
Novel Biopolymer-Based Catalyst for the Multicomponent Synthesis of N-aryl-4-aryl-Substituted Dihydropyridines Derived from Simple and Complex Anilines

Abstract
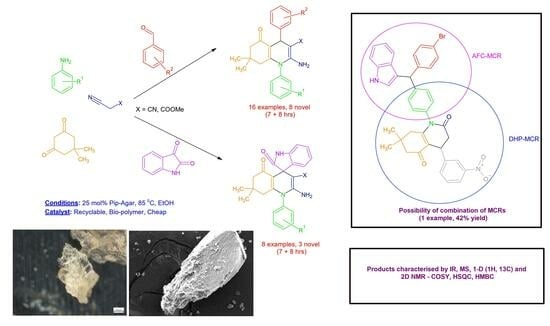
:
1. Introduction
2. Results and Discussion
2.1. Catalyst Screening and Condition Optimisation
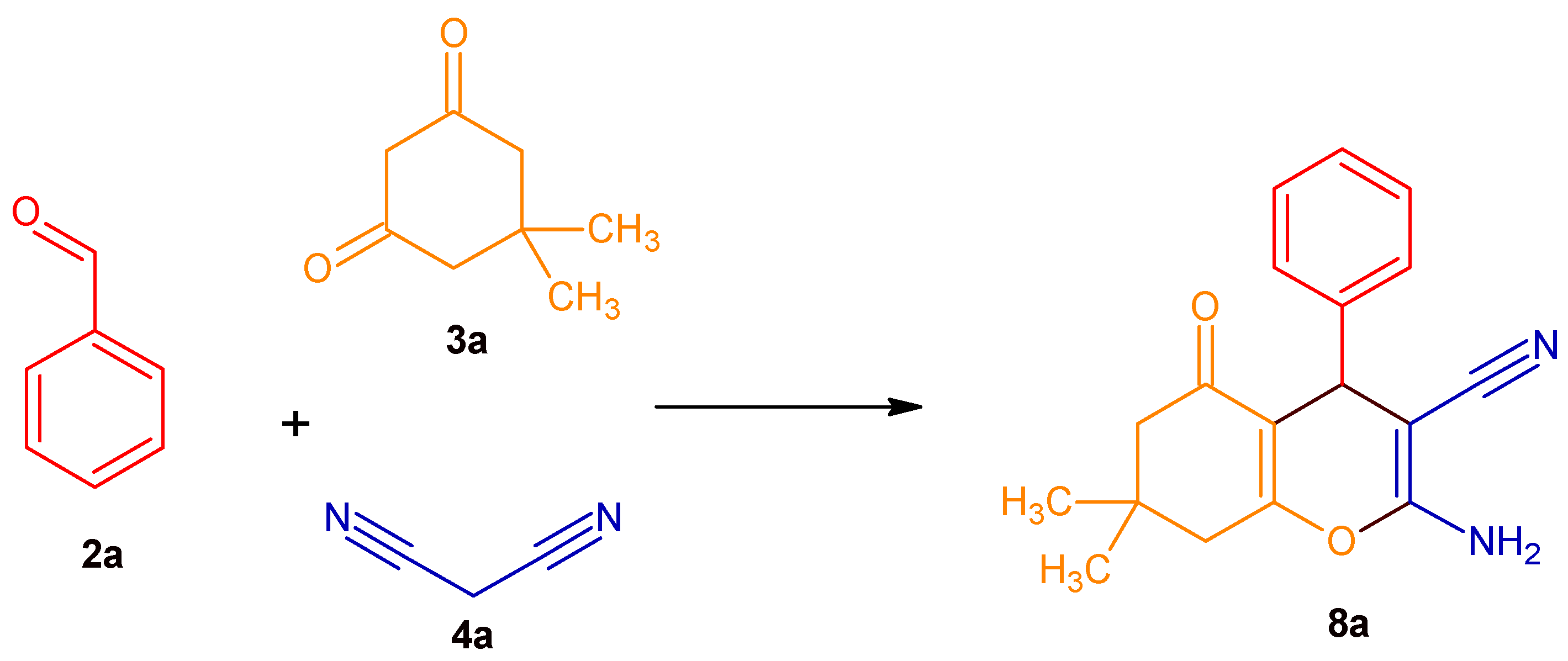
2.2. Expanding the DHP Substrate Scope
2.3. Catalyst Description and Recycling Runs
2.3.1. Optical Microscopy
