER alpha agonist. Neuropharmacology 2004, 47, 1180–1188. [Google Scholar]
- Morelli, M; Carta, AR; Kachroo, A; Schwarzschild, MA. Pathophysiological roles for purines: adenosine, caffeine and urate. Prog. Brain Res 2010, 183, 183–208. [Google Scholar]
- Ikeda, K; Kurokawa, M; Aoyama, S; Kuwana, Y. Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J. Neurochem 2002, 80, 262–270. [Google Scholar]
- Xu, K; Bastia, E; Schwarzschild, M. Therapeutic potential of adenosine A2A receptor antagonists in Parkinson’s disease. Pharmacol. Ther 2005, 105, 267–310. [Google Scholar]
- Azam, F; Ibn-Rajab, IA; Alruiad, AA. Adenosine A2A receptor antagonists as novel anti- Parkinsonian agents: a review of structure-activity relationships. Pharmazie 2009, 64, 771–795. [Google Scholar]
- García, E; Villeda-Hernández, J; Pedraza-Chaverrí, J; Maldonado, PD; Santamaría, A. S-allylcysteine reduces the MPTP-induced striatal cell damage via inhibition of pro-inflammatory cytokine tumor necrosis factor-α and inducible nitric oxide synthase expressions in mice. Phytomedicine 2010, 18, 65–73. [Google Scholar]
- Battaglia, G; Busceti, CL; Pontarelli, F; Biagioni, F; Fornai, F; Paparelli, A; Bruno, V; Ruggieri, S; Nicoletti, F. Protective role of group-II metabotropic glutamate receptors against nigro-striatal degeneration induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. Neuropharmacology 2003, 45, 155–166. [Google Scholar]
- Kim, YK; Lim, HH; Song, YK; Lee, HH; Lim, S; Han, SM; Kim, CJ. Effect of acupuncture on 6-hydroxydopamine-induced nigrostratal dopaminergic neuronal cell death in rats. Neurosci. Lett 2005, 384, 133–138. [Google Scholar]
- Li, XM; Ma, HB; Ma, ZQ; Li, LF; Xu, CL; Qu, R; Ma, SP. Ameliorative and neuroprotective effect in MPTP model of Parkinson’s disease by Zhen-Wu-Tang (ZWT), a traditional Chinese medicine. J. Ethnopharmacol 2010, 130, 19–27. [Google Scholar]
- Kurosaki, R; Muramatsu, Y; Kato, H; Watanabe, Y; Imai, Y; Itoyama, Y; Araki, T. Effect of angiotensin-converting enzyme inhibitor perindopril on interneurons in MPTP-treated mice. Eur. Neuropsychopharmacol 2005, 15, 57–67. [Google Scholar]
- Singh, N; Pillay, V; Choonara, YE. Advances in the treatment of Parkinson’s disease. Prog. Neurobiol 2007, 81, 29–44. [Google Scholar]
- Kotake, Y; Ohta, S. MPP+ analogs acting on mitochondria and inducing neuro-degeneration. Curr. Med. Chem 2003, 10, 2507–2516. [Google Scholar]
- Nagatsu, T. Isoquinoline neurotoxins in the brain and Parkinson’s disease. Neurosci. Res 1997, 29, 99–111. [Google Scholar]
- Mazzio, EA; Soliman, KF. Effects of enhancing mitochondrial oxidative phosphorylation with reducing equivalents and ubiquinone on 1-methyl-4-phenylpyridinium toxicity and complex I-IV damage in neuroblastoma cells. Biochem. Pharmacol 2004, 67, 1167–1184. [Google Scholar]
- Miller, GW; Gainetdinov, RR; Levey, AI; Caron, MG. Dopamine transporters and neuronal injury. Trends Pharmacol. Sci 1999, 20, 424–429. [Google Scholar]
- Del Zompo, M; Piccardi, MP; Ruiu, S; Quartu, M; Gessa, GL; Vaccari, A. Selective MPP+ uptake into synaptic dopamine vesicles: possible involvement in MPTP neurotoxicity. Br. J. Pharmacol 1993, 109, 411–414. [Google Scholar]
- Mazzio, E; Soliman, KF. d-(+)-glucose rescue against 1-methyl-4-phenylpyridinium toxicity through anaerobic glycolysis in neuroblastoma cells. Brain Res 2003, 962, 48–60. [Google Scholar]
- Palacios, JM; Wiederhold, KH. Acute administration of 1-N-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP), a compound producing parkinsonism in humans, stimulates [2–14C]deoxyglucose uptake in the regions of the catecholaminergic cell bodies in the rat and guinea pig brains. Brain Res 1984, 301, 187–191. [Google Scholar]
- Palombo, E; Porrino, LJ; Bankiewicz, KS; Crane, AM; Kopin, IJ; Sokoloff, L. Administration of MPTP acutely increases glucose utilization in the substantia nigra of primates. Brain Res 1988, 453, 227–234. [Google Scholar]
- Schwartzman, RJ; Alexander, GM. Changes in the local cerebral metabolic rate for glucose in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) primate model of Parkinson’s disease. Brain Res 1985, 358, 137–143. [Google Scholar]
- Schwartzman, RJ; Alexander, GM; Ferraro, TN; Grothusen, JR; Stahl, SM. Cerebral metabolism of parkinsonian primates 21 days after MPTP. Exp Neurol 1988, 102, 307–313. [Google Scholar]
- Lagrue, E; Abert, B; Nadal, L; Tabone, L; Bodard, S; Medja, F; Lombes, A; Chalon, S; Castelnau, P. MPTP intoxication in mice: a useful model of Leigh syndrome to study mitochondrial diseases in childhood. Metab. Brain Dis 2009, 24, 321–335. [Google Scholar]
- Drolet, RE; Behrouz, B; Lookingland, KJ; Goudreau, JL. Mice lacking alpha-synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicology 2004, 25, 761–769. [Google Scholar]
- Chan, P; DeLanney, LE; Irwin, I; Langston, JW; Di Monte, D. MPTP-induced ATP loss in mouse brain. Ann. N. Y. Acad. Sci 1992, 648, 306–308. [Google Scholar]
- Koga, K; Mori, A; Ohashi, S; Kurihara, N; Kitagawa, H; Ishikawa, M; Mitsumoto, Y; Nakai, M. 1H MRS identifies lactate rise in the striatum of MPTP-treated C57BL/6 mice. Eur. J. Neurosci 2006, 23, 1077–1081. [Google Scholar]
- Brownell, AL; Jenkins, BG; Elmaleh, DR; Deacon, TW; Spealman, RD; Isacson, O. Combined PET/MRS brain studies show dynamic and long-term physiological changes in a primate model of Parkinson disease. Nat. Med 1998, 4, 1308–1312. [Google Scholar]
- Pastoris, O; Dossena, M; Foppa, P; Catapano, M; Ferrari, R; Dagani, F. Biochemical evaluations in skeletal muscles of primates with MPTP Parkinson-like syndrome. Pharmacol. Res 1995, 31, 361–369. [Google Scholar]
- Singh, Y; Swanson, E; Sokoloski, E; Kutty, RK; Krishna, G. MPTP and MPTP analogs induced cell death in cultured rat hepatocytes involving the formation of pyridinium metabolites. Toxicol. Appl. Pharmacol 1988, 96, 347–359. [Google Scholar]
- Singer, TP; Ramsay, RR; McKeown, K; Trevor, A; Castagnoli, NE, Jr. Mechanism of the neurotoxicity of 1-methyl-4-phenylpyridinium (MPP+) the toxic bioactivation product of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 1988, 49, 17–23. [Google Scholar]
- Scotcher, KP; Irwin, I; DeLanney, LE; Langston, JW; Di Monte, D. Effects of 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on ATP levels of mouse brain synaptosomes. J. Neurochem 1990, 54, 1295–1301. [Google Scholar]
- Di Monte, DA; Wu, EY; Delanney, LE; Irwin, I; Langston, JW. Toxicity of 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine in primary cultures of mouse astrocytes. J. Pharmacol. Exp. Ther 1992, 261, 44–49. [Google Scholar]
- Mazzio, EA; Soliman, YI; Soliman, KF. Variable toxicological response to the loss of OXPHOS through 1-methyl-4-phenylpyridinium-induced mitochondrial damage and anoxia in diverse neural immortal cell lines. Cell Biol. Toxicol 2010, 26, 527–539. [Google Scholar]
- Maruoka, N; Murata, T; Omata, N; Takashima, Y; Fujibayashi, Y; Wada, Y. Topological and chronological features of the impairment of glucose metabolism induced by 1-methyl-4-phenylpyridinium ion (MPP+) in rat brain slices. J. Neural. Transm 2007, 114, 1155–1159. [Google Scholar]
- Clausen, T; Khaldi, A; Zauner, A; Reinert, M; Doppenberg, E; Menzel, M; Soukup, J; Alves, OL; Bullock, MR. Cerebral acid-base homeostasis after severe traumatic brain injury. J. Neurosurg 2005, 103, 597–607. [Google Scholar]
- Woo, CW; Lee, BS; Kim, ST; Kim, KS. Correlation between lactate and neuronal cell damage in the rat brain after focal ischemia: An in vivo 1H magnetic resonance spectroscopic (1H-MRS) study. Acta Radiol 2010, 51, 344–350. [Google Scholar]
- Chow, SL; Rooney, ZJ; Cleary, MA; Clayton, PT; Leonard, JV. The significance of elevated CSF lactate. Arch. Dis. Child 2005, 90, 1188–1189. [Google Scholar]
- Makoroff, KL; Cecil, KM; Care, M; Ball, WS, Jr. Elevated lactate as an early marker of brain injury in inflicted traumatic brain injury. Pediatr. Radiol 2005, 35, 668–676. [Google Scholar]
- Cavus, I; Kasoff, WS; Cassaday, MP; Jacob, R; Gueorguieva, R; Sherwin, RS; Krystal, JH; Spencer, DD; Abi-Saab, WM. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann. Neurol 2005, 57, 226–235. [Google Scholar]
- Brooks, DJ. Imaging approaches to Parkinson disease. J. Nucl. Med 2010, 51, 596–609. [Google Scholar]
- Noda, A; Ohba, H; Kakiuchi, T; Futatsubashi, M; Tsukada, H; Nishimura, S. Age-related changes in cerebral blood flow and glucose metabolism in conscious rhesus monkeys. Brain Res 2002, 936, 76–81. [Google Scholar]
- Cunnane, S; Nugent, S; Roy, M; Courchesne-Loyer, A; Croteau, E; Tremblay, S; Castellano, A; Pifferi, F; Bocti, C; Paquet, N; Begdouri, H; Bentourkia, M; Turcotte, E; Allard, M; Barberger-Gateau, P; Fulop, T; Rapoport, SI. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar]
- Meredith, GE; Totterdell, S; Beales, M; Meshul, CK. Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of Parkinson’s disease. Exp. Neurol 2009, 219, 334–340. [Google Scholar]
- Halestrap, AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans 2010, 38, 841–860. [Google Scholar]
- Bisaglia, M; Soriano, ME; Arduini, I; Mammi, S; Bubacco, L. Molecular characterization of dopamine-derived quinones reactivity toward NADH and glutathione: implications for mitochondrial dysfunction in Parkinson disease. Biochim. Biophys Acta 2010, 1802, 699–706. [Google Scholar]
- Liou, AK; Zhou, Z; Pei, W; Lim, TM; Yin, XM; Chen, J. BimEL up-regulation potentiates AIF translocation and cell death in response to MPTP. FASEB J 2005, 19, 1350–1352. [Google Scholar]
- Halestrap, AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem. Soc. Trans 2006, 34, 232–237. [Google Scholar]
- Bo, J; Ming, BY; Gang, LZ; Lei, C; Jia, AL. Protection by puerarin against MPP+-induced neurotoxicity in PC12 cells mediated by inhibiting mitochondrial dysfunction and caspase-3-like activation. Neurosci. Res 2005, 53, 183–188. [Google Scholar]
- Cappelletti, G; Surrey, T; Maci, R. The Parkinsonism producing neurotoxin MPP+ affects microtubule dynamics by acting as a destabilising factor. FEBS Lett 2005, 579, 4781–4786. [Google Scholar]
- Thomas, B; Beal, MF. Parkinson’s disease. Hum. Mol. Genet 2007, 16, R183–R194. [Google Scholar]
- Rollema, H; de Vries, JB; Damsma, G; Westerink, BH; Kranenborg, GL; Kuhr, WG; Horn, AS. The use of in vivo brain dialysis of dopamine, acetylcholine, amino acids and lactic acid in studies on the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 1988, 49, 503–511. [Google Scholar]
- Ofori, S; Heikkila, RE; Nicklas, WJ. Attenuation by dopamine uptake blockers of the inhibitory effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and some of its analogs on NADH-linked metabolism in mouse neostriatal slices. J. Pharmacol. Exp. Ther 1989, 251, 258–266. [Google Scholar]
- Guillot, TS; Miller, GW. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol. Neurobiol 2009, 39, 149–170. [Google Scholar]
- Tanaka, R; Asaga, H; Takeda, M. Nucleoside triphosphate and cation requirement for dopamine uptake by plain synaptic vesicles isolated from rat cerebrums. Brain Res 1976, 115, 273–283. [Google Scholar]
- Hossain, MM; Filipov, NM. Alteration of dopamine uptake into rat striatal vesicles and synaptosomes caused by an in vitro exposure to atrazine and some of its metabolites. Toxicology 2008, 248, 52–58. [Google Scholar]
- Choi, HJ; Lee, SY; Cho, Y; Hwang, O. Inhibition of vesicular monoamine transporter enhances vulnerability of dopaminergic cells: relevance to Parkinson’s disease. Neurochem. Int 2005, 46, 329–335. [Google Scholar]
- Ren, Y; Liu, W; Jiang, H; Jiang, Q; Feng, J. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J. Biol. Chem 2005, 280, 34105–34112. [Google Scholar]
- Chang, GD; Ramirez, VD. The mechanism of action of MPTP and MPP+ on endogenous dopamine release from the rat corpus striatum superfused in vitro. Brain Res 1986, 368, 134–140. [Google Scholar]
- Kurosaki, R; Muramatsu, Y; Watanabe, H; Michimata, M; Matsubara, M; Imai, Y; Araki, T. Role of dopamine transporter against MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) neurotoxicity in mice. Metab. Brain Dis 2003, 18, 139–146. [Google Scholar]
- Jourdain, S; Morissette, M; Morin, N; Di Paolo, T. Oestrogens prevent loss of dopamine transporter (DAT) and vesicular monoamine transporter (VMAT2) in substantia nigra of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice. J. Neuroendocrinol 2005, 17, 509–517. [Google Scholar]
- Hogan, KA; Staal, RG; Sonsalla, PK. Analysis of VMAT2 binding after methamphetamine or MPTP treatment: disparity between homogenates and vesicle preparations. J. Neurochem 2000, 74, 2217–2220. [Google Scholar]
- Harrington, KA; Augood, SJ; Kingsbury, AE; Foster, OJ; Emson, PC. Dopamine transporter (Dat) and synaptic vesicle amine transporter (VMAT2) gene expression in the substantia nigra of control and Parkinson’s disease. Brain Res. Mol. Brain Res 1996, 36, 157–162. [Google Scholar]
- Frey, KA; Koeppe, RA; Kilbourn, MR; van der Borght, TM; Albin, RL; Gilman, S; Kuhl, DE. Presynaptic monoaminergic vesicles in Parkinson’s disease and normal aging. Ann. Neurol 1996, 40, 873–884. [Google Scholar]
- Reinhard, JF, Jr; Carmichael, SW; Daniels, AJ. Mechanisms of toxicity and cellular resistance to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium in adrenomedullary chromaffin cell cultures. J. Neurochem 1990, 55, 311–320. [Google Scholar]
- Wimalasena, DS; Perera, RP; Heyen, BJ; Balasooriya, IS; Wimalasena, K. Vesicular monoamine transporter substrate/inhibitor activity of MPTP/MPP+ derivatives: A structure-activity study. J. Med. Chem 2008, 51, 760–768. [Google Scholar]
- Przedborski, S. Pathogenesis of nigral cell death in Parkinson’s disease. Parkinsonism Relat. Disord 2005, 11, S3–S7. [Google Scholar]
- Sala, G; Brighina, L; Saracchi, E; Fermi, S; Riva, C; Carrozza, V; Pirovano, M; Ferrarese, C. Vesicular monoamine transporter 2 mRNA levels are reduced in platelets from patients with Parkinson’s disease. J. Neural. Transm 2010, 117, 1093–1098. [Google Scholar]
- Serra, PA; Sciola, L; Delogu, MR; Spano, A; Monaco, G; Miele, E; Rocchitta, G; Miele, M; Migheli, R; Desole, MS. The neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine induces apoptosis in mouse nigrostriatal glia. Relevance to nigral neuronal death and striatal neurochemical changes. J. Biol. Chem 2002, 277, 34451–34461. [Google Scholar]
