
Hybrid Peptide-Alkoxyamine Drugs: A Strategy for the Development of a New Family of Antiplasmodial Drugs
 , , , , , , and
, , , , , , and 
Abstract
:1. Introduction
2. Results
2.1. Drug Design and Docking of Peptide-Alkoxyamine Hybrids onto PLMs I-IV
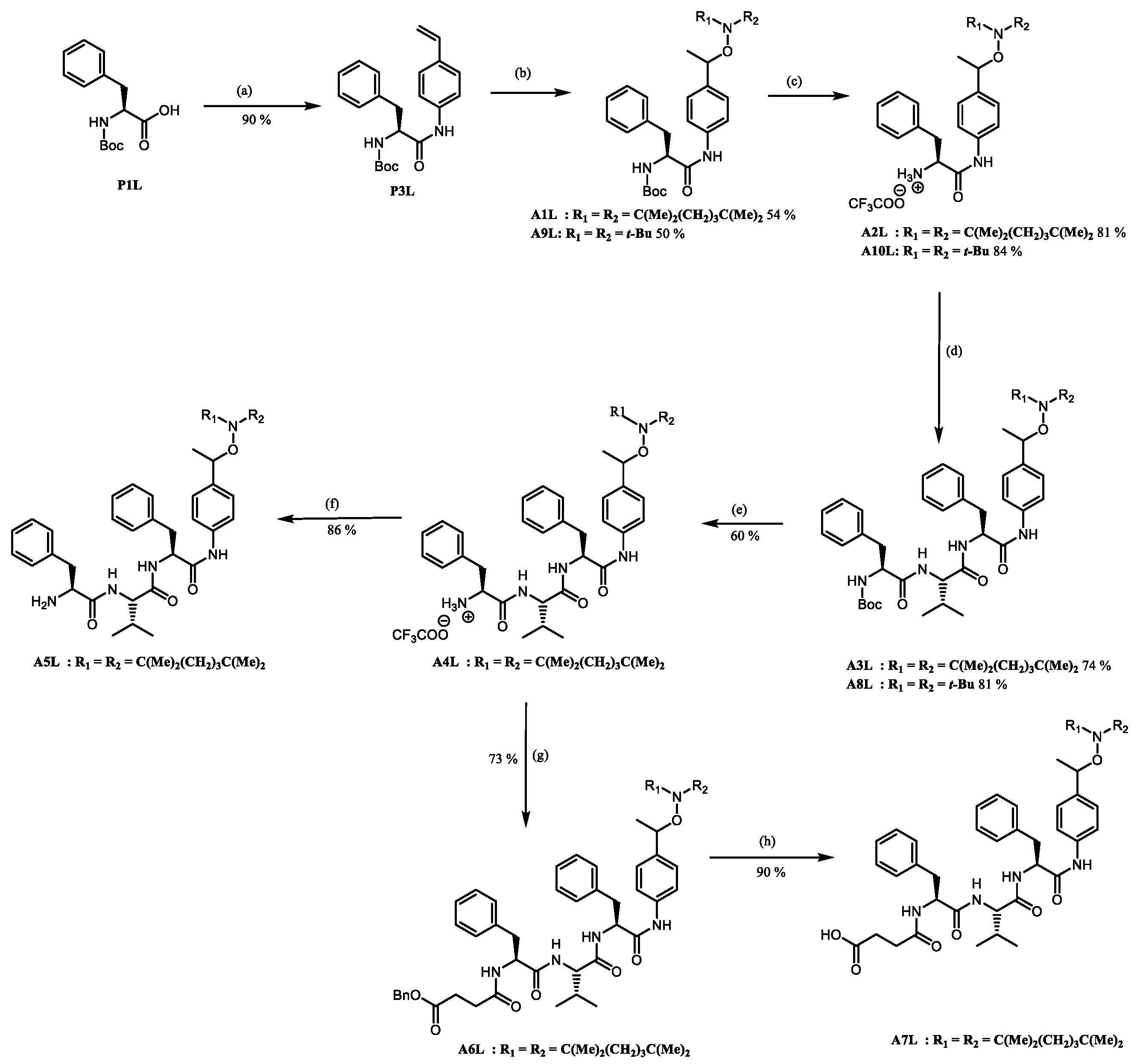
2.2. Preparation of the Peptide-Alkoxyamines Hybrids
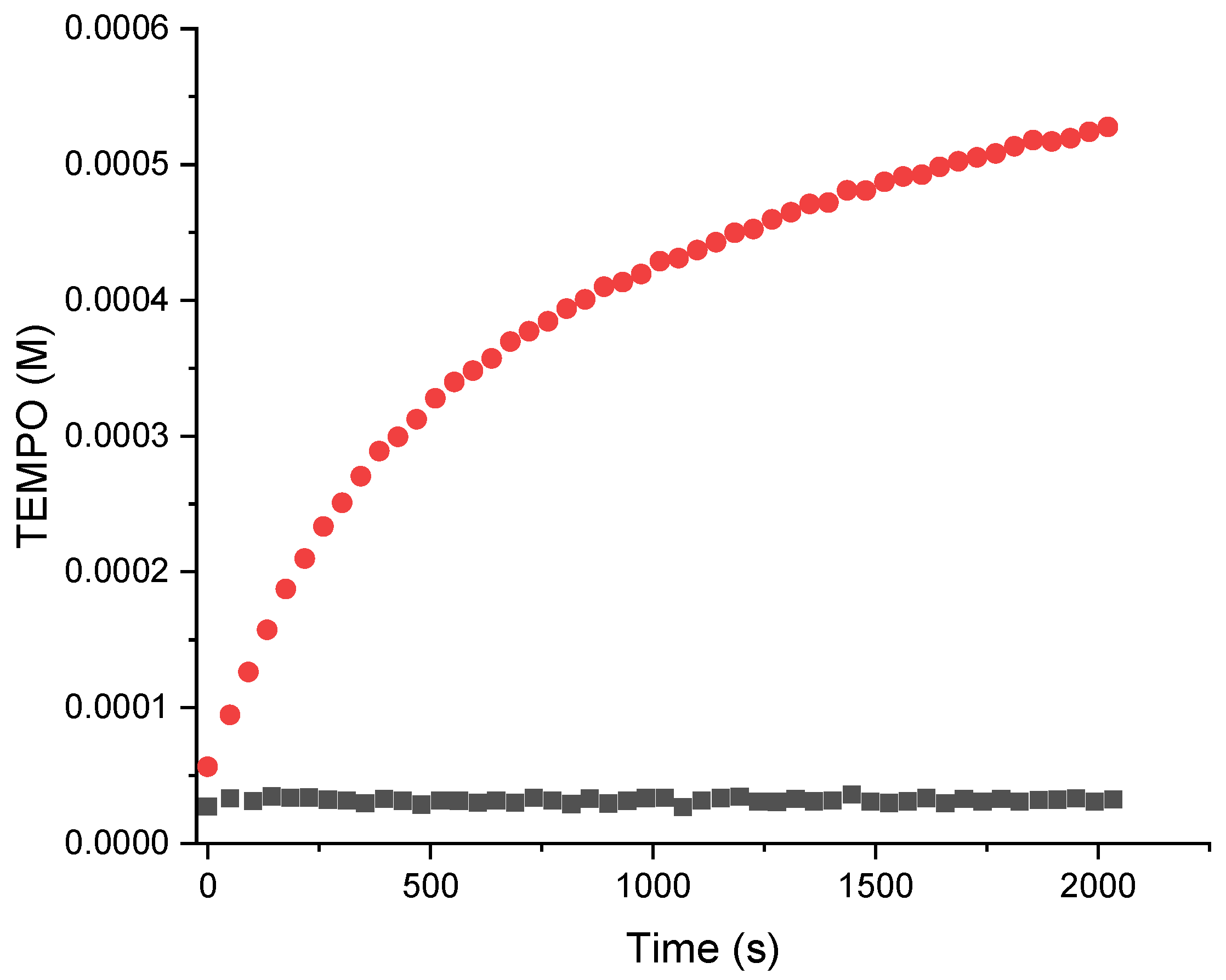
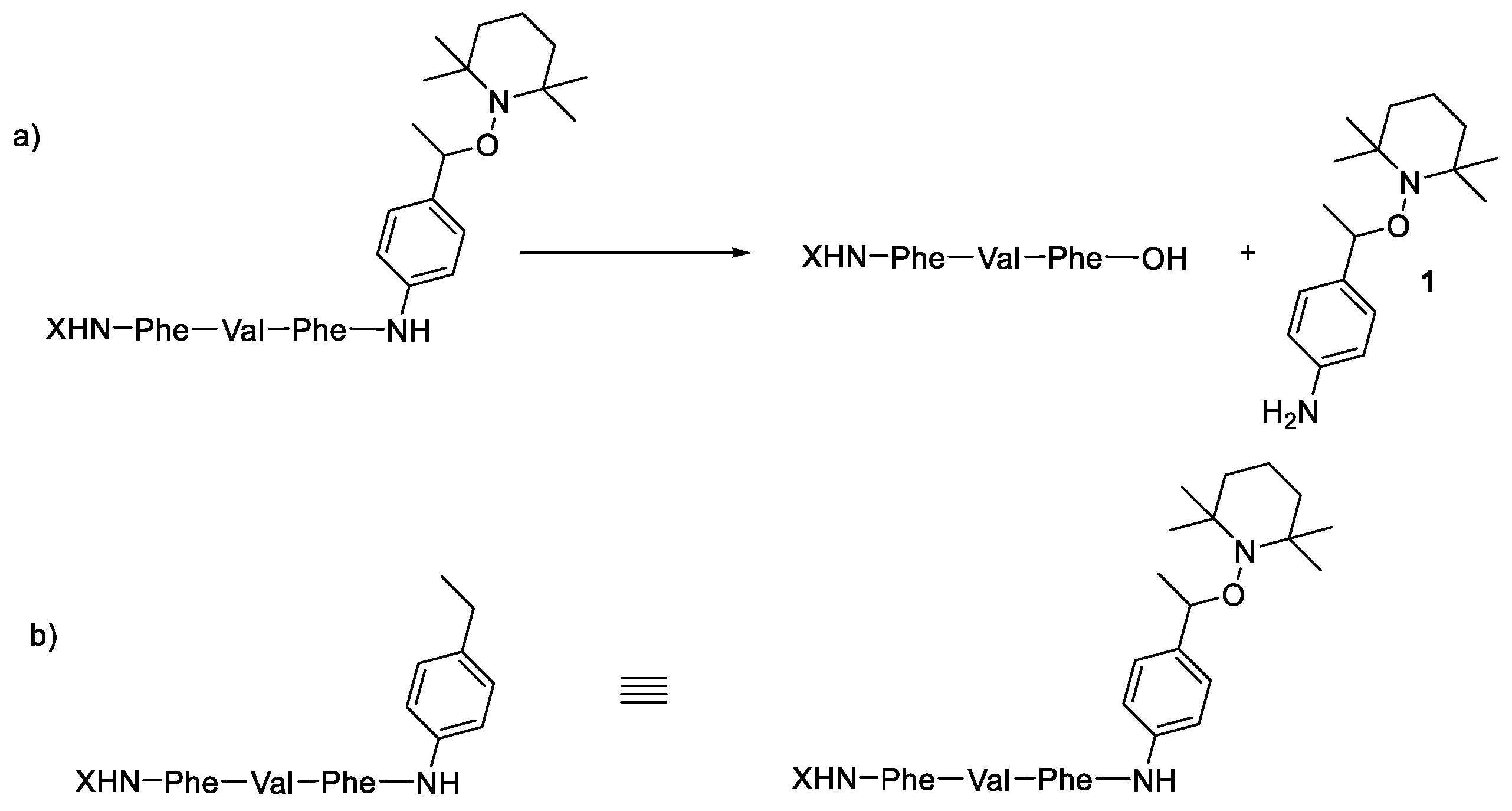
2.3. Kinetic Analysis of Alkoxyamine Bond Homolysis
2.4. Enzymatic Kinetics
2.5. Thermal Homolysis of the Peptide-Alkoxyamine Hybrid A8L in the Presence or Absence of Hemin
2.6. Antimalarial Activity of Peptide-Alkoxyamine Hybrids
2.6.1. Antiproliferative Activity (IC50) against the Artemisinin-Resistant P. falciparum Strain, and Selectivity with Respect to Mammalian Cells (SI)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prodrugs | Structure Relationship e of TEMPO-Based Alkoxyamines | Antiplasmodial Activity a IC50 (µM) | Cytotoxicity b CC50 (µM) | Selectivity Index c CC50/IC50 | |
|---|---|---|---|---|---|
| 1 | A3L |  | 0.30 ± 0.04 | >50 i | >165 |
| 2 | A3D | >50 | >50 | - d | |
| 3 | A4L | >50 | >50 i | - d | |
| 4 | A4D | 2.3 ± 0.3 | 2.6 ± 0.3 | 1.1 | |
| 5 | A5L | >50 | >50 i | - d | |
| 6 | A5D | 2.25 ± 0.17 | 2.20 ± 0.30 | 1.0 | |
| 7 | A6L | 7.2 | >50 i | >6.9 | |
| 8 | A7L | 5.0 ± 3.0 | >50 i | >10 | |
| 9 | 4 f For comparison | H | >>10 e | nd | - d |
| Prodrugs | Structure Relationship e of DBNO-Based Alkoxyamines | Antiplasmodial Activity a IC50 (µM) | Cytotoxicity b CC50 (µM) | Selectivity Index c CC50/IC50 | |
| 10 | A8L |  | 0.27 ± 0.04 | >50 | >185 |
| 11 | A8DL | 0.87 ± 0.04 | >50 | >57 | |
| 12 | A8D | ~50 | >50 | - d | |
| 13 | AW230 | 3.73 ± 0.26 | 9.00 ± 1.20 | 2.4 | |
| 14 | AW231 | 4.48 ± 0.29 | 6.60 ± 0.40 | 1.5 | |
| 15 | ART g | 0.031 ± 0.006 | >50 | >1500 | |