2.3.2. IR Spectrum
2.3.3. Recycling Runs and Hot Filtration Test
2.4. Green Metrics of Model Reaction
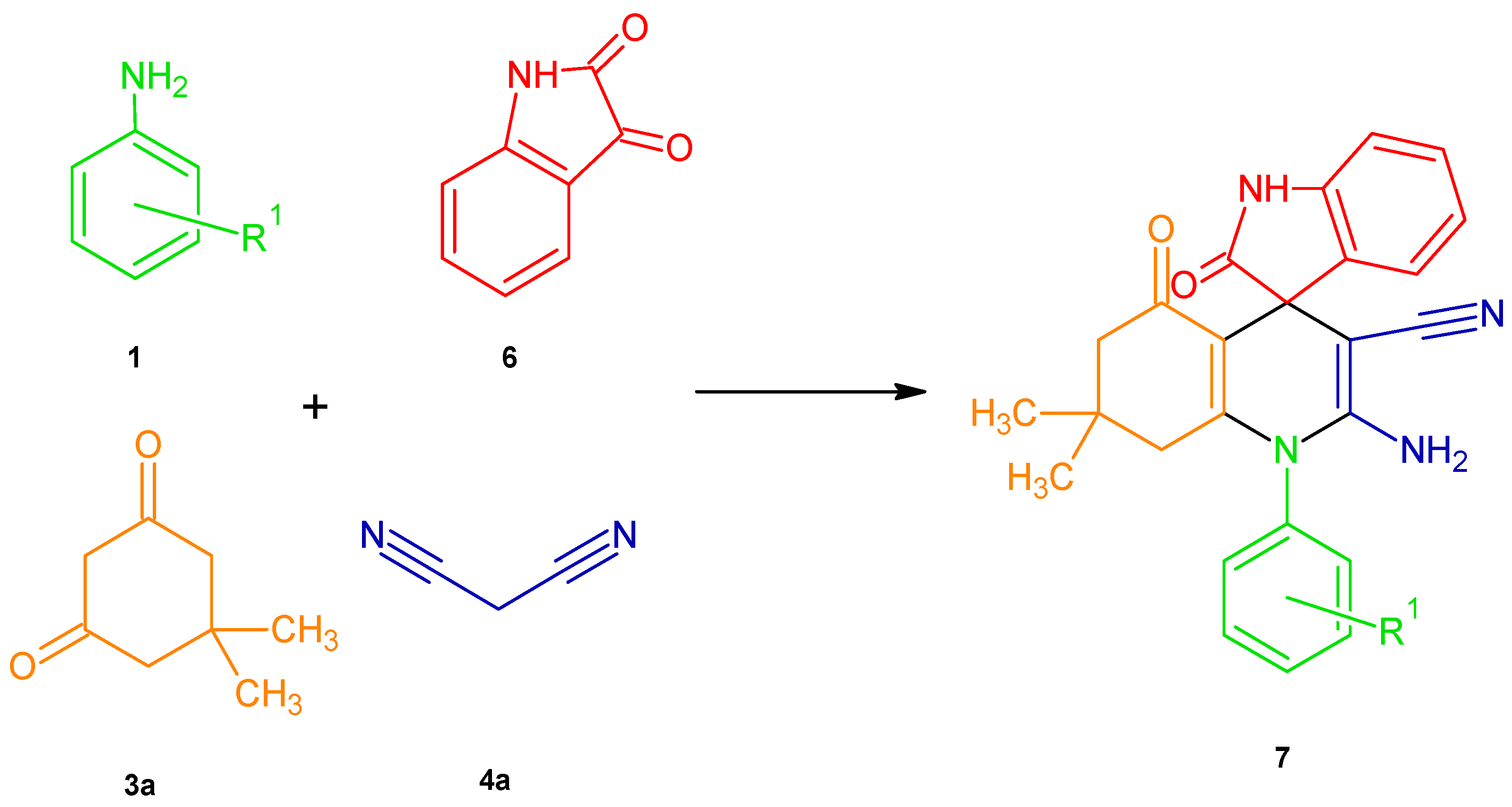
2.5. DHPs Derived from the Products of the Aza-Friedel–Crafts Multicomponent Reaction
2.6. DHP Characterisation by Single and Two-Dimensional NMR Methods
- (1)
- C15 is long-range coupling with H13 only, hence confirming the above methylene assignment. Simultaneously, C10 and C11 are both coupled to H16, but not to H13.