- Asanuma, M; Miyazaki, I; Diaz-Corrales, FJ; Ogawa, N. Quinone formation as dopaminergic neuron-specific oxidative stress in the pathogenesis of sporadic Parkinson’s disease and neurotoxin-induced parkinsonism. Acta Med Okayama 2004, 58, 221–233. [Google Scholar]
- Antunes, F; Nunes, C; Laranjinha, J; Cadenas, E. Redox interactions of nitric oxide with dopamine and its derivatives. Toxicology 2005, 208, 207–212. [Google Scholar]
- Li, HT; Lin, DH; Luo, XY; Zhang, F; Ji, LN; Du, HN; Song, GQ; Hu, J; Zhou, JW; Hu, HY. Inhibition of alpha-synuclein fibrillization by dopamine analogs via reaction with the amino groups of alpha-synuclein. Implication for dopaminergic neurodegeneration. FEBS J 2005, 272, 3661–3672. [Google Scholar]
- Akagawa, M; Ishii, Y; Ishii, T; Shibata, T; Yotsu-Yamashita, M; Suyama, K; Uchida, K. Metal-catalyzed oxidation of protein-bound dopamine. Biochemistry 2006, 45, 15120–15128. [Google Scholar]
- Smythies, J; Galzigna, L. The oxidative metabolism of catecholamines in the brain: a review. Biochim. Biophys Acta 1998, 1380, 159–162. [Google Scholar]
- Zecca, L; Fariello, R; Riederer, P; Sulzer, D; Gatti, A; Tampellini, D. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson’s disease. FEBS Lett 2002, 510, 216–220. [Google Scholar]
- Khan, FH; Sen, T; Maiti, AK; Jana, S; Chatterjee, U; Chakrabarti, S. Inhibition of rat brain mitochondrial electron transport chain activity by dopamine oxidation products during extended in vitro incubation: implications for Parkinson’s disease. Biochim. Biophys Acta 2005, 1741, 65–74. [Google Scholar]
- Halliday, GM; Ophof, A; Broe, M; Jensen, PH; Kettle, E; Fedorow, H; Cartwright, MI; Griffiths, FM; Shepherd, CE; Double, KL. Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson’s disease. Brain 2005, 28, 2654–2664. [Google Scholar]
- García-Molina, F; Fenoll, LG; Morote, JC; García-Ruiz, PA; Rodríguez-López, JN; García-Cánovas, F; Tudela, J. Opposite effects of peroxidase in the initial stages of tyrosinase-catalysed melanin biosynthesis. Int. J. Biochem. Cell Biol 2005, 37, 1179–1196. [Google Scholar]
- Alexi, T; Borlongan, CV; Faull, RL; Williams, CE; Clark, RG; Gluckman, PD; Hughes, PE. Neuroprotective strategies for basal ganglia degeneration: Parkinson’s and Huntington’s diseases. Prog. Neurobiol 2000, 60, 409–740. [Google Scholar]
- Double, KL; Ben-Shachar, D; Youdim, MB; Zecca, L; Riederer, P; Gerlach, M. Influence of neuromelanin on oxidative pathways within the human substantia nigra. Neurotoxicol. Teratol 2002, 24, 621–628. [Google Scholar]
- Gentile, V; Cooper, AJ. Transglutaminases—possible drug targets in human diseases. Curr. Drug Targets CNS Neurol. Disord 2004, 3, 99–104. [Google Scholar]
- Caccamo, D; Currò, M; Condello, S; Ferlazzo, N; Ientile, R. Critical role of transglutaminase and other stress proteins during neurodegenerative processes. Amino Acids 2010, 38, 653–658. [Google Scholar]
- Klivenyi, P; Beal, MF; Ferrante, RJ; Andreassen, OA; Wermer, M; Chin, MR; Bonventre, JV. Mice deficient in group IV cytosolic phospholipase A2 are resistant to MPTP neurotoxicity. J. Neurochem 1998, 71, 2634–2637. [Google Scholar]
- Feng, ZH; Wang, TG; Li, DD; Fung, P; Wilson, BC; Liu, B; Ali, SF; Langenbach, R; Hong, JS. Cyclooxygenase-2-deficient mice are resistant to 1-methyl-4-phenyl1,2,3,6- tetrahydropyridine-induced damage of dopaminergic neurons in the substantia nigra. Neurosci. Lett 2002, 329, 354–358. [Google Scholar]
- Zhang, J; Graham, DG; Montine, TJ; Ho, YS. Enhanced N-methyl-4-phenyl-1,2,3,6- tetrahydropyridine toxicity in mice deficient in CuZn-superoxide dismutase or glutathione peroxidase. J. Neuropathol. Exp. Neurol 2000, 59, 53–61. [Google Scholar]
- St-Pierre, J; Drori, S; Uldry, M; Silvaggi, JM; Rhee, J; Jäger, S; Handschin, C; Zheng, K; Lin, J; Yang, W; Simon, DK; Bachoo, R; Spiegelman, BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar]
- Mohanakumar, KP; Muralikrishnan, D; Thomas, B. Neuroprotection by sodium salicylate against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. Brain Res 2000, 864, 281–290. [Google Scholar]
- Gupta, A; Dhir, A; Kumar, A; Kulkarni, SK. Effect of preferential cyclooxygenase-2 (COX-2) inhibitor against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced striatal lesions in rats: behavioral, biochemical and histological evidences. Indian J. Exp. Biol 2010, 48, 577–585. [Google Scholar]
- Teismann, P; Ferger, B. Inhibition of the cyclooxygenase isoenzymes COX-1 and COX-2 provide neuroprotection in the MPTP-mouse model of Parkinson’s disease. Synapse 2001, 39, 167–174. [Google Scholar]
- Tariq, M; Khan, HA; Al Moutaery, K; Al Deeb, S. Protective effect of quinacrine on striatal dopamine levels in 6-OHDA and MPTP models of Parkinsonism in rodents. Brain Res Bullutin 2001, 54, 77–82. [Google Scholar]
- Nishino, Y; Ando, M; Makino, R; Ueda, K; Okamoto, Y; Kojima, N. Different mechanisms between copper and iron in catecholamines-mediated oxidative DNA damage and disruption of gene expression in vitro. Neurotox Res 2010, in press. [Google Scholar]
- Sayre, LM; Perry, G; Smith, MA. Redox metals and neurodegenerative disease. Curr. Opin. Chem. Biol 1999, 3, 220–225. [Google Scholar]
- Blum, D; Torch, S; Lambeng, N; Nissou, M; Benabid, AL; Sadoul, R; Verna, JM. Molecular pathways involved in the neurotoxicity of 6-OHDA., dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog. Neurobiol 2001, 65, 135–172. [Google Scholar]
- Ebadi, M; Srinivasan, SK; Baxi, MD. Oxidative stress and antioxidant therapy in Parkinson’s disease. Prog. Neurobiol 1996, 48, 1–19. [Google Scholar]
- Annepu, J; Ravindranath, V. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced complex I inhibition is reversed by disulfide reductant, dithiothreitol in mouse brain. Neurosci. Lett 2000, 289, 209–212. [Google Scholar]
- Asanuma, M; Miyazaki, I; Diaz-Corrales, FJ; Ogawa, N. Quinone formation as dopaminergic neuron-specific oxidative stress in the pathogenesis of sporadic Parkinson’s disease and neurotoxin-induced parkinsonism. Acta Med Okayama 2004, 58, 221–233. [Google Scholar]
- Hare, DJ; George, JL; Grimm, R; Wilkins, S; Adlard, PA; Cherny, RA; Bush, AI; Finkelstein, DI; Doble, P. Three-dimensional elemental bio-imaging of Fe, Zn, Cu, Mn and P in a 6-hydroxydopamine lesioned mouse brain. Metallomics 2010, 2, 745–753. [Google Scholar]
- Barnham, KJ; Bush, AI. Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol 2008, 12, 222–228. [Google Scholar]
- Hirsch, EC. Iron transport in Parkinson’s disease. Parkinsonism Relat. Disord 2009, 15, S209–S211. [Google Scholar]
- Jameson, GN; Jameson, RF; Linert, W. New insights into iron release from ferritin: direct observation of the neurotoxin 6-hydroxydopamine entering ferritin and reaching redox equilibrium with the iron core. Org. Biomol. Chem 2004, 2, 2346–2351. [Google Scholar]
- Kobayashi, H; Oikawa, S; Umemura, S; Hirosawa, I; Kawanishi, S. Mechanism of metalmediated DNA damage and apoptosis induced by 6-hydroxydopamine in neuroblastoma SH-SY5Y cells. Free Radic. Res 2008, 42, 651–660. [Google Scholar]
- Gauthier, MA; Eibl, JK; Crispo, JA; Ross, GM. Covalent arylation of metallothionein by oxidized dopamine products: a possible mechanism for zinc-mediated enhancement of dopaminergic neuron survival. Neurotox. Res 2008, 14, 317–328. [Google Scholar]
- Jiang, H; Song, N; Xu, H; Zhang, S; Wang, J; Xie, J. Up-regulation of divalent metal transporter 1 in 6-hydroxydopamine intoxication is IRE/IRP dependent. Cell Res 2010, 20, 345–356. [Google Scholar]
- Nicolaus, BJ. A critical review of the function of neuromelanin and an attempt to provide a unified theory. Med Hypotheses 2005, 65, 791–796. [Google Scholar]
- Koeppen, AH. The history of iron in the brain. J. Neurol. Sci 1995, 134, 1–9. [Google Scholar]
- Kaur, D; Andersen, J. Does cellular iron dysregulation play a causative role in Parkinson’s disease? Ageing Res. Rev 2004, 3, 327–343. [Google Scholar]
- Takanashi, M; Mochizuki, H; Yokomizo, K; Hattori, N; Mori, H; Yamamura, Y; Mizuno, Y. Iron accumulation in the substantia nigra of autosomal recessive juvenile parkinsonism (ARJP). Parkinsonism Relat. Disord 2001, 7, 311–314. [Google Scholar]
- Faucheux, BA; Nillesse, N; Damier, P; Spik, G; Mouatt-Prigent, A; Pierce, A; Leveugle, B; Kubis, N; Hauw, JJ; Agid, Y; et al. Expression of lactoferrin receptors is increased in the mesencephalon of patients with Parkinson disease. Proc. Natl. Acad. Sci USA 1995, 92, 9603–9607. [Google Scholar]
- Jiang, H; Qian, ZM; Xie, JX. Increased DMT1 expression and iron content in MPTP-treated C57BL/6 mice. Sheng Li Xue Bao 2003, 55, 571–576. [Google Scholar]
- Zucca, FA; Giaveri, G; Gallorini, M; Albertini, A; Toscani, M; Pezzoli, G; Lucius, R; Wilms, H; Sulzer, D; Ito, S; Wakamatsu, K; Zecca, L. The neuromelanin of human substantia nigra: physiological and pathogenic aspects. Pigment Cell Res 2004, 17, 610–617. [Google Scholar]
- Zhang, J; Zhang, Y; Wang, J; Cai, P; Luo, C; Qian, Z; Dai, Y; Feng, H. Characterizing iron deposition in Parkinson’s disease using susceptibility-weighted imaging: an in vivo MR study. Brain Res 2010, 1330, 124–130. [Google Scholar]
- Andersen, JK. Iron dysregulation and Parkinson’s disease. J. Alzheimers Dis 2004, 6, S47–S52. [Google Scholar]
- Bou-Abdallah, F; McNally, J; Liu, XX; Melman, A. Oxygen catalyzed mobilization of iron from ferritin by iron(iii) chelate ligands. Chem. Commun 2011, 47, 731–733. [Google Scholar]
- Qian, ZM; Wang, Q. Expression of iron transport proteins and excessive iron accumulation in the brain in neurodegenerative disorders. Brain Res. Rev 1998, 27, 257–267. [Google Scholar]
- Ke, Y; Ming Qian, Z. Iron misregulation in the brain: a primary cause of neurodegenerative disorders. Lancet Neurol 2003, 2, 246–253. [Google Scholar]
- Schipper, HM; Liberman, A; Stopa, EG. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol 1998, 150, 60–68. [Google Scholar]
- Shamoto-Nagai, M; Maruyama, W; Yi, H; Akao, Y; Tribl, F; Gerlach, M; Osawa, T; Riederer, P; Naoi, M. Neuromelanin induces oxidative stress in mitochondria through release of iron: mechanism behind the inhibition of 26S proteasome. J. Neural. Transm 2006, 113, 633–644. [Google Scholar]
- Hirsch, EC. Iron transport in Parkinson’s disease. Parkinsonism Relat. Disord 2009, 15, S209–S211. [Google Scholar]
- Levenson, CW; Cutler, RG; Ladenheim, B; Cadet, JL; Hare, J; Mattson, MP. Role of dietary iron restriction in a mouse model of Parkinson’s disease. Exp. Neurol 2004, 190, 506–514. [Google Scholar]
- Gal, S; Fridkin, M; Amit, T; Zheng, H; Youdim, MB. M30, a novel multifunctional neuroprotective drug with potent iron chelating and brain selective monoamine oxidase-ab inhibitory activity for Parkinson’s disease. J. Neural. Transm. Suppl 2006, 70, 447–456. [Google Scholar]
- Kaur, D; Yantiri, F; Rajagopalan, S; Kumar, J; Mo, JQ; Boonplueang, R; Viswanath, V; Jacobs, R; Yang, L; Beal, MF; DiMonte, D; Volitaskis, I; Ellerby, L; Cherny, RA; Bush, AI; Andersen, JK. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson’s disease. Neuron 2003, 37, 899–909. [Google Scholar]
- Zecca, L; Berg, D; Arzberger, T; Ruprecht, P; Rausch, WD; Musicco, M; Tampellini, D; Riederer, P; Gerlach, M; Becker, G. In vivo detection of iron and neuromelanin by transcranial sonography: a new approach for early detection of substantia nigra damage. Mov. Disord 2005, 20, 1278–1285. [Google Scholar]
- Naoi, M; Maruyama, W. Monoamine oxidase inhibitors as neuroprotective agents in age-dependent neurodegenerative disorders. Curr. Pharm. Des 2010, 16, 2799–2817. [Google Scholar]
- Bortolato, M; Chen, K; Shih, JC. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv. Drug Deliv. Rev 2008, 60, 1527–1533. [Google Scholar]
- Li, SW; Lin, TS; Minteer, S; Burke, WJ. 3,4-Dihydroxyphenylacetaldehyde and hydrogen peroxide generate a hydroxyl radical: possible role in Parkinson’s disease pathogenesis. Brain Res. Mol. Brain Res 2001, 93, 1–7. [Google Scholar]
- Tabner, BJ; Turnbull, S; El-Agnaf, OM; Allsop, D. Formation of hydrogen peroxide and hydroxyl radicals from A(beta) and alpha-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Radic. Biol. Med 2002, 32, 1076–1083. [Google Scholar]
- Matsubara, K; Aoyama, K; Suno, M; Awaya, T. N-methylation underlying Parkinson’s disease. Neurotoxicol. Teratol 2002, 24, 593–598. [Google Scholar]
- Parsons, RB; Smith, SW; Waring, RH; Williams, AC; Ramsden, DB. High expression of nicotinamide N-methyltransferase in patients with idiopathic Parkinson’s disease. Neurosci. Lett 2003, 342, 13–16. [Google Scholar]
- Naoi, M; Maruyama, W; Nagy, GM. Dopamine-derived salsolinol derivatives as endogenous monoamine oxidase inhibitors: occurrence, metabolism and function in human brains. Neurotoxicology 2004, 25, 193–204. [Google Scholar]
- DeCuypere, M; Lu, Y; Miller, DD; LeDoux, MS. Regional distribution of tetrahydroisoquinoline derivatives in rodent, human, and Parkinson’s disease brain. J. Neurochem 2008, 107, 1398–1413. [Google Scholar]
- Antkiewicz-Michaluk, L. Endogenous risk factors in Parkinson’s disease: dopamine and tetrahydroisoquinolines. Pol. J. Pharmacol 2002, 54, 567–572. [Google Scholar]
- Soto-Otero, R; Méndez-Alvarez, E; Sánchez-Sellero, I; Cruz-Landeira, A; López-Rivadulla Lamas, M. Reduction of rat brain levels of the endogenous dopaminergic proneurotoxins 1,2,3,4- tetrahydroisoquinoline and 1,2,3,4-tetrahydro-beta-carboline by cigarette smoke. Neurosci. Lett 2001, 298, 187–190. [Google Scholar]
- Gearhart, DA; Neafsey, EJ; Collins, MA. Phenylethanolamine N-methyltransferase has beta-carboline 2N-methyltransferase activity: hypothetical relevance to Parkinson’s disease. Neurochem. Int 2002, 40, 611–620. [Google Scholar]
- Wu, YN; Johnson, SW. Rotenone potentiates NMDA currents in substantia nigra dopamine neurons. Neurosci. Lett 2007, 421, 96–100. [Google Scholar]
- Gao, WJ; Goldman-Rakic, PS. NMDA receptor-mediated epileptiform persistent activity requires calcium release from intracellular stores in prefrontal neurons. Exp. Neurol 2006, 197, 495–504. [Google Scholar]
- Nizami, S; Lee, VW; Davies, J; Long, P; Jovanovic, JN; Sihra, TS. Presynaptic roles of intracellular Ca(2+) stores in signalling and exocytosis. Biochem. Soc. Trans 2010, 38, 529–535. [Google Scholar]
- Maurois, P; Pages, N; Bac, P; German-Fattal, M; Agnani, G; Delplanque, B; Durlach, J. Threshold to N-methyl-d-aspartate-induced seizures in mice undergoing chronic nutritional magnesium deprivation is lowered in a way partly responsive to acute magnesium and antioxidant administrations. Br. J. Nutr 2009, 101, 317–321. [Google Scholar]