| Peptides and other Comparators | Structure Relationship | Antiplasmodial Activity a IC50 (µM) | Cytotoxicity b CC50 (µM) | Selectivity Index c CC50/IC50 | |
| 16 | P7L |  | >50 | >50 i | - d |
| 17 | P7D | >10 | >50 | - d | |
| 18 | P11L | >10 | >50 i | - d | |
| 19 | P11D | >10 | >50 | - d | |
| 20 | P18L | >5 h | >25 h,i | - d | |
| 21 | P12L | >10 | >50 i | - d | |
| 22 | P10L | >50 | >50 i | - d | |
| Peptides and Other Comparators | Structure Relationship | Antiplasmodial Activity a IC50 (µM) | Cytotoxicity b CC50 (µM) | Selectivity Index c CC50/IC50 | |
| 23 | P21L |  | >50 | >50 i | - d |
| 24 | P21D | >50 | >50 i | - d | |
| 25 | P22L | >10 | >50 i | - d | |
| 26 | P22D | 10.0 ± 0.6 | 11.0 ± 1.6 | 1.1 | |
| 27 | P25L | >10 | >50 i | - d | |
| 28 | P23L | >10 | >50 i | - d | |
| 29 | P24L | >10 | >50 i | - d |
2.6.2. Activity of Peptide-Alkoxyamine Hybrids against Artemisinin-Resistant P. falciparum Parasites at the Quiescent Stage
2.6.3. Moment of Action of Peptide-Alkoxyamine Hybrids Regarding the Erythrocytic Parasite Cell Cycle
3. Discussion
4. Materials and Methods
4.1. Docking
4.2. Enzyme Kinetics of Alkoxyamine Activation
4.3. Thermolysis of A8L in the Absence or in the Presence of Hemin
4.4. Parasite Culture
4.5. Biological Activity of New Compounds
4.6. Quiescent Stage Survival Assay (QSA)
4.7. Microscopic Examination of Parasites upon Drug Exposure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. World Malaria Report 2023; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Ouji, M.; Augereau, J.M.; Paloque, L.; Benoit-Vical, F. Plasmodium falciparum resistance to artemisinin-based combination therapies: A sword of Damocles in the path toward malaria elimination. Parasite 2018, 25, 24. [Google Scholar] [CrossRef] [PubMed]