- (2)
- H20 and 21 are long-range-coupled with C17, but H18 and 19 are not. This may seem strange but, in some instances (especially in para-substituted systems with chemically equivalent hydrogens on either side of the aromatic ring), the HMBC quantum filter removes two-bond correlations (along with single-bond correlations), leaving only three/four-bond correlations. In fact, C3 gives a cross-peak with H2 and 6 (three-bond correlation), while it does not with H1 and 5. In addition, there are two peaks, which are denoted as 20–21 and 18–19 due to three-bond correlations (H20-C21 or H21-C20 for the former and H18-C19 or H19-C18 for the latter).
- (3)
- H2 and 6 and H1 and 5 both give cross-peaks with C2,6 and C1,5, respectively. This may appear incorrect, but what is actually happening is a correlation between H2 and C6 or between C2 and H6 (three bond correlations), similar to what is observed between H/C 20–22.
- (4)
- H1 and 5 give a cross-peak with C4 (three-bond correlation). The identity of C4 is confirmed because it undergoes correlation with H7. Meanwhile, C3 is correlated to the methyl hydrogens H29 and with H2 and 6, as already mentioned.
- (5)
- H7 carries out long-range coupling with both C10 and C11, with C10 also carrying out long-range coupling with H16 (confirming the assignments of methylene hydrogens). H7 also carries out long-range coupling with C9, C8, C23, C4, C2, and 6.
- (6)
- H29s are able to give cross-peaks with C1 and 5 (three-bond correlation).
- (7)
- C15 carries out long-range three-bond coupling with H13 (visible cross-peak in expanded inset image), hence confirming the earlier assignments of the methylene hydrogens.
- (8)
- C8 is involved in long-range coupling with H7 (strongly), H16 (strongly), and with H13 (barely visible).
- (9)
- C9 is involved in long-range coupling with H7 (two-bond correlation) and H25 (three-bond correlation). The two-bond correlation between C9 and H7 is detected because of a small J coupling constant between the two that allows for peaks not to be filtered out by quantum filtering.
- (10)
- C23 is involved in long-range coupling (three-bond), with H7 confirming its identity.
3. Experimental
3.1. General Reaction Procedure
3.2. Pip–Agar Catalyst Preparation and Alkalinity Determination
3.3. Other Catalyst Preparations
3.4. Product Characterization Procedure
3.5. Product Characterization Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, M.S.; Chowdhury, S. Recent developments in solvent-free multicomponent reactions: A perfect synergy for eco-compatible organic synthesis. RSC Adv. 2012, 2, 4547–4592. [Google Scholar] [CrossRef]
- Bosica, G.; Abdilla, R. Recent advances in multicomponent reactions under operationally heterogeneous conditions. Catalysts 2022, 12, 725. [Google Scholar] [CrossRef]
- Paprocki, D.; Madej, A.; Koszelewski, D.; Brodzka, A.; Ostaszewski, R. Multicomponent reactions accelerated by aqueous micelles. Front. Chem. 2018, 6, 502. [Google Scholar] [CrossRef]
- Khelilkordi, Z.; Ziarani, G.M.; Mohajer, F.; Badiei, A.; Sillanpaa, M. Recent advances in the application of magnetic bio-polymers as catalysts in multicomponent reactions. RSC Adv. 2022, 12, 12672–12701. [Google Scholar] [CrossRef] [PubMed]