- Gu, Z; Nakamura, T; Lipton, SA. Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol. Neurobiol 2010, 41, 55–72. [Google Scholar]
- Martínez, A; Portero-Otin, M; Pamplona, R; Ferrer, I. Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol 2010, 20, 281–297. [Google Scholar]
- Adamczyk, A; KaŸmierczak, A; Czapski, GA; Strosznajder, JB. Alpha-synuclein induced cell death in mouse hippocampal (HT22) cells is mediated by nitric oxide-dependent activation of caspase-3. FEBS Lett 2010, 584, 3504–3508. [Google Scholar]
- Adamczyk, A; Czapski, GA; KaŸmierczak, A; Strosznajder, JB. Effect of N-methyl-d-aspartate (NMDA) receptor antagonists on alpha-synuclein-evoked neuronal nitric oxide synthase activation in the rat brain. Pharmacol. Rep 2009, 61, 1078–1085. [Google Scholar]
- Meredith, GE; Totterdell, S; Beales, M; Meshul, CK. Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of Parkinson’s disease. Exp. Neurol 2009, 219, 334–340. [Google Scholar]
- Nimmrich, V; Reymann, KG; Strassburger, M; Schöder, UH; Gross, G; Hahn, A; Schoemaker, H; Wicke, K; Möller, A. Inhibition of calpain prevents NMDA-induced cell death and beta-amyloid-induced synaptic dysfunction in hippocampal slice cultures. Br J Pharmacol 2010, 159 , 1523–1531. [Google Scholar]
- Ma, T; Zhao, Y; Kwak, YD; Yang, Z; Thompson, R; Luo, Z; Xu, H; Liao, FF. Statin’s excitoprotection is mediated by sAPP and the subsequent attenuation of calpain-induced truncation events, likely via rho-ROCK signaling. J. Neurosci 2009, 29, 11226–11236. [Google Scholar]
- Pan, J; Xiao, Q; Sheng, CY; Hong, Z; Yang, HQ; Wang, G; Ding, JQ; Chen, SD. Blockade of the translocation and activation of c-Jun N-terminal kinase 3 (JNK3) attenuates dopaminergic neuronal damage in mouse model of Parkinson’s disease. Neurochem. Int 2009, 54, 418–425. [Google Scholar]
- Wang, AL; Liou, YM; Pawlak, CR; Ho, YJ. Involvement of NMDA receptors in both MPTP-induced neuroinflammation and deficits in episodic-like memory in Wistar rats. Behav. Brain Res 2010, 208, 38–46. [Google Scholar]
- Hald, A; van Beek, J; Lotharius, J. Inflammation in Parkinson’s disease: causative or epiphenomenal? Subcell. Biochem 2007, 42, 249–279. [Google Scholar]
- Kurkowska-Jastrzebska, I; Wrońska, A; Kohutnicka, M; Członkowski, A; Członkowska, A. MHC class II positive microglia and lymphocytic infiltration are present in the substantia nigra and striatum in mouse model of Parkinson’s disease. Acta Neurobiol. Exp 1999, 59, 1–8. [Google Scholar]
- Ouchi, Y; Yoshikawa, E; Sekine, Y; Futatsubashi, M; Kanno, T; Ogusu, T; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol 2005, 57, 168–175. [Google Scholar]
- Nagatsu, T; Sawada, M. Inflammatory process in Parkinson’s disease: role for cytokines. Curr. Pharm. Des 2005, 11, 999–1016. [Google Scholar]
- McGeer, PL; McGeer, EG. Inflammation and the degenerative diseases of aging. Ann. N. Y. Acad. Sci 2004, 1035, 104–116. [Google Scholar]
- Nagatsu, T; Mogi, M; Ichinose, H; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s disease. J. Neural. Transm. Suppl 2000, 60, 277–290. [Google Scholar]
- Sriram, K; Matheson, JM; Benkovic, SA; Miller, DB; Luster, MI; O’Callaghan, JP. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson’s disease. FASEB J 2002, 16, 1474–1476. [Google Scholar]
- Onyango, IG; Tuttle, JB; Bennett, JP, Jr. Activation of p38 and N-acetylcysteine-sensitive c-Jun NH2-terminal kinase signaling cascades is required for induction of apoptosis in Parkinson’s disease cybrids. Mol. Cell Neurosci 2005, 28, 452–461. [Google Scholar]
- Hirsch, EC; Hunot, S; Hartmann, A. Neuroinflammatory processes in Parkinson’s disease. Parkinsonism Relat. Disord 2005, 11, S9–S15. [Google Scholar]
- Tansey, MG; Goldberg, MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis 2010, 37, 510–518. [Google Scholar]
- Zhang, W; Wang, T; Pei, Z; Miller, DS; Wu, X; Block, ML; Wilson, B; Zhang, W; Zhou, Y; Hong, JS; Zhang, J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J 2005, 19, 533–542. [Google Scholar]
- Hirsch, EC; Hunot, S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 2009, 8, 382–397. [Google Scholar]
- Ji, H; Wang, H; Zhang, F; Li, X; Xiang, L; Aiguo, S. PPARγ agonist pioglitazone inhibits microglia inflammation by blocking p38 mitogen-activated protein kinase signaling pathways. Inflamm. Res 2010, 59, 921–929. [Google Scholar]
- Kim, SH; Kim, J; Sharma, RP. Inhibition of p38 and ERK MAP kinases blocks endotoxininduced nitric oxide production and differentially modulates cytokine expression. Pharmacol. Res 2004, 49, 433–439. [Google Scholar]
- Thomas, T; Timmer, M; Cesnulevicius, K; Hitti, E; Kotlyarov, A; Gaestel, M. MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing neuroinflammation—relevance in a mouse model of Parkinson’s disease. J. Neurochem 2008, 105, 2039–2052. [Google Scholar]
- Willesen, MG; Gammeltoft, S; Vaudano, E. Activation of the c-Jun N terminal kinase pathway in an animal model of Parkinson’s disease. Ann. N. Y. Acad. Sci 2002, 973, 237–240. [Google Scholar]
- Lee, DY; Oh, YJ; Jin, BK. Thrombin-activated microglia contribute to death of dopaminergic neurons in rat mesencephalic cultures: dual roles of mitogen-activated protein kinase signaling pathways. Glia 2005, 51, 98–110. [Google Scholar]
- Kao, SJ; Lei, HC; Kuo, CT; Chang, MS; Chen, BC; Chang, YC; Chiu, WT; Lin, CH. Lipoteichoic acid induces nuclear factor-kappaB activation and nitric oxide synthase expression via phosphatidylinositol 3-kinase, Akt, and p38 MAPK in RAW 264.7 macrophages. Imunology 2005, 115, 366–374. [Google Scholar]
- Chio, CC; Chang, YH; Hsu, YW; Chi, KH; Lin, WW. PKA-dependent activation of PKC.; p38 MAPK and IKK in macrophage: implication in the induction of inducible nitric oxide synthase and interleukin-6 by dibutyryl cAMP. Cell Signal 2004, 16, 565–575. [Google Scholar]
- Lee, JK; Choi, SS; Won, JS; Suh, HW. The regulation of inducible nitric oxide synthase gene expression induced by lipopolysaccharide and tumor necrosis factor-alpha in C6 cells: involvement of AP-1 and NFkappaB. Life Sci 2003, 73, 595–609. [Google Scholar]
- Wang, MJ; Lin, WW; Chen, HL; Chang, YH; Ou, HC; Kuo, JS; Hong, JS; Jeng, KC. Silymarin protects dopaminergic neurons against lipopolysaccharide-induced neurotoxicity by inhibiting microglia activation. Eur. J. Neurosci 2002, 16, 2103–2112. [Google Scholar]
- Hua, LL; Zhao, ML; Cosenza, M; Kim, MO; Huang, H; Tanowitz, HB; Brosnan, CF; Lee, SC. Role of mitogen-activated protein kinases in inducible nitric oxide synthase and TNFalpha expression in human fetal astrocytes. J. Neuroimmunol 2002, 126, 180–189. [Google Scholar]
- Gerhard, A; Pavese, N; Hotton, G; Turkheimer, F; Es, M; Hammers, A; Eggert, K; Oertel, W; Banati, RB; Brooks, DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis 2006, 21, 404–412. [Google Scholar]
- Ban, HS; Suzuki, K; Lim, SS; Jung, SH; Lee, S; Ji, J; Lee, HS; Lee, YS; Shin, KH; Ohuchi, K. Inhibition of lipopolysaccharide-induced expression of inducible nitric oxide synthase and tumor necrosis factor-alpha by 2′-hydroxychalcone derivatives in RAW 264.7 cells. Biochem. Pharmacol 2004, 67, 1549–1557. [Google Scholar]
- Wang, W; Ma, C; Mao, Z; Li, M. JNK inhibition as a potential strategy in treating Parkinson’s disease. Drug News Perspect 2004, 17, 646–654. [Google Scholar]
- Saporito, MS; Thomas, BA; Scott, RW. MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J. Neurochem 2000, 75, 1200–1208. [Google Scholar]
- Saporito, MS; Brown, EM; Miller, MS; Carswell, S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons in vivo. J. Pharmacol. Exp. Ther 1999, 288, 421–427. [Google Scholar]
- Kurkowska-Jastrzebska, I; Babiuch, M; Joniec, I; Przybyłkowski, A; Członkowski, A; Członkowska, A. Indomethacin protects against neurodegeneration caused by MPTP intoxication in mice. Int. Immunopharmacol 2002, 2, 1213–1218. [Google Scholar]
- Fahrig, T; Gerlach, I; Horváth, E. A synthetic derivative of the natural product rocaglaol is a potent inhibitor of cytokine-mediated signaling and shows neuroprotective activity in vitro and in animal models of Parkinson’s disease and traumatic brain injury. Mol. Pharmacol 2005, 67, 1544–1555. [Google Scholar]
- Wu, DC; Jackson-Lewis, V; Vila, M; Tieu, K; Teismann, P; Vadseth, C; Choi, DK; Ischiropoulos, H; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci 2002, 22, 1763–1771. [Google Scholar]
- Du, Y; Ma, Z; Lin, S; Dodel, RC; Gao, F; Bales, KR; Triarhou, LC; Chernet, E; Perry, KW; Nelson, DL; Luecke, S; Phebus, LA; Bymaster, FP; Paul, SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci USA 2001, 98, 14669–14674. [Google Scholar]
- Lee, HG; Kim, H; Oh, WK; Yu, KA; Choe, YK; Ahn, JS; Kim, DS; Kim, SH; Dinarello, CA; Kim, K; Yoon, DY. Tetramethoxy hydroxyflavone p7F downregulates inflammatory mediators via the inhibition of nuclear factor kappaB. Ann. N. Y. Acad. Sci 2004, 1030, 555–568. [Google Scholar]
- Anwar, AA; Li, FY; Leake, DS; Ishii, T; Mann, GE; Siow, RC. Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: role of mitogen-activated protein kinases and Nrf2. Free Radic. Biol. Med 2005, 39, 227–236. [Google Scholar]
- Tieu, K; Ischiropoulos, H; Przedborski, S. Nitric oxide and reactive oxygen species in Parkinson’s disease. IUBMB Life 2003, 55, 329–335. [Google Scholar]
- Wu, DC; Teismann, P; Tieu, K; Vila, M; Jackson-Lewis, V; Ischiropoulos, H; Przedborski, S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc. Natl. Acad. Sci USA 2003, 100, 6145–6150. [Google Scholar]
- Dehmer, T; Lindenau, J; Haid, S; Dichgans, J; Schulz, JB. Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J. Neurochem 2000, 74, 2213–2216. [Google Scholar]
- Klivenyi, P; St Clair, D; Wermer, M; Yen, HC; Oberley, T; Yang, L; Beal, MF. Manganese superoxide dismutase overexpression attenuates MPTP toxicity. Neurobiol. Dis 1998, 5, 253–258. [Google Scholar]
- Andreassen, OA; Ferrante, RJ; Dedeoglu, A; Albers, DW; Klivenyi, P; Carlson, EJ; Epstein, CJ; Beal, MF. Mice with a partial deficiency of manganese superoxide dismutase show increased vulnerability to the mitochondrial toxins malonate, 3-nitropropionic acid, and MPTP. Exp. Neurol 2001, 167, 189–195. [Google Scholar]
- Callio, J; Oury, TD; Chu, CT. Manganese superoxide dismutase protects against 6-hydroxydopamine injury in mouse brains. J. Biol. Chem 2005, 280, 18536–18542. [Google Scholar]
- Levites, Y; Weinreb, O; Maor, G; Youdim, MB; Mandel, S. Green tea polyphenol (−)-epigallocatechin-3-gallate prevents N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurodegeneration. J. Neurochem 2001, 78, 1073–1082. [Google Scholar]
- Kurosaki, R; Muramatsu, Y; Michimata, M; Matsubara, M; Kato, H; Imai, Y; Itoyama, Y; Araki, T. Role of nitric oxide synthase against MPTP neurotoxicity in mice. Neurol. Res 2002, 24, 655–662. [Google Scholar]
- Choi, JY; Park, CS; Kim, DJ; Cho, MH; Jin, BK; Pie, JE; Chung, WG. Prevention of nitric oxide-mediated 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease in mice by tea phenolic epigallocatechin 3-gallate. Neurotoxicology 2002, 23, 367–374. [Google Scholar]
- Mazzio, EA; Soliman, KF. Cytoprotection of pyruvic acid and reduced beta-nicotinamide adenine dinucleotide against hydrogen peroxide toxicity in neuroblastoma cells. Neurochem. Res 2003, 28, 733–741. [Google Scholar]
- Mazzio, EA; Reams, RR; Soliman, KF. The role of oxidative stress, impaired glycolysis and mitochondrial respiratory redox failure in the cytotoxic effects of 6-hydroxydopamine in vitro. Brain Res 2004, 1004, 29–44. [Google Scholar]
- Auer, RN. Hypoglycemic brain damage. Metab Brain Dis 2004, 19, 169–175. [Google Scholar]
- Mazzio, E; Soliman, KF. Pyruvic acid cytoprotection against 1-methyl-4-phenylpyridinium, 6-hydroxydopamine and hydrogen peroxide toxicities in vitro. Neurosci. Lett 2003, 337, 77–80. [Google Scholar]
- Gonzalez, SV; Nguyen, NH; Rise, F; Hassel, B. Brain metabolism of exogenous pyruvate. J. Neurochem 2005, 95, 284–293. [Google Scholar]
- Lee, JY; Kim, YH; Koh, JY. Protection by pyruvate against transient forebrain ischemia in rats. J Neurosci 2001, 21, RC171. [Google Scholar]
- Izumi, Y; Zorumski, CF. Neuroprotective effects of pyruvate following NMDA-mediated excitotoxic insults in hippocampal slices. Neurosci. Lett 2010, 478, 131–135. [Google Scholar]
- Cosi, C; Marien, M. Decreases in mouse brain NAD+ and ATP induced by 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine (MPTP): prevention by the poly(ADP-ribose) polymerase inhibitor, benzamide. Brain Res 1998, 809, 58–67. [Google Scholar]
- Cosi, C; Marien, M. Implication of poly (ADP-ribose) polymerase (PARP) in neurodegeneration and brain energy metabolism. Decreases in mouse brain NAD+ and ATP caused by MPTP are prevented by the PARP inhibitor benzamide. Ann. N. Y. Acad. Sci 1999, 890, 227–239. [Google Scholar]
- Iwashita, A; Yamazaki, S; Mihara, K; Hattori, K; Yamamoto, H; Ishida, J; Matsuoka, N; Mutoh, S. Neuroprotective effects of a novel poly(ADP-ribose) polymerase-1 inhibitor, 2-[3-[4-(4-chlorophenyl)-1-piperazinyl] propyl]-4(3H)-quinazolinone (FR255595), in an in vitro model of cell death and in mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. J. Pharmacol. Exp. Ther 2004, 309, 1067–1078. [Google Scholar]
- Yokoyama, H; Kuroiwa, H; Tsukada, T; Uchida, H; Kato, H; Araki, T. Poly(ADPribose) polymerase inhibitor can attenuate the neuronal death after 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine-induced neurotoxicity in mice. J. Neurosci. Res 2010, 88, 1522–1536. [Google Scholar]
- Anderson, DW; Bradbury, KA; Schneider, JS. Broad neuroprotective profile of nicotinamide in different mouse models of MPTP-induced Parkinsonism. Eur. J. Neurosci 2008, 28, 610–617. [Google Scholar]
- Mukherjee, SK; Klaidman, LK; Yasharel, R; Adams, JD, Jr. Increased brain NAD prevents neuronal apoptosis in vivo. Eur. J. Pharmacol 1997, 330, 27–34. [Google Scholar]
- Yokoyama, H; Kuroiwa, H; Tsukada, T; Uchida, H; Kato, H; Araki, T. Poly(ADPribose) polymerase inhibitor can attenuate the neuronal death after 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine-induced neurotoxicity in mice. J. Neurosci. Res 2010, 88, 1522–1536. [Google Scholar]
- Mandir, AS; Simbulan-Rosenthal, CM; Poitras, MF; Lumpkin, JR; Dawson, VL; Smulson, ME; Dawson, TM. A novel in vivo post-translational modification of p53 by PARP-1 in MPTP-induced parkinsonism. J. Neurochem 2002, 83, 186–192. [Google Scholar]
- Duan, W; Zhu, X; Ladenheim, B; Yu, QS; Guo, Z; Oyler, J; Cutler, RG; Cadet, JL; Greig, NH; Mattson, MP. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann. Neurol 2002, 52, 597–606. [Google Scholar]