- Ward, K.E.; Fidock, D.A.; Bridgford, J.L. Plasmodium falciparum resistance to artemisinin-based combination therapies. Curr. Opin. Microbiol. 2022, 69, 102193. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Culleton, R.; Zhang, M.; Ramaprasad, A.; von Seidlein, L.; Zhou, H.; Zhu, G.; Tang, J.; Liu, Y.; Wang, W.; et al. Emergence of Indigenous Artemisinin-Resistant Plasmodium falciparum in Africa. N. Engl. J. Med. 2017, 376, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Uwimana, A.; Legrand, E.; Stokes, B.H.; Ndikumana, J.M.; Warsame, M.; Umulisa, N.; Ngamije, D.; Munyaneza, T.; Mazarati, J.B.; Munguti, K.; et al. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat. Med. 2020, 26, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Reyser, T.; To, T.H.; Egwu, C.; Paloque, L.; Nguyen, M.; Hamouy, A.; Stigliani, J.-L.; Bijani, C.; Augereau, J.-M.; Joly, J.-P.; et al. Alkoxyamines Designed as Potential Drugs against Plasmodium and Schistosoma Parasites. Molecules 2020, 25, 3838. [Google Scholar] [CrossRef] [PubMed]
- Seren, S.; Joly, J.-P.; Voisin, P.; Bouchaud, V.; Audran, G.; Marque, S.R.A.; Mellet, P. Neutrophil Elastase-Activatable Prodrugs Based on an Alkoxyamine Platform to Deliver Alkyl Radicals Cytotoxic to Tumor Cells. J. Med. Chem. 2022, 65, 9253–9266. [Google Scholar] [CrossRef]
- Pagola, S.; Stephens, P.; Bohle, D.; Kosar, A.; Madsen, S. The structure of malaria pigment beta-haematin. Nature 2000, 404, 307–310. [Google Scholar] [CrossRef]
- Meunier, B.; Robert, A. Heme as trigger and target for trioxane-containing antimalarial drugs. Acc. Chem. Res. 2010, 43, 1444–1451. [Google Scholar] [CrossRef]