- Ziarani, G.M.; Kheilkordi, Z.; Mohajer, F.; Badiei, A.; Luque, R. Magnetically recoverable catalysts for the preparation of pyridine derivatives: An overview. RSC Adv. 2021, 11, 17456–17477. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Jamatia, R.; Sarkar, R.; Sarkar, F.K.; Alam, S.; Pal, A.K. One-pot multicomponent reaction: A highly versatile strategy for the construction of valuable nitrogen-containing heterocycles. ChemistrySelect 2022, 7, e202201901. [Google Scholar] [CrossRef]
- Cataldi, M.; Bruno, F. 1,4-dihydropyridines: The multiple personalities of a blockbuster drug family. Transl. Med. 2012, 4, 10–24. [Google Scholar]
- Bosica, G.; Demanuele, K.; Padron, J.M. One-pot multicomponent green Hantzsch synthesis of 1,2-dihydroyridine derivatives with antiproliferative activity. Beilstein J. Org. Chem. 2020, 16, 2862–2869. [Google Scholar] [CrossRef] [PubMed]
- Al-Said, M.S.; Ghorab, M.M.; Al-Dosari, M.S.; Hamed, M.M. Synthesis and in vitro anticancer evaluation of some novel hexahydroquinoline derivatives having a benzenesulfonamide moiety. Eur. J. Med. Chem. 2011, 46, 201–207. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Ragab, F.A.; Heiba, H.I.; Arafa, R.K.; El-Hossary, E.M. In vitro anticancer screening and radiosensitizing evaluation of some new quinolines and pyrimido[4,5-b]quinolines bearing a sulfonamide moiety. Eur. J. Med. Chem. 2010, 45, 3677–3684. [Google Scholar] [CrossRef]
- Han, W.; Inoue, C.; Onizawa, T.; Oriyama, T. Synthesis of N-aryl4-arylhexahydroquinoline derivatives by reaction of cyclic enaminones with arylidenemalononitriles in DMSO. Synthesis 2021, 53, 1495–1502. [Google Scholar]
- Abaszadeh, M.; Seifi, M.; Asadipour, A. MgO as a heterogeneous base catalyst, catalyses multicomponent reaction of cyclic enaminoketones, malononitrile, and aromatic aldehydes, synthesis and reactivity. Synth. React. Inorg. Met. 2016, 46, 512–517. [Google Scholar] [CrossRef]
- Abaszadeh, M.; Seifi, M.; Asadipour, A. Ultrasound promotes one-pot synthesis of 1,4-dihydropyridine and imidazo[1,2-a]quinoline derivatives, catalyzed by ZnO nanoparticles. Res. Chem. Intermed. 2015, 41, 5229–5238. [Google Scholar] [CrossRef]
- Amirheidari, B.; Seifi, M.; Abaszadeh, M. Evaluation of magnetically recyclable nano-Fe3O4 as a green catalyst for the synthesis of mono- and bistetrahydro-4H-chromene and mono and bis-1,4-dihydropyridine derivatives. Res. Chem. Intermed. 2016, 42, 3413–3423. [Google Scholar] [CrossRef]
- Ahadi, N.; Mobinikhaledi, A.; Bodaghifard, M.A. One-pot synthesis of 1,4-dihydropyridines and N-arylquinolines in the presence of copper complex stabilized on MnFe2O4 (MFO) as a novel organic–inorganic hybrid material and magnetically retrievable catalyst. Appl. Organomet. Chem. 2020, 34, 5822–5834. [Google Scholar] [CrossRef]
- Singh, S.K.; Singh, K.N. DBU-catalyzed expeditious and facile multicomponent synthesis of N-arylquinolines under microwave irradiation. Montash. Chem. 2012, 143, 805–808. [Google Scholar] [CrossRef]
- Ghozlan, S.A.S.; Ahmed, A.G.; Abdelhamid, I.A. Regioorientation in the addition reaction of α-substituted cinnamonitrile to enamines utilizing chitosan as a green catalyst: Unambiguous structural characterization using 2D-HMBC NMR spectroscopy. J. Heterocycl. Chem. 2016, 53, 817–823. [Google Scholar] [CrossRef]