- Fukushima, T; Ohta, M; Tanaka, K; Kaneko, S-Y; Maeda, T; Sasaki, A. Niacin metabolism and Parkinson’s disease. Asia Pac. J. Clin. Nutr 2004, 13, S176. [Google Scholar]
- Alston, TA; Abeles, RH. Substrate specificity of nicotinamide methyltransferase isolated from porcine liver. Arch. Biochem. Biophys 1988, 260, 601–618. [Google Scholar]
- Ashihara, H; Crozier, A. Caffeine: a well known but little mentioned compound in plant science. Trends Plant Sci 2001, 6, 407–413. [Google Scholar]
- Koshiishi, C; Kato, A; Yama, S; Crozier, A; Ashihara, H. A new caffeine biosynthetic pathway in tea leaves: utilisation of adenosine released from the S-adenosyl-l-methionine cycle. FEBS Lett 2001, 499, 50–54. [Google Scholar]
- Góngora-Alfaro, JL. Caffeine as a preventive drug for Parkinson’s disease: epidemiologic evidence and experimental support. Rev. Neurol 2010, 50, 221–229. [Google Scholar]
- Singh, K; Singh, S; Singhal, NK; Sharma, A; Parmar, D; Singh, MP. Nicotine- and caffeinemediated changes in gene expression patterns of MPTP-lesioned mouse striatum: Implications in neuroprotection mechanism. Chem. Biol. Interact 2010, 185, 81–93. [Google Scholar]
- Upmeier, B; Gross, W; Köster, S; Barz, W. Purification and properties of S-adenosyl-l-methionine: nicotinic acid-N-methyltransferase from cell suspension cultures of Glycine max L. Arch. Biochem. Biophys 1988, 262, 445–454. [Google Scholar]
- Oyanagi, K. The nature of the parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam and magnesium deficiency. Parkinsonism Relat. Disord 2005, 11, S17–S23. [Google Scholar]
- Barbiroli, B; Martinelli, P; Patuelli, A; Lodi, R; Iotti, S; Cortelli, P; Montagna, P. Phosphorus magnetic resonance spectroscopy in multiple system atrophy and Parkinson’s disease. Mov. Disord 1999, 14, 430–435. [Google Scholar]
- Philippu, A; Matthaei, H; Lentzen, H. Uptake of dopamine into fractions of pig caudate nucleus homogenates. Naunyn Schmiedebergs Arch. Pharmacol 1975, 287, 181–190. [Google Scholar]
- Schümann, HJ; Althoff, B. Effects of calcium and phosphate on catecholamines, ATP and dopamine beta-hydroxylase of chromaffin medullary granules. Naunyn Schmiedebergs Arch. Pharmacol 1976, 293, 67–74. [Google Scholar]
- Baker, PF; Knight, DE. Gaining access to the site of exocytosis in bovine adrenal medullary cells. J. Physiol 1980, 76, 497–504. [Google Scholar]
- Yang, YC; Lee, CH; Kuo, CC. Ionic flow enhances low-affinity binding: a revised mechanistic view into Mg2+ block of NMDA receptors. J. Physiol 2010, 588, 633–650. [Google Scholar]
- Safar, MM; Abdallah, DM; Arafa, NM; Abdel-Aziz, MT. Magnesium supplementation enhances the anticonvulsant potential of valproate in pentylenetetrazol-treated rats. Brain Res 2010, 1334, 58–64. [Google Scholar]
- Lin, JY; Chung, SY; Lin, M; Cheng, FC. Effects of magnesium sulfate on energy metabolites and glutamate in the cortex during focal cerebral ischemia and reperfusion in the gerbil monitored by a dual-probe microdialysis technique. Life Sci 2002, 71, 803–811. [Google Scholar]
- Johnson, S. Micronutrient accumulation and depletion in schizophrenia, epilepsy, autism and Parkinson’s disease? Med Hypotheses 2001, 56, 641–645. [Google Scholar]
- Brosnan, JT; Jacobs, RL; Stead, LM; Brosnan, ME. Methylation demand: a key determinant of homocysteine metabolism. Acta Biochim. Pol 2004, 51, 405–413. [Google Scholar]
- Zoccolella, S; Lamberti, P; Armenise, E; de Mari, M; Lamberti, SV; Mastronardi, R; Fraddosio, A; Iliceto, G; Livrea, P. Plasma homocysteine levels in Parkinson’s disease: role of antiparkinsonian medications. Parkinsonism Relat. Disord 2005, 11, 131–133. [Google Scholar]
- Lamberti, P; Zoccolella, S; Armenise, E; Lamberti, SV; Fraddosio, A; de Mari, M; Iliceto, G; Livrea, P. Hyperhomocysteinemia in L-dopa treated Parkinson’s disease patients: effect of cobalamin and folate administration. Eur. J. Neurol 2005, 12, 365–368. [Google Scholar]
- McCully, KS. Chemical pathology of homocysteine. IV. Excitotoxicity, oxidative stress, endothelial dysfunction, and inflammation. Ann. Clin. Lab. Sci 2009, 39, 219–232. [Google Scholar]
- dos Santos, EF; Busanello, EN; Miglioranza, A; Zanatta, A; Barchak, AG; Vargas, CR; Saute, J; Rosa, C; Carrion, MJ; Camargo, D; Dalbem, A; da Costa, JC; de Sousa Miguel, SR; de Mello Rieder, CR; Wajner, M. Evidence that folic acid deficiency is a major determinant of hyperhomocysteinemia in Parkinson’s disease. Metab. Brain Dis 2009, 24, 257–269. [Google Scholar]
- Miller, JW. Homocysteine, folate deficiency, and Parkinson’s disease. Nutr. Rev 2002, 60, 410–413. [Google Scholar]
- Duan, W; Ladenheim, B; Cutler, RG; Kruman, II; Cadet, JL; Mattson, MP. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. J. Neurochem 2002, 80, 101–110. [Google Scholar]
- Yagisawa, M; Okawa, N; Shigematsu, N; Nakata, R. Effects of intravenous betaine on methionine-loading-induced plasma homocysteine elevation in rats. J. Nutr. Biochem 2004, 15, 666–671. [Google Scholar]
- Kharbanda, KK; Rogers, DD, II; Mailliard, ME; Siford, GL; Barak, AJ; Beckenhauer, HC; Sorrell, MF; Tuma, DJ. Role of elevated S-adenosylhomocysteine in rat hepatocyte apoptosis: protection by betaine. Biochem. Pharmacol 2005, 70, 1883–1890. [Google Scholar]
- Yeh, Y-Y; Lim, H-S; Yeh, S-M; Picciano, MF. Garlic extract attenuates hyperhomocysteinemia caused by folic acid deficiency in the rat. Nutr. Res 2005, 25, 93–102. [Google Scholar]
- Sled, VD; Rudnitzky, NI; Hatefi, Y; Ohnishi, T. Thermodynamic analysis of flavin in mitochondrial NADH:ubiquinone oxidoreductase (complex I). Biochemistry 1994, 33, 10069–10075. [Google Scholar]
- Gerards, M; van den Bosch, BJ; Danhauser, K; Serre, V; van Weeghel, M; Wanders, RJ; Nicolaes, GA; Sluiter, W; Schoonderwoerd, K; Scholte, HR; Prokisch, H; Rötig, A; de Coo, IF; Smeets, HJ. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene. Brain 2011, 134, 210–219. [Google Scholar]
- Bar-Meir, M; Elpeleg, ON; Saada, A. Effect of various agents on adenosine triphosphate synthesis in mitochondrial complex I deficiency. J. Pediatr 2001, 139, 868–870. [Google Scholar]
- Griebel, V; Krägeloh-Mann, I; Ruitenbeek, W; Trijbels, JM; Paulus, W. A mitochondrial myopathy in an infant with lactic acidosis. Dev. Med. Child Neurol 1990, 32, 528–531. [Google Scholar]
- Antozzi, C; Garavaglia, B; Mora, M; Rimoldi, M; Morandi, L; Ursino, E; DiDonato, S. Late-onset riboflavin-responsive myopathy with combined multiple acyl coenzyme A dehydrogenase and respiratory chain deficiency. Neurology 1994, 44, 2153–2158. [Google Scholar]
- Ogle, RF; Christodoulou, J; Fagan, E; Blok, RB; Kirby, DM; Seller, KL; Dahl, HH; Thorburn, DR. Mitochondrial myopathy with tRNA(Leu(UUR)) mutation and complex I deficiency responsive to riboflavin. J. Pediatr 1997, 130, 138–145. [Google Scholar]
- Kerr, DS. Treatment of mitochondrial electron transport chain disorders: a review of clinical trials over the past decade. Mol. Genet. Metab 2010, 99, 246–255. [Google Scholar]
- Jia, H; Liu, Z; Li, X; Feng, Z; Hao, J; Li, X; Shen, W; Zhang, H; Liu, J. Synergistic anti-Parkinsonism activity of high doses of B vitamins in a chronic cellular model. Neurobiol Aging 2010, 31, 636–646. [Google Scholar]
- Brownell, AL; Jenkins, BG; Isacson, O. Dopamine imaging markers and predictive mathematical models for progressive degeneration in Parkinson’s disease. Biomed. Pharmacother 1999, 53, 131–140. [Google Scholar]
- Matthews, RT; Ferrante, RJ; Klivenyi, P; Yang, L; Klein, AM; Mueller, G; Kaddurah- Daouk, R; Beal, MF. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp. Neurol 1999, 157, 142–149. [Google Scholar]
- McCarty, MF. The therapeutic potential of glucose tolerance factor. Med Hypotheses 1980, 6, 1177–1189. [Google Scholar]
- McCarty, MF. High-dose biotin, an inducer of glucokinase expression, may synergize with chromium picolinate to enable a definitive nutritional therapy for type II diabetes. Med Hypotheses 1999, 52, 401–406. [Google Scholar]
- Aguilar, MV; Jiménez-Jiménez, FJ; Molina, JA; Meseguer, I; Mateos-Vega, CJ; González- Muñoz, MJ; de Bustos, F; Gómez-Escalonilla, C; Ort-Pareja, M; Zurdo, M; Martínez-Para, MC. Cerebrospinal fluid selenium and chromium levels in patients with Parkinson’s disease. J. Neural Transm 1998, 105, 1245–1251. [Google Scholar]
- Greggio, E; Bergantino, E; Carter, D; Ahmad, R; Costin, GE; Hearing, VJ; Clarimon, J; Singleton, A; Eerola, J; Hellström, O; Tienari, PJ; Miller, DW; Beilina, A; Bubacco, L; Cookson, MR. Tyrosinase exacerbates dopamine toxicity but is not genetically associated with Parkinson’s disease. J. Neurochem 2005, 93, 246–256. [Google Scholar]
- Boissy, RE; Visscher, M; DeLong, MA. DeoxyArbutin: a novel reversible tyrosinase inhibitor with effective in vivo skin lightening potency. Exp. Dermatol 2005, 14, 601–608. [Google Scholar]
- Galeazzi, MA. Behavior of polyphenoloxidases in food. Arch. Lat. Nutr 1984, 34, 269–289. [Google Scholar]
- Matheis, G; Belitz, HD. Studies on enzymic browning of potatoes (Solanum tuberosum). III. Kinetics of potato phenoloxidase (EC 1.14.18.1 monophenol, dihydroxyphenylalanine: oxygenoxidoreductase). Z. Lebensm. Unters. Forsch 1977, 163, 191–195. [Google Scholar]
- Henderson, HM; Eskin, NA; Pinsky, C; Bose, R; Ashique, AM. Pyridine and other coal tar constituents as inhibitors of potato polyphenol oxidase: a non-animal model for neurochemical studies. Life Sci 1992, 51, PL207–210. [Google Scholar]
- Khatib, S; Nerya, O; Musa, R; Shmuel, M; Tamir, S; Vaya, J. Chalcones as potent tyrosinase inhibitors: the importance of a 2,4-substituted resorcinol moiety. Bioorg. Med. Chem 2005, 13, 433–441. [Google Scholar]
- Nerya, O; Musa, R; Khatib, S; Tamir, S; Vaya, J. Chalcones as potent tyrosinase inhibitors: the effect of hydroxyl positions and numbers. Phytochemistry 2004, 65, 1389–1395. [Google Scholar]
- Lee, NK; Son, KH; Chang, HW; Kang, SS; Park, H; Heo, MY; Kim, HP. Prenylated flavonoids as tyrosinase inhibitors. Arch. Pharm. Res 2004, 27, 1132–1135. [Google Scholar]
- Kim, SJ; Son, KH; Chang, HW; Kang, SS; Kim, HP. Tyrosinase inhibitory prenylated flavonoids from Sophora flavescens. Biol. Pharm Bullutin 2003, 26, 1348–1350. [Google Scholar]
- Son, JK; Park, JS; Kim, JA; Kim, Y; Chung, SR; Lee, SH. Prenylated flavonoids from the roots of Sophora flavescens with tyrosinase inhibitory activity. Planta Med 2003, 69, 559–561. [Google Scholar]
- Tan, C; Zhu, W; Lu, Y. Aloin, cinnamic acid and sophorcarpidine are potent inhibitors of tyrosinase. Chin. Med. J. (Engl ) 2002, 115, 1859–1862. [Google Scholar]
- Shi, Y; Chen, Q-X; Wang, Q; Song, KK; Qiu, L. Inhibitory effects of cinnamic acid and its derivatives on the diphenolase activity of mushroom (Agaricus bisporus) tyrosinase. Food Chem 2005, 92, 707–712. [Google Scholar]
- Nerya, O; Vaya, J; Musa, R; Izrael, S; Ben-Arie, R; Tamir, S. Glabrene and isoliquiritigenin as tyrosinase inhibitors from licorice roots. J. Agric. Food Chem 2003, 51, 1201–1207. [Google Scholar]
- Fu, B; Li, H; Wang, X; Lee, FS; Cui, S. Isolation and identification of flavonoids in licorice and a study of their inhibitory effects on tyrosinase. J. Agric. Food Chem 2005, 53, 7408–7414. [Google Scholar]
- Xie, LP; Chen, QX; Huang, H; Wang, HZ; Zhang, RQ. Inhibitory effects of some flavonoids on the activity of mushroom tyrosinase. Biochemistry 2003, 68, 487–491. [Google Scholar]
- Masamoto, Y; Murata, Y; Baba, K; Shimoishi, Y; Tada, M; Takahata, K. Inhibitory effects of esculetin on melanin biosynthesis. Biol. Pharm Bullutin 2004, 27, 422–425. [Google Scholar]
- Chen, QX; Ke, LN; Song, KK; Huang, H; Liu, XD. Inhibitory effects of hexylresorcinol and dodecylresorcinol on mushroom (Agaricus bisporus) tyrosinase. Protein J 2004, 23, 135–141. [Google Scholar]
- Shin, NH; Ryu, SY; Choi, EJ; Kang, SH; Chang, IM; Min, KR; Kim, Y. Oxyresveratrol as the potent inhibitor on dopa oxidase activity of mushroom tyrosinase. Biochem. Biophys. Res. Commun 1998, 243, 801–803. [Google Scholar]
- Ohguchi, K; Tanaka, T; Iliya, I; Ito, T; Iinuma, M; Matsumoto, K; Akao, Y; Nozawa, Y. Gnetol as a potent tyrosinase inhibitor from genus Gnetum. Biosci. Biotechnol. Biochem 2003, 67, 663–665. [Google Scholar]
- Kim, DS; Park, SH; Kwon, SB; Li, K; Youn, SW; Park, KC. (−)-Epigallocatechin-3- gallate and hinokitiol reduce melanin synthesis via decreased MITF production. Arch. Pharm. Res 2004, 27, 334–339. [Google Scholar]
- No, JK; Soung, DY; Kim, YJ; Shim, KH; Jun, YS; Rhee, SH; Yokozawa, T; Chung, HY. Inhibition of tyrosinase by green tea components. Life Sci 1999, 65, PL241–246. [Google Scholar]
- Zocca, F; Lomolino, G; Lante, A. Antibrowning potential of Brassicacaea processing water. Bioresour. Technol 2010, 101, 3791–3795. [Google Scholar]
- Negishi, O; Ozawa, T. Inhibition of enzymatic browning and protection of sulfhydryl enzymes by thiol compounds. Phytochemistry 2000, 54, 481–487. [Google Scholar]
- Nagai, T; Suzuki, N. Partial purification of polyphenol oxidase from Chinese cabbage Brassica rapa L. J. Agric. Food Chem 2001, 49, 3922–3926. [Google Scholar]
- Yang, CP; Fujita, S; Kohno, K; Kusubayashi, A; Ashrafuzzaman, M; Hayashi, N. Partial purification and characterization of polyphenol oxidase from banana (Musa sapientum L.) peel. J. Agric. Food Chem 2001, 49, 1446–1449. [Google Scholar]
- Pérez-Gilabert, M; García-Carmona, F. Dimethyl sulfide, a volatile flavor constituent, is a slow-binding inhibitor of tyrosinase. Biochem. Biophys. Res. Commun 2001, 285, 257–261. [Google Scholar]
- Graf, E; Empson, KL; Eaton, JW. Phytic acid. A natural antioxidant. J. Biol. Chem 1987, 262, 11647–11650. [Google Scholar]
- Kubo, I; Kinst-Hori, I; Nihei, K; Soria, F; Takasaki, M; Calderón, JS; Céspedes, CL. Tyrosinase inhibitors from galls of Rhus javanica leaves and their effects on insects. Z. Naturforsch C 2003, 58, 719–725. [Google Scholar]
- Sasaki, K; Yoshizaki, F. Nobiletin as a tyrosinase inhibitor from the peel of Citrus fruit. Biol. Pharm Bullutin 2002, 25, 806–808. [Google Scholar]
- Kubo, I; Kinst-Hori, I. Flavonols from saffron flower: tyrosinase inhibitory activity and inhibition mechanism. J. Agric. Food Chem 1999, 47, 4121–4125. [Google Scholar]
- Kubo, I; Kinst-Hori, I; Chaudhuri, SK; Kubo, Y; Sánchez, Y; Ogura, T. Flavonols from Heterotheca inuloides: tyrosinase inhibitory activity and structural criteria. Bioorg. Med. Chem 2000, 8, 1749–1755. [Google Scholar]