- Egwu, C.O.; Tsamesidis, I.; Perio, P.; Augereau, J.M.; Benoit-Vical, F.; Reybier, K. Superoxide: A major role in the mechanism of action of essential antimalarial drugs. Free Radic. Biol. Med. 2021, 167, 271–275. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, H.; Gerhard, G.S. Heme synthesis increases artemisinin-induced radical formation and cytotoxicity that can be suppressed by superoxide scavengers. Chem. Biol. Interact. 2010, 186, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Nasamu, A.S.; Polino, A.J.; Istvan, E.S.; Goldberg, D.E. Malaria parasite plasmepsins: More than just plain old degradative pepsins. J. Biol. Chem. 2020, 295, 8425–8441. [Google Scholar] [CrossRef]
- Lisauskaite, M.; Nixon, G.L.; Woodley, C.M.; Berry, N.G.; Coninckx, A.; Qie, L.C.; Leung, S.C.; Taramelli, D.; Basilico, N.; Parapini, S.; et al. Design, synthesis and modelling of photoreactive chemical probes for investigating target engagement of plasmepsin IX and X in Plasmodium falciparum. RSC Chem. Biol. 2024, 5, 19–29. [Google Scholar] [CrossRef]
- Lowe, M.A.; Cardenas, A.; Valentin, J.P.; Zhu, Z.; Abendroth, J.; Castro, J.L.; Class, R.; Delaunois, A.; Fleurance, R.; Gerets, H.; et al. Discovery and Characterization of Potent, Efficacious, Orally Available Antimalarial Plasmepsin X Inhibitors and Preclinical Safety Assessment of UCB7362. J. Med. Chem. 2022, 65, 14121–14143. [Google Scholar] [CrossRef] [PubMed]
- Cheuka, P.M.; Dziwornu, G.; Okombo, J.; Chibale, K. Plasmepsin Inhibitors in Antimalarial Drug Discovery: Medicinal Chemistry and Target Validation (2000 to Present). J. Med. Chem. 2020, 63, 4445–4467. [Google Scholar] [CrossRef] [PubMed]
- Tyas, L.; Gluzman, I.; Moon, R.P.; Rupp, K.; Westling, J.; Ridley, R.G.; Kay, J.; Goldberg, D.E.; Berry, C. Naturally-occurring and recombinant forms of the aspartic proteinases plasmepsins I and II from the human malaria parasite Plasmodium falciparum. FEBS Lett. 1999, 454, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Dechy-Cabaret, O.; Benoit-Vical, F.; Robert, A.; Meunier, B. Preparation