- Abaszadeh, M.; Seifi, M. Crown ether complex cation ionic liquids: Synthesis and catalytic applications for the synthesis of tetrahydro-4H-chromene and 1,4-dihydropyridine derivatives. J. Sulf. Chem. 2017, 38, 440–449. [Google Scholar] [CrossRef]
- Sanaei-Rad, S.; Saeidiroshan, H.; Mirhosseini-Eshkevari, B.; Ali Ghasemzadeh, M. Hexamethylenetetramine-based ionic liquid anchored onto the metal–organic framework MIL-101(Cr) as a superior and reusable heterogeneous catalyst for the preparation of hexahydroquinolines. Res. Chem. Intermed. 2021, 47, 2143–2159. [Google Scholar] [CrossRef]
- Gao, S.; Tsai, C.H.; Tseng, C.; Yao, C. Fluoride ion catalyzed multicomponent reactions for efficient synthesis of 4H-chromene and N-arylquinoline derivatives in aqueous media. Tetrahedron 2008, 64, 9143–9149. [Google Scholar] [CrossRef]
- Kang, S.R.; Lee, Y.R. Efficient one-pot synthesis of spirooxindole derivatives bearing hexahydroquinolines using multicomponent reactions catalyzed by ethylenediamine diacetate. Synthesis 2013, 45, 2593–2599. [Google Scholar] [CrossRef]
- Khodaei, M.M.; Bahrami, K.; Farrokhi, A. Amberlite IRA-400 (OH−) as a catalyst in the preparation of 4H-benzo[b]pyrans in aqueous media. Synth. Commun. 2010, 40, 1492–1499. [Google Scholar] [CrossRef]
- Bosica, G.; Abdilla, R. Piperazine-Amberlyst®15-catalysed synthesis of 2-amino-4H-chromenes, chromeno[2,3-b]pyridines and chromeno[2,3-d]pyrimidines. Tetrahedron Green Chem. 2023, 1, 100011. [Google Scholar] [CrossRef]
- Jimenez-Gonzalez, C.; Ponder, C.S.; Broxterman, Q.B.; Manley, J.B. Using the right green yardstick: Why process mass intensity is used in the pharmaceutical industry to drive more sustainable processes. Org. Process Res. Dev. 2011, 15, 912–917. [Google Scholar] [CrossRef]
- Bosica, G.; Abdilla, R. A regioselective one-pot aza-Friedel–Crafts reaction for primary, secondary and tertiary anilines using a heterogeneous catalyst. Green Chem. 2017, 19, 5683–5690. [Google Scholar] [CrossRef]
- Sabaqian, S.; Nemati, F.; Heravi, M.M.; Nahzomi, H.T. Copper(I) iodide supported on modified cellulose-based nano-magnetite composite as a biodegradable catalyst for the synthesis of 1,2,3-triazoles. Appl. Organomet. Chem. 2016, 31, e3660. [Google Scholar] [CrossRef]
- Daisy, S.L.; Mary, A.C.C.; Devi, K.; Prabha, S.S.; Rajendran, S.; Zahirullah, S.S. Magnesium oxide nanoparticles—Synthesis and characterisation. Int. J. Nano Corr. Sci. Eng. 2015, 2, 64–69. [Google Scholar]
- Enache, D.F.; Vasile, E.; Simonescu, C.M.; Culita, D.; Vasile, E.; Oprea, O.; Pandele, A.M.; Razvan, A.; Dumitru, F.; Nechifor, G. Schiff base-functionalised mesoporous silicas (MCM-41, HMS) as Pb(II) adsorbents. RSC Adv. 2018, 8, 176–189. [Google Scholar] [CrossRef]
- Ghorbani-Choghamarani, A.; Hajjami, M.; Tahmasbi, B.; Noori, N. Boehmite silica sulfuric acid: As a new acidic material and reusable heterogeneous nanocatalyst for the various organic oxidation reactions. J. Iran. Chem. Soc. 2016, 13, 2193–2202. [Google Scholar] [CrossRef]
- Habte, L.; Shiferaw, N.; Mulatu, D.; Thenepalli, T.; Chilakala, R.; Ahn, J.W. Synthesis of nano-calcium oxide from waste eggshell by sol-gel method. Sustainability 2019, 11, 3196. [Google Scholar] [CrossRef]