- Masuda, T; Yamashita, D; Takeda, Y; Yonemori, S. Screening for tyrosinase inhibitors among extracts of seashore plants and identification of potent inhibitors from Garcinia subelliptica. Biosci. Biotechnol. Biochem 2005, 69, 197–201. [Google Scholar]
- Gómez-Cordovés, C; Bartolomé, B; Vieira, W; Virador, VM. Effects of wine phenolics and sorghum tannins on tyrosinase activity and growth of melanoma cells. J. Agric. Food Chem 2001, 49, 1620–1624. [Google Scholar]
- An, BJ; Kwak, JH; Son, JH; Park, JM; Lee, JY; Park, TS; Kim, SY; Kim, YS; Jo, C; Byun, MW. Physiological activity of irradiated green tea polyphenol on the human skin. Am. J. Chin. Med 2005, 33, 535–546. [Google Scholar]
- Shoji, T; Masumoto, S; Moriichi, N; Kobori, M; Kanda, T; Shinmoto, H; Tsushida, T. Procyanidin trimers to pentamers fractionated from apple inhibit melanogenesis in B16 mouse melanoma cells. J. Agric. Food Chem 2005, 53, 6105–6111. [Google Scholar]
- Yamakoshi, J; Otsuka, F; Sano, A; Tokutake, S; Saito, M; Kikuchi, M; Kubota, Y. Lightening effect on ultraviolet-induced pigmentation of guinea pig skin by oral administration of a proanthocyanidin-rich extract from grape seeds. Pigment Cell Res 2003, 16, 629–638. [Google Scholar]
- Kubo, I; Kinst-Hori, I; Kubo, Y; Yamagiwa, Y; Kamikawa, T; Haraguchi, H. Molecular design of antibrowning agents. J. Agric. Food Chem 2000, 48, 1393–1399. [Google Scholar]
- Roh, JS; Han, JY; Kim, JH; Hwang, JK. Inhibitory effects of active compounds isolated from safflower (Carthamus tinctorius L.) seeds for melanogenesis. Biol. Pharm Bullutin 2004, 27, 1976–1978. [Google Scholar]
- Kubo, I; Chen, QX; Nihei, K; Calderón, JS; Céspedes, CL. Tyrosinase inhibition kinetics of anisic acid. Z. Naturforsch C 2003, 58, 713–718. [Google Scholar]
- Kubo, I; Kinst-Hori, I. Tyrosinase inhibitory activity of the olive oil flavor compounds. J. Agric. Food Chem 1999, 47, 4574–4578. [Google Scholar]
- Kim, HP; Son, KH; Chang, HW; Kang, SS. Anti-inflammatory plant flavonoids and cellular action mechanisms. J. Pharmacol. Sci 2004, 96, 229–245. [Google Scholar]
- Francis, JA; Rumbeiha, W; Nair, MG. Constituents in Easter lily flowers with medicinal activity. Life Sci 2004, 76, 671–683. [Google Scholar]
- Hou, DX; Fujii, M; Terahara, N; Yoshimoto, M. Molecular Mechanisms Behind the Chemopreventive Effects of Anthocyanidins. J. Biomed. Biotechnol 2004, 5, 321–325. [Google Scholar]
- Seeram, NP; Zhang, Y; Nair, MG. Inhibition of proliferation of human cancer cells and cyclooxygenase enzymes by anthocyanidins and catechins. Nutr Cancer 2003, 46, 101–106. [Google Scholar]
- Woo, KJ; Jeong, YJ; Inoue, H; Park, JW; Kwon, TK. Chrysin suppresses lipopolysaccharide-induced cyclooxygenase-2 expression through the inhibition of nuclear factor for IL-6 (NF-IL6) DNA-binding activity. FEBS Lett 2005, 579, 705–711. [Google Scholar]
- Liang, YC; Huang, YT; Tsai, SH; Lin-Shiau, SY; Chen, CF; Lin, JK. Suppression of inducible cyclooxygenase and inducible nitric oxide synthase by apigenin and related flavonoids in mouse macrophages. Carcinogenesis 1999, 20, 1945–1952. [Google Scholar]
- Raso, GM; Meli, R; Di Carlo, G; Pacilio, M; Di Carlo, R. Inhibition of inducible nitric oxide synthase and cyclooxygenase-2 expression by flavonoids in macrophage J774A.1. Life Sci 2001, 68, 921–931. [Google Scholar]
- Chi, YS; Cheon, BS; Kim, HP. Effect of wogonin, a plant flavone from Scutellaria radix, on the suppression of cyclooxygenase-2 and the induction of inducible nitric oxide synthase in lipopolysaccharide-treated RAW 264.7 cells. Biochem. Pharmacol 2001, 61, 1195–1203. [Google Scholar]
- O’Leary, KA; de Pascual-Tereasa, S; Needs, PW; Bao, YP; O’Brien, NM; Williamson, G. Effect of flavonoids and vitamin E on cyclooxygenase-2 (COX-2) transcription. Mutat. Res 2004, 551, 245–254. [Google Scholar]
- Mutoh, M; Takahashi, M; Fukuda, K; Komatsu, H; Enya, T; Matsushima-Hibiya, Y; Mutoh, H; Sugimura, T; Wakabayashi, K. Suppression by flavonoids of cyclooxygenase-2 promoterdependent transcriptional activity in colon cancer cells: structure-activity relationship. Jpn. J. Cancer Res 2000, 91, 686–691. [Google Scholar]
- Huss, U; Ringbom, T; Perera, P; Bohlin, L; Vasänge, M. Screening of ubiquitous plant constituents for COX-2 inhibition with a scintillation proximity based assay. J. Nat. Prod 2002, 65, 1517–1521. [Google Scholar]
- Seaver, B; Smith, JR. Inhibition of COX isoforms by nutraceuticals. J. Herb. Pharmacother 2004, 4, 11–18. [Google Scholar]
- Murias, M; Handler, N; Erker, T; Pleban, K; Ecker, G; Saiko, P; Szekeres, T; Jäger, W. Resveratrol analogues as selective cyclooxygenase-2 inhibitors: synthesis and structure-activity relationship. Bioorg. Med. Chem 2004, 12, 5571–5578. [Google Scholar]
- Chi, YS; Jong, HG; Son, KH; Chang, HW; Kang, SS; Kim, HP. Effects of naturally occurring prenylated flavonoids on enzymes metabolizing arachidonic acid: cyclooxygenases and lipoxygenases. Biochem. Pharmacol 2001, 62, 1185–1191. [Google Scholar]
- Selvam, C; Jachak, SM; Bhutani, KK. Cyclooxygenase inhibitory flavonoids from the stem bark of Semecarpus anacardium Linn. Phytother. Res 2004, 18, 582–584. [Google Scholar]
- Yoo, SW; Kim, JS; Kang, SS; Son, KH; Chang, HW; Kim, HP; Bae, K; Lee, CO. Constituents of the fruits and leaves of Euodia daniellii. Arch. Pharm. Res 2002, 25, 824–830. [Google Scholar]
- Kim, HP; Mani, I; Iversen, L; Ziboh, VA. Effects of naturally-occurring flavonoids and biflavonoids on epidermal cyclooxygenase and lipoxygenase from guinea-pigs. Prostaglandins Leukot. Essent Fatty Acids 1998, 58, 17–24. [Google Scholar]
- Chen, Y; Yang, L; Lee, TJ. Oroxylin A inhibition of lipopolysaccharide-induced iNOS and COX-2 gene expression via suppression of nuclear factor-kappaB activation. Biochem. Pharmacol 2000, 59, 1445–1457. [Google Scholar]
- Rossi, A; Ligresti, A; Longo, R; Russo, A; Borrelli, F; Sautebin, L. The inhibitory effect of propolis and caffeic acid phenethyl ester on cyclooxygenase activity in J774 macrophages. Phytomedicine 2002, 9, 530–535. [Google Scholar] [Green Version]
- You, KM; Jong, HG; Kim, HP. Inhibition of cyclooxygenase/lipoxygenase from human platelets by polyhydroxylated/methoxylated flavonoids isolated from medicinal plants. Arch. Pharm. Res 1999, 22, 18–24. [Google Scholar]
- Prasad, NS; Raghavendra, R; Lokesh, BR; Naidu, KA. Spice phenolics inhibit human PMNL 5-lipoxygenase. Prostaglandins Leukot. Essent Fatty Acids 2004, 70, 521–528. [Google Scholar]
- O’Prey, J; Brown, J; Fleming, J; Harrison, PR. Effects of dietary flavonoids on major signal transduction pathways in human epithelial cells. Biochem. Pharmacol 2003, 66, 2075–2088. [Google Scholar]
- Hsieh, RJ; German, JB; Kinsella, JE. Relative inhibitory potencies of flavonoids on 12-lipoxygenase of fish gill. Lipids 1988, 23, 322–326. [Google Scholar]
- Sekiya, K; Okuda, H; Arichi, S. Selective inhibition of platelet lipoxygenase by esculetin. Biochim. Biophys Acta 1982, 713, 68–72. [Google Scholar]
- Sadik, CD; Sies, H; Schewe, T. Inhibition of 15-lipoxygenases by flavonoids: structure-activity relations and mode of action. Biochem. Pharmacol 2003, 65, 773–781. [Google Scholar]
- Nakadate, T; Yamamoto, S; Aizu, E; Kato, R. Effects of flavonoids and antioxidants on 12-Otetradecanoyl- phorbol-13-acetate-caused epidermal ornithine decarboxylase induction and tumor promotion in relation to lipoxygenase inhibition by these compounds. Gann 1984, 75, 214–222. [Google Scholar]
- Fylaktakidou, KC; Hadjipavlou-Litina, DJ; Litinas, KE; Nicolaides, DN. Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr. Pharm. Des 2004, 10, 3813–3833. [Google Scholar]
- Malterud, KE; Rydland, KM. Inhibitors of 15-lipoxygenase from orange peel. J. Agric. Food Chem 2000, 48, 5576–5580. [Google Scholar]
- Rui, YC. Advances in pharmacological studies of silymarin. Mem. Inst Oswaldo Cruz 1991, 86, 79–85. [Google Scholar]
- Robak, J; Duniec, Z; Rzadkowska-Bodalska, H; Olechnowicz-Stepień, W; Cisowski, W. The effect of some flavonoids on non-enzymatic lipid oxidation and enzymatic oxidation of arachidonic acid. Pol. J. Pharmacol. Pharm 1986, 38, 483–491. [Google Scholar]
- Ferrándiz, ML; Nair, AG; Alcaraz, MJ. Inhibition of sheep platelet arachidonate metabolism by flavonoids from Spanish and Indian medicinal herbs. Pharmazie 1990, 45, 206–208. [Google Scholar]
- Yoshimoto, T; Furukawa, M; Yamamoto, S; Horie, T; Watanabe-Kohno, S. Flavonoids: potent inhibitors of arachidonate 5-lipoxygenase. Biochem. Biophys. Res. Commun 1983, 116, 612–618. [Google Scholar]
- Schewe, T; Kühn, H; Sies, H. Flavonoids of cocoa inhibit recombinant human 5-lipoxygenase. J. Nutr 2002, 132, 1825–1829. [Google Scholar]
- Oomah, BD; Corbé, A; Balasubramanian, P. Antioxidant and anti-inflammatory activities of bean (Phaseolus vulgaris L.) hulls. J. Agric. Food. Chem 2010, 58, 8225–8230. [Google Scholar]
- Abad, MJ; Bermejo, P; Villar, A. The activity of flavonoids extracted from Tanacetum microphyllum DC. (Compositae) on soybean lipoxygenase and prostaglandin synthetase. Gen. Pharmacol 1995, 26, 815–819. [Google Scholar]
- Mattammal, MB; Strong, R; Lakshmi, VM; Chung, HD; Stephenson, AH. Prostaglandin H synthetase-mediated metabolism of dopamine: implication for Parkinson’s disease. J. Neurochem 1995, 64, 1645–1654. [Google Scholar]
- Zhou, LE; Wang, WJ; Bai, JY; Cheng, GF. Effects of ginkgolide B on arachidonic acid metabolizing enzymes and level of intracellular calcium in rat polymorphonuclear leukocytes. Yao Xue Xue Bao 2001, 36, 92–95. [Google Scholar]
- Kim, HR; Pham, HT; Ziboh, VA. Flavonoids differentially inhibit guinea pig epidermal cytosolic phospholipase A2. Prostaglandins Leukot. Essent Fatty Acids 2001, 65, 281–286. [Google Scholar]
- Grataroli, R; Léonardi, J; Charbonnier, M; Lafont, R; Lafont, H; Nalbone, G. Effects of dietary corn oil and salmon oil on lipids and prostaglandin E2 in rat gastric mucosa. Lipids 1988, 23, 666–670. [Google Scholar]
- Grataroli, R; Vamecq, J; Poupaert, JH; Léonardi, J; Termine, E; Lafont, H; Nalbone, G. Effects of dietary n−6/n−3 ratios on lipid and prostaglandin E2 metabolism in rat gastric mucosa. J. Lipid Mediat 1992, 5, 227–236. [Google Scholar]
- Han, CK; Son, MJ; Chang, HW; Chi, YS; Park, H; Kim, HP. Inhibition of prostaglandin production by a structurally-optimized flavonoid derivative, 2′,4′,7-trimethoxyflavone and cellular action mechanism. Biol. Pharm Bullutin 2005, 28, 1366–1370. [Google Scholar]
- Tanaka, S; Sato, T; Akimoto, N; Yano, M; Ito, A. Prevention of UVB-induced photoinflammation and photoaging by a polymethoxy flavonoid, nobiletin, in human keratinocytes in vivo and in vitro. Biochem. Pharmacol 2004, 68, 433–439. [Google Scholar]
- Ticli, FK; Hage, LI; Cambraia, RS; Pereira, PS; Magro, AJ; Fontes, MR; Stábeli, RG; Giglio, JR; França, SC; Soares, AM; Sampaio, SV. Rosmarinic acid, a new snake venom phospholipase A2 inhibitor from Cordia verbenacea (Boraginaceae): antiserum action potentiation and molecular interaction. Toxicon 2005, 46, 318–327. [Google Scholar]
- Adam, O. Dietary fatty acids and immune reactions in synovial tissue. Eur. J. Med. Res 2003, 8, 381–387. [Google Scholar]
- Shao, ZH; Vanden Hoek, TL; Li, CQ; Schumacker, PT; Becker, LB; Chan, KC; Qin, Y; Yin, JJ; Yuan, CS. Synergistic effect of Scutellaria baicalensis and grape seed proanthocyanidins on scavenging reactive oxygen species in vitro. Am. J. Chin. Med 2004, 32, 89–95. [Google Scholar]
- Dew, TP; Day, AJ; Morgan, MR. Xanthine oxidase activity in vitro: effects of food extracts and components. J. Agric. Food Chem 2005, 53, 6510–6515. [Google Scholar]
- Lin, JK; Chen, PC; Ho, CT; Lin-Shiau, SY. Inhibition of xanthine oxidase and suppression of intracellular reactive oxygen species in HL-60 cells by theaflavin-3,3′-digallate, (−)-epigallocatechin-3-gallate, and propyl gallate. J. Agric. Food Chem 2000, 48, 2736–2743. [Google Scholar]
- Kurisawa, M; Chung, JE; Uyama, H; Kobayashi, S. Oxidative coupling of epigallocatechin gallate amplifies antioxidant activity and inhibits xanthine oxidase activity. Chem. Commun 2004, 7, 294–295. [Google Scholar]
- van Hoorn, DE; Nijveldt, RJ; van Leeuwen, PA; Hofman, Z; M’Rabet, L; de Bont, DB; van Norren, K. Accurate prediction of xanthine oxidase inhibition based on the structure of flavonoids. Eur. J. Pharmacol 2002, 451, 111–118. [Google Scholar]
- Selloum, L; Reichl, S; Müller, M; Sebihi, L; Arnhold, J. Effects of flavonols on the generation of superoxide anion radicals by xanthine oxidase and stimulated neutrophils. Arch. Biochem. Biophys 2001, 395, 49–56. [Google Scholar]
- Nagao, A; Seki, M; Kobayashi, H. Inhibition of xanthine oxidase by flavonoids. Biosci. Biotechnol. Biochem 1999, 63, 1787–1790. [Google Scholar]
- Iio, M; Ono, Y; Kai, S; Fukumoto, M. Effects of flavonoids on xanthine oxidation as well as on cytochrome c reduction by milk xanthine oxidase. J. Nutr. Sci. Vitaminol 1986, 32, 635–642. [Google Scholar]
- Zhu, JX; Wang, Y; Kong, LD; Yang, C; Zhang, X. Effects of Biota orientalis extract and its flavonoid constituents, quercetin and rutin on serum uric acid levels in oxonate-induced mice and xanthine dehydrogenase and xanthine oxidase activities in mouse liver. J. Ethnopharmacol 2004, 93, 133–140. [Google Scholar]
- Moridani, MY; Pourahmad, J; Bui, H; Siraki, A; O’Brien, PJ. Dietary flavonoid iron complexes as cytoprotective superoxide radical scavengers. Free Radic. Biol. Med 2003, 34, 243–253. [Google Scholar]
- Foppoli, C; Coccia, R; Cini, C; Rosei, MA. Catecholamines oxidation by xanthine oxidase. Biochim. Biophys Acta 1997, 1334, 200–206. [Google Scholar]
- Day, AJ; Bao, Y; Morgan, MR; Williamson, G. Conjugation position of quercetin glucuronides and effect on biological activity. Free Radic. Biol. Med 2000, 29, 1234–1243. [Google Scholar]
- Lin, CM; Chen, CS; Chen, CT; Liang, YC; Lin, JK. Molecular modeling of flavonoids that inhibits xanthine oxidase. Biochem. Biophys. Res. Commun 2002, 294, 167–172. [Google Scholar]
- Beiler, JM; Martin, GJ. The inhibition of xanthine oxidase by flavonoids and related compounds. J. Biol. Chem 1951, 192, 831–834. [Google Scholar]
- Yoshizumi, K; Nishioka, N; Tsuji, T. Xanthine oxidase inhibitory activity and hypouricemia effect of propolis in rats. Yakugaku Zasshi 2005, 125, 315–321. [Google Scholar]
- Russo, A; Longo, R; Vanella, A. Antioxidant activity of propolis: role of caffeic acid phenethyl ester and galangin. Fitoterapia 2002, 73, S21–S29. [Google Scholar]