and antimalarial activities of “trioxaquines”, new modular molecules with a trioxane skeleton linked to a 4-aminoquinoline. Chembiochem 2000, 1, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Ouji, M.; Nguyen, M.; Mustiere, R.; Jimenez, T.; Augereau, J.M.; Benoit-Vical, F.; Deraeve, C. Novel molecule combinations and corresponding hybrids targeting artemisinin-resistant Plasmodium falciparum parasites. Bioorg. Med. Chem. Lett. 2021, 39, 127884. [Google Scholar] [CrossRef]
- Embo-Ibouanga, A.W.; Nguyen, M.; Joly, J.-P.; Coustets, M.; Augereau, J.-M.; Paloque, L.; Vanthuyne, N.; Bikanga, R.; Robert, A.; Benoit-Vical, F.; et al. Peptide-Alkoxyamine drugs. An Innovative Approach to Fight Against Schistosomiasis: “Digging their Graves with their Forks”. Pathogens, 2024; in revision. [Google Scholar]
- Marque, S.; Le Mercier, C.; Tordo, P.; Fischer, H. Factors Influencing the C−O−Bond Homolysis of Trialkylhydroxylamines. Macromolecules 2000, 33, 4403–4410. [Google Scholar] [CrossRef]
- Traylor, T.G.; Ciccone, J.P. Mechanism of reactions of hydrogen peroxide and hydroperoxides with iron(III) porphyrins. Effects of hydroperoxide structure on kinetics. J. Am. Chem. Soc. 1989, 111, 8413–8420. [Google Scholar] [CrossRef]
- Meunier, B. Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- Siqueira-Neto, J.L.; Wicht, K.J.; Chibale, K.; Burrows, J.N.; Fidock, D.A.; Winzeler, E.A. Antimalarial drug discovery: Progress and approaches. Nat. Rev. Drug Discov. 2023, 22, 807–826. [Google Scholar] [CrossRef]
- Witkowski, B.; Lelievre, J.; Barragan, M.J.; Laurent, V.; Su, X.Z.; Berry, A.; Benoit-Vical, F. Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob. Agents Chemother. 2010, 54, 1872–1877. [Google Scholar] [CrossRef]