- Onyszko, M.; Markowska-Szczupak, A.; Rakoczy, R.; Paszkiewicz, O.; Janusz, J.; Gorgon-Kuza, A.; Wenelska, K.; Mijowska, E. The cellulose fibers functionalized with star-like zinc oxide nanoparticles with boosted antibacterial performance for hygienic products. Sci. Rep. 2022, 12, 1321–1323. [Google Scholar] [CrossRef] [PubMed]
- Maheswari, C.S.; Shanmugapriya, C.; Revathy, K.; Lalitha, A. SnO2 nanoparticles as an efficient heterogeneous catalyst for the synthesis of 2H-indazolo[2,1-b]phthalazine-triones. J. Nanostruct. Chem. 2017, 7, 283–291. [Google Scholar] [CrossRef]
- Thanh, N.D.; Hai, D.S.; Huyen, L.T.; Thuy, V.T.T.; Tung, D.T.; Van, H.T.K.; Toan, V.N.; Giang, N.T.K.; Tri, N.M. Fe3O4-MNPs@MMT-K10: A reusable catalyst for synthesis of propargyl 4-aryl-4H-pyran-3-carboxyles via one-pot three-component reaction under microwave-assisted solvent-free conditions. Res. Chem. Intermed. 2023, 49, 525–555. [Google Scholar] [CrossRef]
- Zhu, S.; Zhu, Y.; Gao, X.; Mo, T.; Zhu, Y.; Li, Y. Production of bioadditives from glycerol esterification over zirconia supported heteropolyacids. Bioresour. Technol. 2013, 130, 41–45. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry a | Catalyst b | Amount of Catalyst | Temperature (°C)/Time (hr) | Solvent/Amount (mL) | Molar Ratio 1a/2a/3a/4a | Yield of 5a (%) c |
| 1 | Pip-A15 (2.72 mmol/g) | 20 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1 | 64% |
| 2 | 20 mol% | 85/2 + 7 | EtOH/2 | 1:1:1:1 | 47% | |
| 3 | 20 mol% | 85/7 + 1 | EtOH/2 | 1:1:1:1 | 11% | |
| 4 | 20 mol% + 0.2 g 4 Å MS f | 85/7 + 7 | EtOH/2 | 1:1:1:1 | 71% | |
| 5 | 20 mol% + 0.4 g 4 Å MS f | 85/7 + 7 | EtOH/2 | 1:1:1:1 | 52% | |
| 6 | 30 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1 | 63% | |
| 7 | 30 mol% | 85/7 + 16 | EtOH/2 | 1:1:1:1 | 74% | |
| 8 | Ethylenediamine-A15 (2 mmol/g) | 25 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1 | 63% |
| 9 | Morpholine-A15 (2.00 mmol/g) | 20 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1 | - |
| 10 | Pip–Dowex (2.4 mmol/g) | 20 mol% | 85/7 + 16 | EtOH/2 | 1:1:1:1 | 67% |
| 11 | 25 mol% | 85/7 + 16 | EtOH/2 | 1:1:1:1 | 70% | |
| 12 | 25 mol% | 95/7 + 16 | i-PrOH/2 | 1:1:1:1 | 65% | |
| 13 | Pip–Dowex (2.1 mmol/g) | 40 mol% | 85/7 + 7 85/7 + 12 | EtOH/2 | 1:1:1:1 | 66% 69% |
| 14 | 40 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 61% | |
| 15 | 30 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 67% | |
| 16 | 30 mol% | 85/7 + 7 | EtOH/2 | 1:1.3:1:1.3 | 82% g | |
| 17 | Cell-NH2 (0.5 mmol/g) | 5 mol% | 90/4 + 4 | EtOH/2 | 1:1:1:1 | 48% |
| 18 | 20 mol% | 85/7 + 16 | EtOH/2 | 1:1:1:1 | 46% | |
| 19 | A21 (dry) | 0.2 g | 85/7 + 8 | EtOH/2 | 1:1:1:1 | 53% |
| 20 | A26 (dry) | 0.3 g | 85/7 + 14 | EtOH/2 | 1:1:1:1 | 22% |
| 21 | MgO | 40 mol% | 85/7 + 4 | EtOH/2 | 1:1:1:1 | 57% |
| 22 | 20 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1 | 61% | |
| 23 | MgO-SiO2 (63–200 um) (4 mmol/g) | 10 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1 | 42% |
| 24 | 20 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1 | 46% | |
| 25 | MgO-nano-MCM41 (4.5 mmol/g) | 20 mol% | 85/7 + 16 | EtOH/2 | 1:1:1:1 | 60% |
| 26 | CaO–Boehmite (20% w/w, 3.6 mmol/g) | 10 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1 | 54% |