- Tapia, A; Rodriguez, J; Theoduloz, C; Lopez, S; Feresin, GE; Schmeda-Hirschmann, G. Free radical scavengers and antioxidants from Baccharis grisebachii. J. Ethnopharmacol 2004, 95, 155–161. [Google Scholar]
- Huang, Y; Tsang, SY; Yao, X; Chen, ZY. Biological properties of baicalein in cardiovascular system. Curr. Drug Targets Cardiovasc. Haematol. Disord 2005, 5, 177–184. [Google Scholar]
- Shieh, DE; Liu, LT; Lin, CC. Antioxidant and free radical scavenging effects of baicalein, baicalin and wogonin. Anticancer Res 2000, 20, 2861–2865. [Google Scholar]
- Chang, WS; Lee, YJ; Lu, FJ; Chiang, HC. Inhibitory effects of flavonoids on xanthine oxidase. Anticancer Res 1993, 13, 2165–2170. [Google Scholar]
- Varga, Z; Ujhelyi, L; Kiss, A; Balla, J; Czompa, A; Antus, S. Effect of silybin on phorbol myristate actetate-induced protein kinase C translocation NADPH oxidase activity and apoptosis in human neutrophils. Phytomedicine 2004, 11, 206–212. [Google Scholar]
- Sheu, SY; Lai, CH; Chiang, HC. Inhibition of xanthine oxidase by purpurogallin and silymarin group. Anticancer Res 1998, 18, 263–267. [Google Scholar]
- Łuczaj, W; Skrzydlewska, E. Antioxidative properties of black tea. Prev. Med 2005, 40, 910–918. [Google Scholar]
- Wang, Y; Zhu, JX; Kong, LD; Yang, C; Cheng, CH; Zhang, X. Administration of procyanidins from grape seeds reduces serum uric acid levels and decreases hepatic xanthine dehydrogenase/oxidase activities in oxonate-treated mice. Basic Clin. Pharmacol. Toxicol 2004, 94, 232–237. [Google Scholar]
- Packer, L; Rimbach, G; Virgili, F. Antioxidant activity and biologic properties of a procyanidin-rich extract from pine (Pinus maritima) bark, pycnogenol. Free Radic. Biol. Med 1999, 27, 704–724. [Google Scholar]
- Moini, H; Guo, Q; Packer, L. Enzyme inhibition and protein-binding action of the procyanidinrich french maritime pine bark extract, pycnogenol: effect on xanthine oxidase. J. Agric. Food Chem 2000, 48, 5630–5639. [Google Scholar]
- Moini, H; Guo, Q; Packe, L. Xanthine oxidase and xanthine dehydrogenase inhibition by the procyanidin-rich French maritime pine bark extract, pycnogenol: a protein binding effect. Adv. Exp. Med. Biol 2002, 505, 141–149. [Google Scholar]
- Acquaviva, R; Russo, A; Galvano, F; Galvano, G; Barcellona, ML; Li Volti, G; Vanella, A. Cyanidin and cyanidin 3-O-beta-d-glucoside as DNA cleavage protectors and antioxidants. Cell Biol. Toxicol 2003, 19, 243–252. [Google Scholar]
- Cioffi, G; D’Auria, M; Braca, A; Mendez, J; Castillo, A; Morelli, I; de Simone, F; de Tommasi, N. Antioxidant and free-radical scavenging activity of constituents of the leaves of Tachigalia paniculata. J. Nat. Prod 2002, 65, 1526–1529. [Google Scholar]
- Robak, J; Gryglewski, RJ. Flavonoids are scavengers of superoxide anions. Biochem. Pharmacol 1988, 37, 837–841. [Google Scholar]
- Ignatov, S; Shishniashvili, D; Ge, B; Scheller, FW; Lisdat, F. Amperometric biosensor based on a functionalized gold electrode for the detection of antioxidants. Biosens. Bioelectron 2002, 17, 191–199. [Google Scholar]
- Marfak, A; Trouillas, P; Allais, DP; Champavier, Y; Calliste, CA; Duroux, JL. Radiolysis of kaempferol in water/methanol mixtures. Evaluation of antioxidant activity of kaempferol and products formed. J. Agric. Food Chem 2003, 51, 1270–1277. [Google Scholar]
- Lu, Y; Foo, LY. Antioxidant activities of polyphenols from sage (Salvia officinalis). Food Chem 2001, 75, 197–202. [Google Scholar]
- Beyer, G; Melzig, MF. Effects of selected flavonoids and caffeic acid derivatives on hypoxanthine-xanthine oxidase-induced toxicity in cultivated human cells. Planta Med 2003, 69, 1125–1129. [Google Scholar]
- Park, YH; Han, DW; Suh, H; Ryu, GH; Hyon, SH; Cho, BK; Park, JC. Protective effects of green tea polyphenol against reactive oxygen species-induced oxidative stress in cultured rat calvarial osteoblast. Cell Biol. Toxicol 2003, 19, 325–337. [Google Scholar]
- Rah, DK; Han, DW; Baek, HS; Hyon, SH; Park, JC. Prevention of reactive oxygen species-induced oxidative stress in human microvascular endothelial cells by green tea polyphenol. Toxicol. Lett 2005, 155, 269–275. [Google Scholar]
- Liu, H; Yang, XL; Wang, Y; Tang, XQ; Jiang, DY; Xu, HB. Protective effects of scutellarin on superoxide-induced oxidative stress in rat cortical synaptosomes. Acta Pharmacol. Sin 2003, 24, 1113–1117. [Google Scholar]
- Taubert, D; Breitenbach, T; Lazar, A; Censarek, P; Harlfinger, S; Berkels, R; Klaus, W; Roesen, R. Reaction rate constants of superoxide scavenging by plant antioxidants. Free Radic. Biol. Med 2003, 35, 1599–1607. [Google Scholar]
- Cheel, J; Theoduloz, C; Rodríguez, J; Schmeda-Hirschmann, G. Free radical scavengers and antioxidants from Lemongrass (Cymbopogon citratus (DC.) Stapf.). J. Agric Food Chem 2005, 53, 2511–2517. [Google Scholar]
- Moridani, MY; O’Brien, PJ. Iron complexes of deferiprone and dietary plant catechols as cytoprotective superoxide radical scavengers. Biochem. Pharmacol 2001, 62, 1579–1585. [Google Scholar]
- Shi, H; Zhao, B; Xin, W. Scavenging effects of baicalin on free radicals and its protection on erythrocyte membrane from free radical injury. Biochem. Mol. Biol. Int 1995, 35, 981–994. [Google Scholar]
- Toyo’oka, T; Kashiwazaki, T; Kato, M. On-line screening methods for antioxidants scavenging superoxide anion radical and hydrogen peroxide by liquid chromatography with indirect chemiluminescence detection. Talanta 2003, 60, 467–475. [Google Scholar]
- Stinefelt, B; Leonard, SS; Blemings, KP; Shi, X; Klandorf, H. Free radical scavenging, DNA protection, and inhibition of lipid peroxidation mediated by uric acid. Ann. Clin. Lab. Sci 2005, 35, 37–45. [Google Scholar]
- Mishra, B; Priyadarsini, KI; Kumar, MS; Unnikrishnan, MK; Mohan, H. Effect of O-glycosilation on the antioxidant activity and free radical reactions of a plant flavonoid, chrysoeriol. Bioorg. Med. Chem 2003, 11, 2677–2685. [Google Scholar]
- Candan, F. Effect of Rhus coriaria L. (Anacardiaceae) on superoxide radical scavenging and xanthine oxidase activity. J. Enzyme Inhib. Med. Chem 2003, 18, 59–62. [Google Scholar]
- Ozgová, S; Hermánek, J; Gut, I. Different antioxidant effects of polyphenols on lipid peroxidation and hydroxyl radicals in the NADPH-, Fe-ascorbate- and Fe-microsomal systems. Biochem. Pharmacol 2003, 66, 1127–1137. [Google Scholar]
- Wei, IH; Wu, YC; Wen, CY; Shieh, JY. Green tea polyphenol (−)-epigallocatechin gallate attenuates the neuronal NADPH-d/nNOS expression in the nodose ganglion of acute hypoxic rats. Brain Res 2004, 999, 73–80. [Google Scholar]
- Chen, S; Deng, PS; Swiderek, K; Li, M; Chan, SI. Interaction of flavones and their bromoacetyl derivatives with NAD(P)H:quinone acceptor oxidoreductase. Mol. Pharmacol 1995, 47, 419–424. [Google Scholar]
- Liu, XF; Liu, ML; Iyanagi, T; Legesse, K; Lee, TD; Chen, SA. Inhibition of rat liver NAD(P)H:quinone acceptor oxidoreductase (DT-diaphorase) by flavonoids isolated from the Chinese herb scutellariae radix (Huang Qin). Mol. Pharmacol 1990, 37, 911–915. [Google Scholar]
- Tamura, M; Kagawa, S; Tsuruo, Y; Ishimura, K; Morita, K. Effects of flavonoid compounds on the activity of NADPH diaphorase prepared from the mouse brain. Jpn. J. Pharmacol 1994, 65, 371–373. [Google Scholar]
- Terland, O; Flatmark, T; Tangerås, A; Grønberg, M. Dopamine oxidation generates an oxidative stress mediated by dopamine semiquinone and unrelated to reactive oxygen species. J. Mol. Cell Cardiol 1997, 29, 1731–1738. [Google Scholar]
- Mocchegiani, E; Bertoni-Freddari, C; Marcellini, F; Malavolta, M. Brain, aging and neurodegeneration: role of zinc ion availability. Prog. Neurobiol 2005, 75, 367–390. [Google Scholar]
- Kukreja, RC; Loesser, KE; Kearns, AA; Naseem, SA; Hess, ML. Protective effects of histidine during ischemia-reperfusion in isolated perfused rat hearts. Am. J. Physiol 1993, 264, H1370–H1381. [Google Scholar]
- Obata, T; Aomine, M; Yamanaka, Y. Protective effect of histidine on iron (II)-induced hydroxyl radical generation in rat hearts. J. Physiol Paris 1999, 93, 213–218. [Google Scholar]
- Obata, T; Inada, T. Protective effect of histidine on MPP+-induced hydroxyl radical generation in rat striatum. Brain Res 1999, 817, 206–208. [Google Scholar]
- Lundvig, D; Lindersson, E; Jensen, PH. Pathogenic effects of alpha-synuclein aggregation. Brain Res. Mol. Brain Res 2005, 134, 3–17. [Google Scholar]
- O’Dowd, Y; Driss, F; Dang, PM; Elbim, C; Gougerot-Pocidalo, MA; Pasquier, C; El-Benna, J. Antioxidant effect of hydroxytyrosol, a polyphenol from olive oil: scavenging of hydrogen peroxide but not superoxide anion produced by human neutrophils. Biochem. Pharmacol 2004, 68, 2003–2008. [Google Scholar]
- Everse, J; Coates, PW. Role of peroxidases in Parkinson disease: a hypothesis. Free Radic. Biol. Med 2005, 38, 1296–1310. [Google Scholar]
- Thiruchelvam, M; Prokopenko, O; Cory-Slechta, DA; Buckley, B; Mirochnitchenko, O. Overexpression of superoxide dismutase or glutathione peroxidase protects against the paraquat + maneb-induced Parkinson disease phenotype. J. Biol. Chem 2005, 280, 22530–22539. [Google Scholar]
- Fornai, F; Carrì, MT; Ferri, A; Paolucci, E; Prisco, S; Bernardi, G; Rotilio, G; Mercuri, NB. Resistance to striatal dopamine depletion induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice expressing human mutant Cu,Zn superoxide dismutase. Neurosci. Lett 2002, 325, 124–128. [Google Scholar]
- Zhao, Y; Gao, Z; Li, H; Xu, H. Hemin/nitrite/H2O2 induces brain homogenate oxidation and nitration: effects of some flavonoids. Biochim. Biophys Acta 2004, 1675, 105–112. [Google Scholar]
- Klivenyi, P; Andreassen, OA; Ferrante, RJ; Dedeoglu, A; Mueller, G; Lancelot, E; Bogdanov, M; Andersen, JK; Jiang, D; Beal, MF. Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4- phenyl-1,2,5,6-tetrahydropyridine. J. Neurosci 2000, 20, 1–7. [Google Scholar]
- Biswas, SK; McClure, D; Jimenez, LA; Megson, IL; Rahman, I. Curcumin induces glutathione biosynthesis and inhibits NF-kappaB activation and interleukin-8 release in alveolar epithelial cells: mechanism of free radical scavenging activity. Antioxid. Redox Signal 2005, 7, 32–41. [Google Scholar]
- Ferrari, CK. Functional foods, herbs and nutraceuticals: towards biochemical mechanisms of healthy aging. Biogerontology 2004, 5, 275–289. [Google Scholar]
- Mansouri, A; Makris, DP; Kefalas, P. Determination of hydrogen peroxide scavenging activity of cinnamic and benzoic acids employing a highly sensitive peroxyoxalate chemiluminescencebased assay: structure-activity relationships. J. Pharm. Biomed. Anal 2005, 39, 22–26. [Google Scholar]
- Yilmaz, Y; Toledo, RT. Major flavonoids in grape seeds and skins: antioxidant capacity of catechin, epicatechin, and gallic acid. J. Agric. Food Chem 2004, 52, 255–260. [Google Scholar]
- Sroka, Z; Cisowski, W. Hydrogen peroxide scavenging, antioxidant and anti-radical activity of some phenolic acids. Food Chem. Toxicol 2003, 41, 753–758. [Google Scholar]
- Vanzani, P; Rossetto, M; Rigo, A; Vrhovsek, U; Mattivi, F; D’Amato, E; Scarpa, M. Major phytochemicals in apple cultivars: contribution to peroxyl radical trapping efficiency. J. Agric. Food Chem 2005, 53, 3377–3382. [Google Scholar]
- López-Alarcón, C; Lissi, E. Interaction of pyrogallol red with peroxyl radicals. A basis for a simple methodology for the evaluation of antioxidant capabilities. Free Radic. Res 2005, 39, 729–736. [Google Scholar]
- Mazzio, EA; Reams, RR; Soliman, KF. The role of oxidative stress, impaired glycolysis and mitochondrial respiratory redox failure in the cytotoxic effects of 6-hydroxydopamine in vitro. Brain Res 2004, 1004, 29–44. [Google Scholar]
- Kabuto, H; Nishizawa, M; Tada, M; Higashio, C; Shishibori, T; Kohno, M. Zingerone [4-(4-hydroxy-3-methoxyphenyl)-2-butanone] prevents 6-hydroxydopamine-induced dopamine depression in mouse striatum and increases superoxide scavenging activity in serum. Neurochem. Res 2005, 30, 325–332. [Google Scholar]
- Caillet, S; Yu, H; Lessard, S; Lamoureux, G; Ajdukovic, D; Lacroix, M. Fenton reaction applied for screening natural antioxidants. Food Chem 2007, 100, 542–552. [Google Scholar]
- van Acker, SA; van den Berg, DJ; Tromp, MN; Griffioen, DH; van Bennekom, WP; van der Vijgh, WJ; Bast, A. Structural aspects of antioxidant activity of flavonoids. Free Radic. Biol. Med 1996, 20, 331–342. [Google Scholar]
- Brown, JE; Khodr, H; Hider, RC; Rice-Evans, CA. Structural dependence of flavonoid interactions with Cu2+ ions: implications for their antioxidant properties. Biochem. J 1998, 330, 1173–1178. [Google Scholar]
- Arora, A; Nair, MG; Strasburg, GM. Structure-activity relationships for antioxidant activities of a series of flavonoids in a liposomal system. Free Radic. Biol. Med 1998, 24, 1355–1363. [Google Scholar]
- Fernandez, MT; Mira, ML; Florêncio, MH; Jennings, KR. Iron and copper chelation by flavonoids: an electrospray mass spectrometry study. J. Inorg. Biochem 2002, 92, 105–111. [Google Scholar]
- Cheng, IF; Breen, K. On the ability of four flavonoids, baicilein, luteolin, naringenin, and quercetin, to suppress the Fenton reaction of the iron-ATP complex. Biometals 2000, 13, 77–83. [Google Scholar]
- Aherne, SA; O’Brien, NM. Mechanism of protection by the flavonoids, quercetin and rutin, against tert-butylhydroperoxide- and menadione-induced DNA single strand breaks in Caco-2 cells. Free Radic. Biol. Med 2000, 29, 507–514. [Google Scholar]
- Mahakunakorn, P; Tohda, M; Murakami, Y; Matsumoto, K; Watanabe, H. Antioxidant and free radical-scavenging activity of Choto-san and its related constituents. Biol. Pharm Bullutin 2004, 27, 38–46. [Google Scholar]
- Yoshida, H; Ishikawa, T; Hosoai, H; Suzukawa, M; Ayaori, M; Hisada, T; Sawada, S; Yonemura, A; Higashi, K; Ito, T; Nakajima, K; Yamashita, T; Tomiyasu, K; Nishiwaki, M; Ohsuzu, F; Nakamura, H. Inhibitory effect of tea flavonoids on the ability of cells to oxidize low density lipoprotein. Biochem. Pharmacol 1999, 58, 1695–1703. [Google Scholar]
- O’Coinceanainn, M; Bonnely, S; Baderschneider, B; Hynes, MJ. Reaction of iron(III) with theaflavin: complexation and oxidative products. J. Inorg. Biochem 2004, 98, 657–663. [Google Scholar]
- Fraga, CG; Oteiza, PI. Iron toxicity and antioxidant nutrients. Toxicology 2002, 180, 23–32. [Google Scholar]
- Hynes, MJ; Coinceanainn, MO. The kinetics and mechanisms of the reaction of iron(III) with gallic acid, gallic acid methyl ester and catechin. J. Inorg. Biochem 2001, 85, 131–142. [Google Scholar]
- Borsari, M; Gabbi, C; Ghelfi, F; Grandi, R; Saladini, M; Severi, S; Borella, F. Silybin, a new iron-chelating agent. J. Inorg. Biochem 2001, 85, 123–129. [Google Scholar]
- Kostyuk, VA; Potapovich, AI. Antiradical and chelating effects in flavonoid protection against silica-induced cell injury. Arch. Biochem. Biophys 1998, 355, 43–48. [Google Scholar]
- Schipper, HM. Heme oxygenase expression in human central nervous system disorders. Free Radic. Biol. Med 2004, 37, 1995–2011. [Google Scholar]
- Abate, A; Yang, G; Wong, RJ; Schroder, H; Stevenson, DK; Dennery, PA. Apigenin decreases hemin-mediated heme oxygenase-1 induction. Free Radic. Biol. Med 2005, 39, 711–718. [Google Scholar]
- Kantengwa, S; Polla, BS. Flavonoids, but not protein kinase C inhibitors, prevent stress protein synthesis during erythrophagocytosis. Biochem. Biophys. Res. Commun 1991, 180, 308–314. [Google Scholar]
- Zatta, P; Lucchini, R; van Rensburg, SJ; Taylor, A. The role of metals in neurodegenerative processes: aluminum, manganese, and zinc. Brain Res Bullutin 2003, 62, 15–28. [Google Scholar]
- Schweizer, U; Bräuer, AU; Köhrle, J; Nitsch, R; Savaskan, NE. Selenium and brain function: a poorly recognized liaison. Brain Res. Brain Res. Rev 2004, 45, 164–178. [Google Scholar]
- Johnson, S. Is Parkinson’s disease the heterozygote form of Wilson’s disease: PD = 1/2 WD? Med Hypotheses 2001, 56, 171–173. [Google Scholar]
- Sziráki, I; Mohanakumar, KP; Rauhala, P; Kim, HG; Yeh, KJ; Chiueh, CC. Manganese: a transition metal protects nigrostriatal neurons from oxidative stress in the iron-induced animal model of Parkinsonism. Neuroscience 1998, 85, 1101–1111. [Google Scholar]
- Cuajungco, MP; Lees, GJ. Zinc metabolism in the brain: relevance to human neurodegenerative disorders. Neurobiol. Dis 1997, 4, 137–169. [Google Scholar]
- Bush, AI. Metals and neuroscience. Curr. Opin. Chem. Biol 2000, 4, 184–191. [Google Scholar]
- Zago, MP; Mackenzie, GG; Adamo, AM; Keen, CL; Oteiza, PI. Differential modulation of MAP kinases by zinc deficiency in IMR-32 cells: role of H2O2. Antioxid. Redox Signal 2005, 7, 1773–1782. [Google Scholar]
- Blonska, M; Bronikowska, J; Pietsz, G; Czuba, ZP; Scheller, S; Krol, W. Effects of ethanol extract of propolis (EEP) and its flavones on inducible gene expression in J774A.1 macrophages. J. Ethnopharmacol 2004, 91, 25–30. [Google Scholar]
- Cho, H; Yun, CW; Park, WK; Kong, JY; Kim, KS; Park, Y; Lee, S; Kim, BK. Modulation of the activity of pro-inflammatory enzymes, COX-2 and iNOS.; by chrysin derivatives. Pharmacol. Res 2004, 49, 37–43. [Google Scholar]
- Chen, JC; Ho, FM; Pei-Dawn, LC; Chen, CP; Jeng, KC; Hsu, HB; Lee, ST; Wen, TW; Lin, WW. Inhibition of iNOS gene expression by quercetin is mediated by the inhibition of IkappaB kinase, nuclear factor-kappa B and STAT1, and depends on heme oxygenase-1 induction in mouse BV-2 microglia. Eur. J. Pharmacol 2005, 521, 9–20. [Google Scholar]
- Martínez-Flórez, S; Gutiérrez-Fernández, B; Sánchez-Campos, S; González-Gallego, J; Tuñón, MJ. Quercetin attenuates nuclear factor-kappaB activation and nitric oxide production in interleukin-1beta-activated rat hepatocytes. J. Nutr 2005, 135, 1359–1365. [Google Scholar]
- Jung, WJ; Sung, MK. Effects of major dietary antioxidants on inflammatory markers of RAW 264.7 macrophages. Biofactors 2004, 21, 113–117. [Google Scholar]
- Cho, SY; Park, SJ; Kwon, MJ; Jeong, TS; Bok, SH; Choi, WY; Jeong, WI; Ryu, SY; Do, SH; Lee, CS; Song, JC; Jeong, KS. Quercetin suppresses proinflammatory cytokines production through MAP kinases andNF-kappaB pathway in lipopolysaccharide-stimulated macrophage. Mol. Cell Biochem 2003, 243, 153–160. [Google Scholar]
- van Meeteren, ME; Hendriks, JJ; Dijkstra, CD; van Tol, EA. Dietary compounds prevent oxidative damage and nitric oxide production by cells involved in demyelinating disease. Biochem. Pharmacol 2004, 67, 967–975. [Google Scholar]
- Scuro, LS; Simioni, PU; Grabriel, DL; Saviani, EE; Modolo, LV; Tamashiro, WM; Salgado, I. Suppression of nitric oxide production in mouse macrophages by soybean flavonoids accumulated in response to nitroprusside and fungal elicitation. BMC Biochem 2004, 21, 5. [Google Scholar]
- Hu, C; Kitts, DD. Luteolin and luteolin-7-O-glucoside from dandelion flower suppress iNOS and COX-2 in RAW264.7 cells. Mol. Cell Biochem 2004, 265, 107–113. [Google Scholar]
- Kim, SJ; Park, H; Kim, HP. Inhibition of nitric oxide production from lipopolysaccharidetreated RAW 264.7 cells by synthetic flavones: structure-activity relationship and action mechanism. Arch. Pharm. Res 2004, 27, 937–943. [Google Scholar]
- Matsuda, H; Morikawa, T; Ando, S; Toguchida, I; Yoshikawa, M. Structural requirements of flavonoids for nitric oxide production inhibitory activity and mechanism of action. Bioorg. Med. Chem 2003, 11, 1995–2000. [Google Scholar]
- Rojas, J; Payá, M; Devesa, I; Dominguez, JN; Ferrándiz, ML. Therapeutic administration of 3,4,5-trimethoxy-4′-fluorochalcone, a selective inhibitor of iNOS expression, attenuates the development of adjuvant-induced arthritis in rats. Naunyn Schmiedebergs Arch. Pharmacol 2003, 368, 225–233. [Google Scholar]
- Ko, HH; Tsao, LT; Yu, KL; Liu, CT; Wang, JP; Lin, CN. Structure-activity relationship studies on chalcone derivatives. The potent inhibition of chemical mediators release. Bioorg. Med. Chem 2003, 11, 105–111. [Google Scholar]
- Chiu, FL; Lin, JK. HPLC analysis of naturally occurring methylated catechins, 3″- and 4″-methyl-epigallocatechin gallate, in various fresh tea leaves and commercial teas and their potent inhibitory effects on inducible nitric oxide synthase in macrophages. J. Agric. Food Chem 2005, 53, 7035–7042. [Google Scholar]
- Sutherland, BA; Shaw, OM; Clarkson, AN; Jackson, DN; Sammut, IA; Appleton, I. Neuroprotective effects of (−)-epigallocatechin gallate following hypoxia-ischemia-induced brain damage: novel mechanisms of action. FASEB J 2005, 19, 258–260. [Google Scholar]
- Singh, R; Ahmed, S; Islam, N; Goldberg, VM; Haqqi, TM. Epigallocatechin-3-gallate inhibits interleukin-1beta-induced expression of nitric oxide synthase and production of nitric oxide in human chondrocytes: suppression of nuclear factor kappaB activation by degradation of the inhibitor of nuclear factor kappaB. Arthritis Rheum 2002, 46, 2079–2086. [Google Scholar]
- Lee, SH; Seo, GS; Sohn, DH. Inhibition of lipopolysaccharide-induced expression of inducible nitric oxide synthase by butein in RAW 264.7 cells. Biochem. Biophys. Res. Commun 2004, 323, 125–132. [Google Scholar]
- Sautebin, L; Rossi, A; Serraino, I; Dugo, P; Di Paola, R; Mondello, L; Genovese, T; Britti, D; Peli, A; Dugo, G; Caputi, AP; Cuzzocrea, S. Effect of anthocyanins contained in a blackberry extract on the circulatory failure and multiple organ dysfunction caused by endotoxin in the rat. Planta Med 2004, 70, 745–752. [Google Scholar]
- Liu, H; Yang, X; Tang, R; Liu, J; Xu, H. Effect of scutellarin on nitric oxide production in early stages of neuron damage induced by hydrogen peroxide. Pharmacol. Res 2005, 51, 205–210. [Google Scholar]
- Lin, CM; Huang, ST; Liang, YC; Lin, MS; Shih, CM; Chang, YC; Chen, TY; Chen, CT. Isovitexin suppresses lipopolysaccharide-mediated inducible nitric oxide synthase through inhibition of NF-kappa B in mouse macrophages. Planta Med 2005, 71, 748–753. [Google Scholar]
- Sakata, K; Hirose, Y; Qiao, Z; Tanaka, T; Mori, H. Inhibition of inducible isoforms of cyclooxygenase and nitric oxide synthase by flavonoid hesperidin in mouse macrophage cell line. Cancer Lett 2003, 199, 139–145. [Google Scholar]
- Kanno, S; Shouji, A; Tomizawa, A; Hiura, T; Osanai, Y; Ujibe, M; Obara, Y; Nakahata, N; Ishikawa, M. Inhibitory effect of naringin on lipopolysaccharide (LPS)-induced endotoxin shock in mice and nitric oxide production in RAW 264.7 macrophages. Life Sci 2006, 78, 673–681. [Google Scholar]
- Lin, HY; Shen, SC; Chen, YC. Anti-inflammatory effect of heme oxygenase 1: glycosylation and nitric oxide inhibition in macrophages. J. Cell Physiol 2005, 202, 579–590. [Google Scholar]
- Chen, CJ; Raung, SL; Liao, SL; Chen, SY. Inhibition of inducible nitric oxide synthase expression by baicalein in endotoxin/cytokine-stimulated microglia. Biochem. Pharmacol 2004, 67, 957–965. [Google Scholar]
- Lin, HY; Juan, SH; Shen, SC; Hsu, FL; Chen, YC. Inhibition of lipopolysaccharideinduced nitric oxide production by flavonoids in RAW264.7 macrophages involves heme oxygenase-1. Biochem. Pharmacol 2003, 66, 1821–1832. [Google Scholar]
- Schümann, J; Prockl, J; Kiemer, AK; Vollmar, AM; Bang, R; Tiegs, G. Silibinin protects mice from T cell-dependent liver injury. J. Hepatol 2003, 39, 333–340. [Google Scholar]
- Kang, JS; Jeon, YJ; Kim, HM; Han, SH; Yang, KH. Inhibition of inducible nitric-oxide synthase expression by silymarin in lipopolysaccharide-stimulated macrophages. J. Pharmacol. Exp. Ther 2002, 302, 138–144. [Google Scholar]
- Banerjee, T; van der Vliet, A; Ziboh, VA. Downregulation of COX-2 and iNOS by amentoflavone and quercetin in A549 human lung adenocarcinoma cell line. Prostaglandins Leukot. Essent Fatty Acids 2002, 66, 485–492. [Google Scholar]
- Takahashi, T; Takasuka, N; Iigo, M; Baba, M; Nishino, H; Tsuda, H; Okuyama, T. Isoliquiritigenin, a flavonoid from licorice, reduces prostaglandin E2 and nitric oxide, causes apoptosis, and suppresses aberrant crypt foci development. Cancer Sci 2004, 95, 448–453. [Google Scholar]
- Lee, H; Kim, YO; Kim, H; Kim, SY; Noh, HS; Kang, SS; Cho, GJ; Choi, WS; Suk, K. lavonoid wogonin from medicinal herb is neuroprotective by inhibiting inflammatory activation of microglia. FASEB J 2003, 17, 1943–1944. [Google Scholar]
- Markovic, M; Miljkovic, Dj; Trajkovic, V. Regulation of inducible nitric oxide synthase by cAMP-elevating phospho-diesterase inhibitors. Curr. Drug Targets Inflamm Allergy 2003, 2, 63–79. [Google Scholar]
- Hulley, P; Hartikka, J; Abdel’Al, S; Engels, P; Buerki, HR; Wiederhold, KH; Müller, T; Kelly, P; Lowe, D; Lübbert, H. Inhibitors of type IV phosphodiesterases reduce the toxicity of MPTP in substantia nigra neurons in vivo. Eur. J. Neurosci 1995, 7, 2431–2440. [Google Scholar]
- Yu, SM; Cheng, ZJ; Kuo, SC. Endothelium-dependent relaxation of rat aorta by butein, a novel cyclic AMP-specific phosphodiesterase inhibitor. Eur. J. Pharmacol 1995, 280, 69–77. [Google Scholar]
- Girotti, C; Ginet, M; Demarne, FC; Lagarde, M; Géloën, A. Lipolytic activity of cirsimarin extracted from Microtea debilis. Planta Med 2005, 71, 1170–1172. [Google Scholar]
- Dell’agli, M; Bellosta, S; Rizzi, L; Galli, GV; Canavesi, M; Rota, F; Parente, R; Bosisio, E; Romeo, S. A structure-activity study for the inhibition of metalloproteinase-9 activity and gene expression by analogues of gallocatechin-3-gallate. Cell Mol. Life Sci 2005, 62, 2896–2903. [Google Scholar]
- Ko, WC; Shih, CM; Lai, YH; Chen, JH; Huang, HL. Inhibitory effects of flavonoids on phosphodiesterase isozymes from guinea pig and their structure-activity relationships. Biochem. Pharmacol 2004, 68, 2087–2094. [Google Scholar]
- Paliyath, G; Poovaiah, BW. Identification of naturally occurring calmodulin inhibitors in plants and their effects on calcium- and calmodulin-promoted protein phosphorylation. Plant Cell Physiol 1985, 26, 201–209. [Google Scholar]
- Campos-Toimil, M; Lugnier, C; Droy-Lefaix, MT; Takeda, K. Inhibition of type 4 phosphodiesterase by rolipram and Ginkgo biloba extract (EGb 761) decreases agonist-induced rises in internal calcium in human endothelial cells. Arterioscler. Thromb. Vasc. Biol 2000, 20, E34–E40. [Google Scholar]
- Nichols, MR; Morimoto, BH. Differential inhibition of multiple cAMP phosphodiesterase isozymes by isoflavones and tyrphostins. Mol. Pharmacol 2000, 57, 738–745. [Google Scholar]
- Satake, N; Imanishi, M; Shibata, S. Increased nitroglycerin-induced relaxation by genistein in rat aortic rings. Eur. J. Pharmacol 1999, 377, 193–197. [Google Scholar]
- Saponara, R; Bosisio, E. Inhibition of cAMP-phosphodiesterase by biflavones of Ginkgo biloba in rat adipose tissue. J. Nat. Prod 1998, 61, 1386–1387. [Google Scholar]
- Kuppusamy, UR; Das, NP. Effects of flavonoids on cyclic AMP phosphodiesterase and lipid mobilization in rat adipocytes. Biochem. Pharmacol 1992, 44, 1307–1315. [Google Scholar]
- Orallo, F; Camiña, M; Alvarez, E; Basaran, H; Lugnier, C. Implication of cyclic nucleotide phosphodiesterase inhibition in the vasorelaxant activity of the citrus-fruits flavonoid (+/−)-naringenin. Planta Med 2005, 71, 99–107. [Google Scholar]
- Kumar, S; Sarkar, A; Sundar, D. Controlling aggregation propensity in A53T mutant of alphasynuclein causing Parkinson’s disease. Biochem. Biophys. Res. Commun 2009, 387, 305–309. [Google Scholar]
- McCormack, AL; Mak, SK; Shenasa, M; Langston, WJ; Forno, LS; Di Monte, DA. Pathologic modifications of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated squirrel monkeys. J. Neuropathol. Exp. Neurol 2008, 67, 793–802. [Google Scholar]
- Ulusoy, A; Febbraro, F; Jensen, PH; Kirik, D; Romero-Ramos, M. Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur. J. Neurosci 2010, 32, 409–422. [Google Scholar]
- Paxinou, E; Chen, Q; Weisse, M; Giasson, BI; Norris, EH; Rueter, SM; Trojanowski, JQ; Lee, VM; Ischiropoulos, H. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci 2001, 21, 8053–8061. [Google Scholar]
- Santner, A; Uversky, VN. Metalloproteomics and metal toxicology of α-synuclein. Metallomics 2010, 2, 378–392. [Google Scholar]
- Perez, RG; Hastings, TG. Could a loss of alpha-synuclein function put dopaminergic neurons at risk? J. Neurochem 2004, 89, 1318–1324. [Google Scholar]
- Volles, MJ; Lansbury, PT, Jr. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41, 4595–4602. [Google Scholar]
- Parihar, MS; Parihar, A; Fujita, M; Hashimoto, M; Ghafourifar, P. Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int J. Biochem. Cell Biol 2009, 41, 2015–2024. [Google Scholar]
- Reindl, W; Yuan, J; Krämer, A; Strebhardt, K; Berg, T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol 2008, 15, 459–466. [Google Scholar]
- Mbefo, MK; Paleologou, KE; Boucharaba, A; Oueslati, A; Schell, H; Fournier, M; Olschewski, D; Yin, G; Zweckstetter, M; Masliah, E; Kahle, PJ; Hirling, H; Lashuel, HA. Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem 2010, 285, 2807–2822. [Google Scholar]
- Cozza, G; Bonvini, P; Zorzi, E; Poletto, G; Pagano, MA; Sarno, S; Donella-Deana, A; Zagotto, G; Rosolen, A; Pinna, LA; Meggio, F; Moro, S. Identification of ellagic acid as potent inhibitor of protein kinase CK2: a successful example of a virtual screening application. J. Med. Chem 2006, 49, 2363–2366. [Google Scholar]