| 27 | SnO-MK30 (1 mmol/g) | 10 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 52% |
| 28 | ZnO–Cellulose | 0.1 g | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 67% |
| 29 | 0.125 g | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 57% | |
| 30 | 0.08 g | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 43% | |
| 31 | WSi-MK30 (10% w/w) | 0.1 g (0.3 mol%) | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 65% |
| 32 | PW-MK30 (10% w/w) | 0.1 g (0.3 mol%) | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 59% |
| 33 | MK30-nano-Fe3O4 | 0.1 g | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 70% |
| 34 | 0.1 g | 85/7 + 7 | EtOH/2 | 1:1.3:1:1.3 | 79% g | |
| 35 | 0.125 g | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 67% | |
| 36 | 0.075 g | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 61% | |
| 37 | 0.1 g | 85/7 + 7 | EtOH/H2O (7:3)/2 | 1:1:1:1.2 | 65% | |
| 38 | 0.1 g + 0.1 g 4 Å MS f | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 70% | |
| 39 | 0.1 g | 85/12 + 7 | EtOH/2 | 1:1:1:1.2 | 72% | |
| 40 | 0.1 g | 85/7 | EtOH/2 | 1:1:1:1.2 | 70% h | |
| 41 | Pip–Agar (1.10 mmol/g) | 25 mol% | 85/7 + 7 | EtOH/2 | 1:1.3:1:1.3 | 80% g |
| 42 | 20 mol% | 85/7 + 7 | EtOH/2 | 1:1.3:1:1.3 | 76% g | |
| 43 | 25 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 70% | |
| 44 | 85/7 + 8 | EtOH/2 | 1:1:1:1.2 | 80% i | ||
| 45 | 17 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 61% | |
| 46 | Ethylenediamine diacetate–Agar (1 mmol/g) | 20 mol% | 85/7 + 7 | EtOH/2 | 1:1:1:1.2 | 65% |
| 47 | Hexamine–Agar (1 mmol/g) | 20 mol% | 85/7 + 16 | EtOH/4 | 1:1:1:1.2 | 44% |
| 48 | DABCO–Agar (1 mmol/g) | 20 mol% | 85/7 + 16 | EtOH/4 | 1:1:1:1.2 | 68% |
 | |||||
|---|---|---|---|---|---|
| Entry a | Amine (1) | Aldehyde (2) | Diketone (3) | Active-Methylene Compound (4) | Yield % (Time, hrs) b [Product Code] |
| 1 | C6H5NH2 (1a) | 4-Me-C6H4CHO (2b) |  (3a) | X = CN (4a) | 71% (7 + 8) [5b] |
| 2 | 1a | 4-Cl-C6H4CHO (2c) | 3a | 4a | 78% (7 + 8) [5c] |
| 3 | 1a | 2,4-Cl2-C6H3CHO (2d) | 3a | 4a | 80% (7 + 8) [5d] |
| 4 | 1a | 4-F-C6H4CHO (2e) | 3a | 4a | 72% (7 + 8) [5e] |
| 5 | 1a |  (2f) | 3a | 4a | 49% (7 + 8) [5f] |
| 6 | 1a | 3-NO2-C6H4CHO (2g) | 3a | 4a | 79% (7 + 8) [5g] |
| 7 | 1a | 4-OCH3-C6H4CHO (2h) | 3a | 4a | 58% (7 + 8) [5h] |
| 8 (Novel) | 1a | 2d | 3a | X = COOMe (4b) | 25% (7 + 8) [5i] |
| 9 (Novel) | 3-Me-C6H4NH2 (1b) | 2g | 3a | 4a | 84% (7 + 8) [5j] |
| 10 (Novel) | 3-Cl-C6H4NH2 (1c) | 2d | 3a | 4a | 83% (7 + 8) [5k] |
| 11 (Novel) | 3-NO2-C6H4NH2 (1d) | 2d | 3a | 4a | 84% (7 + 8) [5l] |
| 12 (Novel) | 4-OCH3-C6H4NH2 (1e) | 3-Me-C6H4CHO (2i) | 3a | 4a | 45% (7 + 8) [5m] |
| 13 (Novel) | BnNH2 (1f) | 2d | 3a | 4a | 10% (7 + 8) [5n] |
| 14 | c-hexylNH2 (1g) | 2g | 3a | 4a | −(7 + 5) c [5o] |
| 15 (Novel) |  (1h) | 4-CN-C6H4CHO (2j) | 3a | 4a | 53% (7 + 8) [5p] |
| 16 (Novel) |  (1i) | 2d | 3a | 4a | 23% (7 + 9) [5q] |
| 17 | 1a | 2a |  (3b) | 4a | −(7 + 2) d [5r] |
| 18 | 1a | 2b |  (3c) | 4a | 65% (7 + 8) [5s] |
| 19 | 1a | 2c | 3c | 4a | 64% (7 + 8) [5t] |
 | ||
|---|---|---|
| Entry a | Aniline R1 (Reactant Code) | Yield % (Time, hrs) b [Product Code] |
| 1 | H (1a) | 78% (7 + 8) [7a] |
| 2 (Novel) | 3-Me- (1b) | 19% (7 + 8) [7b] |