- Chen, L; Feany, MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci 2005, 8, 657–663. [Google Scholar]
- Chinta, SJ; Mallajosyula, JK; Rane, A; Andersen, JK. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett 2010, 486, 235–239. [Google Scholar]
- Mandel, SA; Fishman-Jacob, T; Youdim, MB. Modeling sporadic Parkinson’s disease by silencing the ubiquitin E3 ligase component, SKP1A. Parkinsonism Relat. Disord 2009, 15, S148–S151. [Google Scholar]
- Yasuda, T; Mochizuki, H. The regulatory role of α-synuclein and parkin in neuronal cell apoptosis; possible implications for the pathogenesis of Parkinson’s disease. Apoptosis 2010, 15, 1312–1321. [Google Scholar]
- Xie, W; Li, X; Li, C; Zhu, W; Jankovic, J; Le, W. Proteasome inhibition modeling nigral neuron degeneration in Parkinson’s disease. J. Neurochem 2010, 115, 188–199. [Google Scholar]
- Hyun, DH; Lee, M; Halliwell, B; Jenner, P. Proteasomal inhibition causes the formation of protein aggregates containing a wide range of proteins, including nitrated proteins. J. Neurochem 2003, 86, 363–373. [Google Scholar]
- Zhu, W; Xie, W; Pan, T; Xu, P; Fridkin, M; Zheng, H; Jankovic, J; Youdim, MB; Le, W. Prevention and restoration of lactacystin-induced nigrostriatal dopamine neuron degeneration by novel brain-permeable iron chelators. FASEB J 2007, 21, 3835–3844. [Google Scholar]
- Zhou, ZD; Lim, TM. Dopamine (DA) induced irreversible proteasome inhibition via DA derived quinones. Free Radic. Res 2009, 43, 417–430. [Google Scholar]
- McNaught, KS; Jnobaptiste, R; Jackson, T; Jengelley, TA. The pattern of neuronal loss and survival may reflect differential expression of proteasome activators in Parkinson’s disease. Synapse 2010, 64, 241–250. [Google Scholar]
- Olanow, CW. The pathogenesis of cell death in Parkinson’s disease—2007. Mov. Disord 2007, 22, S335–S342. [Google Scholar]
- Chang, CR; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci 2010, 1201, 34–39. [Google Scholar]
- Lee, JY; Nagano, Y; Taylor, JP; Lim, KL; Yao, TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol 2010, 189, 671–679. [Google Scholar]
- Renna, M; Jimenez-Sanchez, M; Sarkar, S; Rubinsztein, DC. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J. Biol. Chem 2010, 285, 11061–11067. [Google Scholar]
- Cherra, SJ, III; Kulich, SM; Uechi, G; Balasubramani, M; Mountzouris, J; Day, BW; Chu, CT. Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol 2010, 190, 533–539. [Google Scholar]
- Sevlever, D; Jiang, P; Yen, SH. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 2008, 47, 9678–9687. [Google Scholar]
- Murgas Torrazza, R; Suryawan, A; Gazzaneo, MC; Orellana, RA; Frank, JW; Nguyen, HV; Fiorotto, ML; El-Kadi, S; Davis, TA. Leucine supplementation of a low-protein meal increases skeletal muscle and visceral tissue protein synthesis in neonatal pigs by stimulating mTOR-dependent translation initiation. J. Nutr 2010, 140, 2145–5212. [Google Scholar]
- Bauchart-Thevret, C; Cui, L; Wu, G; Burrin, DG. Arginine-induced stimulation of protein synthesis and survival in IPEC-J2 cells is mediated by mTOR but not nitric oxide. Am. J. Physiol. Endocrinol. Metab 2010, 299, E899–E909. [Google Scholar]
- Kim, E. Mechanisms of amino acid sensing in mTOR signaling pathway. Nutr. Res. Pract 2009, 3, 64–71. [Google Scholar]
- Machiya, Y; Hara, S; Arawaka, S; Fukushima, S; Sato, H; Sakamoto, M; Koyama, S; Kato, T. Phosphorylated {alpha}-Synuclein at Ser-129 Is Targeted to the Proteasome Pathway in a Ubiquitin-independent Manner. Biol. Chem 2010, 285, 40732–40744. [Google Scholar]
- Crews, L; Spencer, B; Desplats, P; Patrick, C; Paulino, A; Rockenstein, E; Hansen, L; Adame, A; Galasko, D; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 2010, 5, e9313. [Google Scholar]
- Tang, FY; Chiang, EP; Pai, MH. Consumption of S-Allylcysteine Inhibits the Growth of Human Non-Small-Cell Lung Carcinoma in a Mouse Xenograft Model. J Agric Food Chem 2010, in press. [Google Scholar]
- Petrovski, G; Das, DK. Does autophagy take a front seat in lifespan extension? J. Cell Mol. Med 2010, 14, 2543–2551. [Google Scholar]
- Zhou, H; Luo, Y; Huang, S. Updates of mTOR inhibitors. Anticancer Agents Med. Chem 2010, 10, 571–581. [Google Scholar]
- Lee, YK; Lee, WS; Kim, GS; Park, OJ. Anthocyanins are novel AMPKα1 stimulators that suppress tumor growth by inhibiting mTOR phosphorylation. Oncol. Rep 2010, 24, 1471–1477. [Google Scholar]
- Lee, YK; Park, SY; Kim, YM; Kim, DC; Lee, WS; Surh, YJ; Park, OJ. Suppression of mTOR via Akt-dependent and -independent mechanisms in selenium-treated colon cancer cells: involvement of AMPKalpha1. Carcinogenesis 2010, 31, 1092–1099. [Google Scholar]
- Tang, FY; Cho, HJ; Pai, MH; Chen, YH. Concomitant supplementation of lycopene and eicosapentaenoic acid inhibits the proliferation of human colon cancer cells. J. Nutr. Biochem 2009, 20, 426–434. [Google Scholar]
- Balgi, AD; Fonseca, BD; Donohue, E; Tsang, TC; Lajoie, P; Proud, CG; Nabi, IR; Roberge, M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One 2009, 4, e7124. [Google Scholar]
- Bruno, P; Calastretti, A; Priulla, M; Asnaghi, L; Scarlatti, F; Nicolin, A; Canti, G. Cell survival under nutrient stress is dependent on metabolic conditions regulated by Akt and not by autophagic vacuoles. Cell Signal 2007, 19, 2118–2126. [Google Scholar]





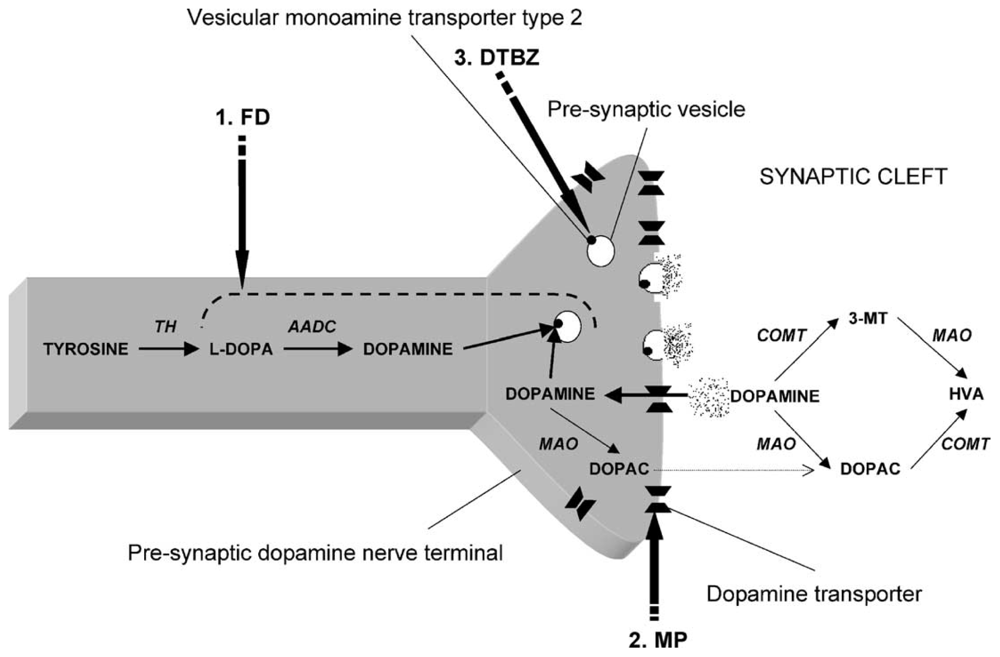{kind=link}
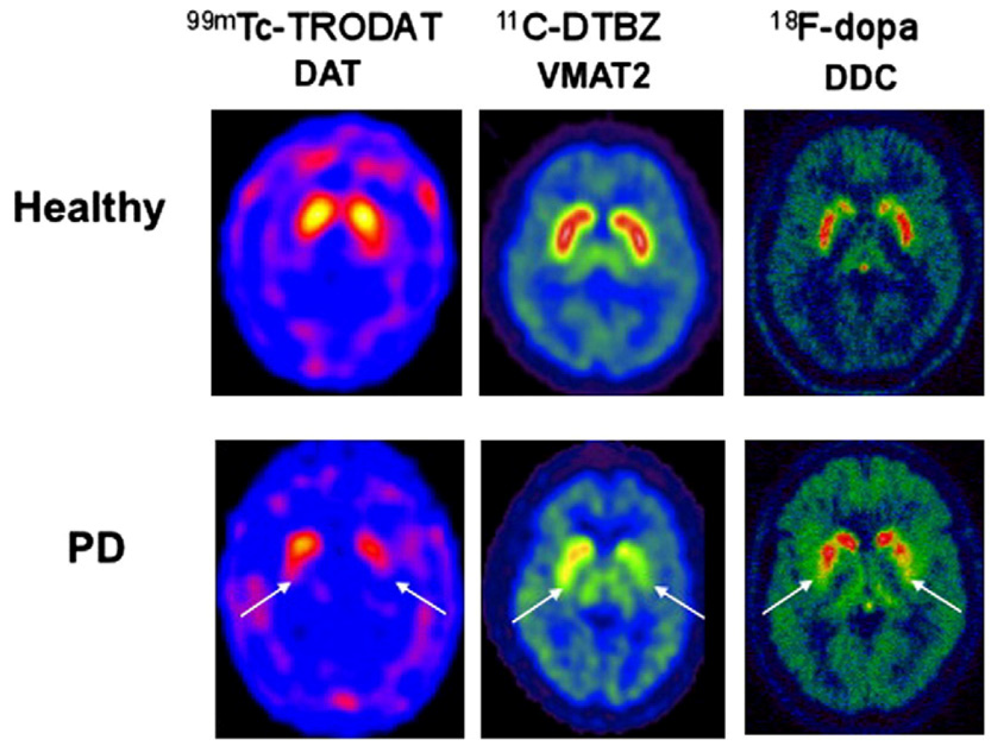{kind=link}
{kind=link}
{kind=link}
| Tyrosinase Inhibitors | Reference |
|---|---|
| Tetrahydroxychalcones, Butein | [307,308] |
| Prenylated flavonoids, Sanggenon D | [309] |
| Sophoraflavanone G, Kuraridin, Kurarinone, Norkurarinol | [310,311] |
| Cinnamic acid, Aloin, Sophorcarpidine | [312,313] |
| Glabrene/Licorice, licuraside, isoliquiritin and licochalcone | [314,315] |
| Quercetin, Galangin, Morin, Fisetin, Luteolin, Apigenin, | [316] |
| Esculetin | [317] |
| Hexylresorcinol, Dodecylresorcinol | [318] |
| Oxyresveratrol | [319] |
| Gnetol | [320] |
| (−)-Epigallocatechin-3-gallate, Hinokitiol (beta-thujaplicin), Kojic acid | [321,322] |
| Reduced glutathione, cysteine, thiol compounds, ascorbic acid, acetic acid | [323–326] |
| Dimethylsulfide | [327] |
| Phytic acid | [328] |
| Tannic acid | [329] |
| Nobiletin | [330] |
| Kaempferol | [331,332] |
| Extract of hibiscus, carex pumila, and garcinia subelliptica | [333] |
| Wine phenolics | [334] |
| Green tea | [335] |
| Procyanidins, Grape seed extract | [336,337] |
| Gallic acid derivatives | [338] |
| Safflower | [339] |
| Aisic acid | [340] |
| Olive oil constituents | [341] |
| Cycloxygenase I/II Inhibitors | Reference |
|---|---|
| Quercetin, Kampferol, Chrysin and Galangin | [342,343] |
| Anthocyanins, Delphinidin, Cyanidin, Malvidin | [344,345] |
| Galangin, Morin, Apigenin, Rutin, Catechin, EGCG, Quercetin, Chrysin, Flavones, Luteolin, Tectorigenin, Bilobetin, Nobiletin, Fisetin, Naringenin, Quercetin, Lonchocarpol, Tomentosanol and Wogonin | [346–349] |
| Quercetin, Quercetin 3-glucuronide, Quercetin 3′-sulfate 3′-methylquercetin 3-glucuronide | [350,351] |
| Ursolic acid, Eugenol, Pyrogallol and Cinnamaldehyde | [352] |
| Ipriflavone, Resveratrol, MSV-60, Amentoflavone, Ruscus extract | [353,354] |
| Notoginseng Prenylated flavonoids, Morusin, Kuwanon C, Sanggenon, Kazinol, Kuraridin, Kurarinone, Sophoraflavanone G | [355] |
| Butein and 7,3′,4′-trihydroxy flavone | [356] |
| Coumarins, Bergapten | [357] |
| Amentoflavone | [358] |
| Oroxylin A | [359] |
| Caffeic acid Phenethyl Ester and Propolis | [360] |
| Lipoxygenase Inhibitors | Reference |
|---|---|
| Luteolin, Baicalein, Fisetin, Quercetin, Eugenol, Curcumin, Cinnamaldehyde, Piperine, Capsaicin, Allyl sulfide, Oroxylin A, Wogonin | [361–364] |
| Morin, Galangin, Kaempherol, Taxifolin, EGCG, Esculetin, Propyl gallate | [365–367] |
| Coumarin, 7-hydroxy-derivative, Fraxetin, Daphnetin, Coumarin derivatives | [368] |
| Amentoflavone | [369] |
| Kuraridin, Sophoroflavonone G, Kenusanone A, Psoralidin | [370] |
| 3,5,6,7,3′,4′-hexamethoxyflavone, Sinensetin, Nobiletin, Tangeretin, Rhamnetin Tetramethylscutellarein, 6,7,8,3′,4′-heptamethoxyflavone, Hesperidin, Ferulic acid | [371] |
| Sophoraflavanone G, Quercetin, Kenusanone A | [372] |
| Circiliol, Hypolatein, Sideritloflavone | [373] |
| Silymarin | [374] |
| Bean (Phaseolus vulgaris L.) hulls | [375] |
| Cirsiliol, Hypolaetin, Hypolaetin-8-O-beta-d-glucoside, Gossypetin, Gossypin, Hibifolin, Leucocyanidol | [376,377] |
| Oroxylin A, Baicalein, Wogonin | [378] |
| Procyanidins | [379] |
| Quercetin glycosides | [380] |
| Entaureidin and 5,3′-dihydroxy-4′-methoxy-7-carbomethoxyflavonol | [381] |
| Phospholipase A2 Inhibitors | Reference |
|---|---|
| Quercetin. Kaempferol, Myrecetin, Kaempferole-3-galactoside, Scutellarein, Ochnaflavone, Amentoflavone, Ginkgetin, Isoginkgetin, Morelloflavone, Bilobetin, Prenylated flavonoids | [342] |
| Ginkolide | [378] |
| Amentoflavone, Ginkgetin | [379] |
| Fish oil, Evening primrose oil | [380,381] |
| 2′,4′,7-trimethoxyflavone | [382] |
| Nobiletin | [383] |
| Rosmarinic acid | [384] |
| Omega-3 fatty acids | [385] |
| Xanthine Oxidase Inhibitors | Reference |
|---|---|
| Skull Cap (Scutellaria baicalensis (SbE)), Grape seed proanthocyanidins | [386] |
| Hesperitin, Theaflavin-3,3′-digallate, Cranberry juice | [387–389] |
| Chrysin, Phloretin, Luteolin, Kaempferol, Quercetin, Myrecetin, Galagin, Apigenin, Morin, Isorhamnetin, Fisetin, Rutin | [390–395] |
| EGCG, 4-t-butylcatechol, Catechin, Fisetin, Luteolin, Raxifolin | [395,396] |
| Quercetin glycosides | [397] |
| Apigenin, Quercetin, Isovitexin | [398] |
| Hydroxyl or Methyl Chalcones (i.e., 3,3,4,4-tetrahydroxychalcone), Esculetin, 4-methylumbelliferone | [399] |
| Propolis, Caffeic acid phenetyl ester, Chrysin, Galangin | [400,401] |
| 5,7,4′-Trihydroxy-6-methoxyflavone p-coumaric acid derivatives drupanin, 4-acetyl-3,5-diprenylcinnamic acid, trans-ferulic acid O-hexan-3-onyl-ether | [402] |
| Baicalein, Wogonin, Baicalin | [403–405] |
| Pycnogenol, Silymarin, Silybin, Silybin flavones, Purpurogallin | [406,407] |
| Black Tea | [408] |
| Procyanidins, Pygnogenol | [409–412] |
| Anthocyanins, Cyanidin, Cyanidin 3-O-beta-d-glucoside | [413] |
| Myricetin Glycosides | [414] |
| Xanthine Oxidase and Superoxide Scavengers | Reference |
|---|---|
| EGCG, EGC, Pyrogallol, Catechin, Luteolin, Myrecetin, Rutin, Apigenin, Quercetin, Hesperitin, Naringenin, Biochanin, Retinol, Daidzein, Genestein, 4-t-butylcatechol, Taxifolin, Fisetin, Kaempferol, 5,7,4′-trihydroxy-6-methoxyflavone | [395,402,415–417] |
| Caffeic acid, Rosmarinic acid, Salvianolic acid, Sage | [418] |
| Apigenin, Quercetin, Diosmin | [419] |
| Green tea polyphenolics, Theaflavin, EGCG | [388,389,420,421] |
| Scutellarin | [422] |
| Oligomeric proanthocyanidins, EGCG, Delphinidin, Myrecetin, Gallic acid, Caffeic acid, Fisetin, Quercetin, Catechin, Epicatechin | [423] |
| Galangin/Caffeic acid phenethyl ester, Propolis, Caffeic, Chlorogenic acid, Gallic acid | [401,424,425] |
| Baicalein, Baicalin, Morin | [404,426,427] |
| Uric acid | [428] |
| Chrysoeriol ± glycoside | [429] |
| Anacardiaceae spice | [430] |
| Myrecetin, Fisetin, Quercetin | [431] |
| Peroxide Scavengers | Reference |
|---|---|
| Acacetin, Dihydrorobinetin, Fisetin, Isorhamnetin, Robinetin, Myricitrin, Hyperoside | [450] |
| Resveratrol, Catechin, Gallocatechin | [451,452] |
| Pygnogenol, Pyrogallol, Gallic acid, Anthocyanidins | [452,453] |
| Gallic acid, Trolox, Kaempferol | [454] |
| Vanillic/Caffeic acids | [450] |
| Baicalein | [448] |
| Hydroxytyrosol | [442] |
| Iron Reducing/Chelating Compounds | Reference |
|---|---|
| Rutin, Morin, Rosemary, Sage, Oregano | [457] |
| Phytic acid, Brown rice bran, Tannic acid | [449] |
| Apigenin, Diosmin, Phloretin, Fisetin, Taxifolin, Naringenin | [458] |
| Quercetin, Rutin, Myrecetin, Luteolin, Epicatechin Caffeic acid, Catechin, Kaempferol, Naringenin, Baicilein | [459–464] |
| Theaflavin, Theaflavin Digallate | [455,465,466] |
| Vitamin E, Zinc | [467] |
| Gallic Acid | [468] |
| Silymarin, Silybin | [469] |
| Rutin | [470] |
| MAPK/NF-κB/iNOS/COX-2 (−) | Reference |
|---|---|
| Selenium, Zinc | [429,435] |
| Chrysin, Quercetin, Galangin, Propolis or its derivatives | [481–486] |
| Apigenin | [487,488] |
| Luteolin | [487,489,490] |
| Diosmetin, 3-hydroxyflavone, Pillion,4′,7′-dihydroxyflavone, Ayanin, Luteolin, Tectochrysin, 3′,4′-dihydroxyflavone, Tamarixetin, Genestein, Kaempferol, Izalpinin, Ombuine, Biochanin, Tectorigenin, Daidzein, 7-hydroxyflavone, Rhamnetin, flavone, EGCG, Mearnsetin, Liquiritigenin, Myrecetin | [491] |
| Hydroxychalcones | [228,492,493] |
| EGCG/Green tea | [494–496] |
| Butein | [497] |
| Anthocyanins | [344,498] |
| 5,6,3′,5′-tetramethoxy 7,4′-hydroxyflavone, Artemisia Absinthium, Wormwood, Blackwalnut | [235] |
| Scutellarin | [499] |
| Isovitexin | [500] |
| Naringin, Hesperitin and Naringenin | [501–503] |
| Baicalein | [504,505] |
| Silibinin, Silymarin | [225,506,507] |
| Amentoflavone | [508] |
| Licorice | [509] |
| Wogonin | [510] |
| Curcumin, Luteolin, Wognonin, Kaempferol, Nobiletin, Bilobetin | [342] |
| Phosphodiesterase Inhibitors | Reference |
|---|---|
| Butein | [513] |
| Cirsimarin | [514] |
| Grape Skins, Anthocyanin, Malvidin | [515] |
| Diosmetin, Luteolin, Apigenin, Quercetin, Myrecetin | [516] |
| (+)-Catechin, Caffeic acid | [517] |
| Gingko Biloba | [518] |
| Biochanin A, Tyrphostin, Diadzein | [519] |
| Theophylline | [520] |
| Amentoflavone, Bilobetin, Sequoiaflavone, Ginkgetin, Isoginkgetin | [521] |
| Scutellarein, Phloretin, Naringenin | [522,523] |
© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mazzio, E.A.; Close, F.; Soliman, K.F.A. The Biochemical and Cellular Basis for Nutraceutical Strategies to Attenuate Neurodegeneration in Parkinson’s Disease. Int. J. Mol. Sci. 2011, 12, 506-569. https://doi.org/10.3390/ijms12010506
Mazzio EA, Close F, Soliman KFA. The Biochemical and Cellular Basis for Nutraceutical Strategies to Attenuate Neurodegeneration in Parkinson’s Disease. International Journal of Molecular Sciences. 2011; 12(1):506-569. https://doi.org/10.3390/ijms12010506
Chicago/Turabian StyleMazzio, Elizabeth A., Fran Close, and Karam F.A. Soliman. 2011. "The Biochemical and Cellular Basis for Nutraceutical Strategies to Attenuate Neurodegeneration in Parkinson’s Disease" International Journal of Molecular Sciences 12, no. 1: 506-569. https://doi.org/10.3390/ijms12010506