| 3 (Novel) | 3-NO2- (1d) | 79% (7 + 8) [7c] |
| 4 | 4-OCH3- (1e) | 60% (7 + 8) [7d] |
| 5 | 4-Cl- (1j) | 68% (7 + 8) [7e] |
| 6 | 4-Br- (1k) | 72% (7 + 8) [7f] |
| 7 | 4-Cl- (1l) | 20% (7 + 8) [7g] |
| 8 |  (1h) | - [7h] |
| 9 (Novel) | 3-Cl- (1c) | 53% (7 + 8) [7i] |
|
Total mass of waste = mass of starting materials − (mass of product + mass of recovered catalyst) Total mass of reactants = 0.00125 × (140.18 + 93.13 + 106.12) = 0.507 g Total mass of catalyst = 0.28 g (complete mass recovery) Total mass of solvent = 2 mL × 0.789 g/mL = 1.578 g Total mass of product = 0.80 × 369.46 × 0.00125 = 0.369 g Hence, |
| Paper Authors (Year) [Reference] | Conditions | Main Advantages | Main Drawbacks |
|---|---|---|---|
| Singh, S.K. et al. (2012) [16] | 5 mol% DBU MW (140/120 W) EtOH, 80 °C, 3–5 min 92–99%, 23 examples | Short reaction time, excellent yields | Homogeneous catalyst (non-recoverable) No substituted anilines and no isatin used Pre-formed pure enaminone |
| Abaszadeh, M. et al. (2015) [12] | 55 mol% MgO EtOH, 17–43 min 87–92%, 18 examples | Short reaction time, excellent yields | No recycling runs Metallic catalyst No substituted anilines and no isatin used Preformed enaminone |
| Abaszadeh, M. et al. (2015) [13] | 10 mol% ZnO EtOH, 80 °C, 27–55 min 88–92%, 18 examples | Short reaction time, excellent yields Recyclable | Metallic catalyst No substituted anilines and no isatin used Preformed pure enaminone |
| Abaszadeh, M. et al. (2016) [14] | 10 mol% Fe3O4 EtOH, 80 °C, 4–15 min 87–95%, 20 examples | Short reaction time, excellent yields | Metallic catalyst No substituted anilines and no isatin used Preformed pure enaminone No recycling runs |
| Abasadeh, M. et al. (2016) [18] | 30 mol% Crown ether EtOH/H2O, reflux 12–20 min 87–91%, 16 examples | Short reaction time, excellent yields | No substituted anilines and no isatin used No recycling runs Preformed pure enaminone |
| Sanaei-Rad, S. et al. (2021) [19] | MIL-101(Cr) Neat, reflux 10–20 min 92–98%, 14 examples | Short reaction time, excellent yields, recyclable catalyst, one-pot method | Toxic metal containing catalyst (Cr) Hazardous solvents in catalyst preparation No heteroaromatic aldehydes and no isatin used |
| This study | 25 mol% pip–agar EtOH, 7 + 8 hrs 27 examples | Cheap metal-free easy-to-prepare biopolymer catalyst Recyclable Union of MCRs possible Use of substituted anilines and isatin One-pot method | Long reaction times |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosica, G.; Abdilla, R. Novel Biopolymer-Based Catalyst for the Multicomponent Synthesis of N-aryl-4-aryl-Substituted Dihydropyridines Derived from Simple and Complex Anilines. Molecules 2024, 29, 1884. https://doi.org/10.3390/molecules29081884
Bosica G, Abdilla R. Novel Biopolymer-Based Catalyst for the Multicomponent Synthesis of N-aryl-4-aryl-Substituted Dihydropyridines Derived from Simple and Complex Anilines. Molecules. 2024; 29(8):1884. https://doi.org/10.3390/molecules29081884
Chicago/Turabian StyleBosica, Giovanna, and Roderick Abdilla. 2024. "Novel Biopolymer-Based Catalyst for the Multicomponent Synthesis of N-aryl-4-aryl-Substituted Dihydropyridines Derived from Simple and Complex Anilines" Molecules 29, no. 8: 1884. https://doi.org/10.3390/molecules29081884