- Smilkstein, M.; Sriwilaijaroen, N.; Kelly, J.X.; Wilairat, P.; Riscoe, M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [Google Scholar] [CrossRef] [PubMed]
- Paloque, L.; Ramadani, A.P.; Mercereau-Puijalon, O.; Augereau, J.M.; Benoit-Vical, F. Plasmodium falciparum: Multifaceted resistance to artemisinins. Malar. J. 2016, 15, 149. [Google Scholar] [CrossRef] [PubMed]
- Reyser, T.; Paloque, L.; Ouji, M.; Nguyen, M.; Menard, S.; Witkowski, B.; Augereau, J.M.; Benoit-Vical, F. Identification of compounds active against quiescent artemisinin-resistant Plasmodium falciparum parasites via the quiescent-stage survival assay (QSA). J. Antimicrob. Chemother. 2020, 75, 2826–2834. [Google Scholar] [CrossRef]
- Nixon, G.L.; Moss, D.M.; Shone, A.E.; Lalloo, D.G.; Fisher, N.; O’Neill, P.M.; Ward, S.A.; Biagini, G.A. Antimalarial pharmacology and therapeutics of atovaquone. J. Antimicrob. Chemother. 2013, 68, 977–985. [Google Scholar] [CrossRef]
- Boissier, J.; Cosledan, F.; Robert, A.; Meunier, B. In vitro activities of trioxaquines against Schistosoma mansoni. Antimicrob. Agents Chemother. 2009, 53, 4903–4906. [Google Scholar] [CrossRef]
- Portela, J.; Boissier, J.; Gourbal, B.; Pradines, V.; Colliere, V.; Cosledan, F.; Meunier, B.; Robert, A. Antischistosomal activity of trioxaquines: In vivo efficacy and mechanism of action on Schistosoma mansoni. PLoS Negl. Trop. Dis. 2012, 6, e1474. [Google Scholar] [CrossRef] [PubMed]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]












| Peptide-Alkoxyamine Hybrids | T (°C) a | kd (10−4 s−1) a,b,c | Ea (kJ/mol) b,d | T (°C) e | kd (10−4 s−1) b,c,e | Ea (kJ/mol) b,d,e |
|---|---|---|---|---|---|---|
| A8L | 97.7 | 22.1 | 120.9 | 75.0 | 1.7 | 120.9 |
| A8DL | 91.3 | 19.0 | 119.3 | 83 | 10 | 118.4 |
| A9L | 107.9 | 72.3 | 120.5 | 81 | 6.7 | 118.9 |
| A8D | 91.1 | 10 | 121.1 | 90.2 | 22.0 | 118.5 |
| A9D | 89 | 9.7 | 120.8 | 85.2 | 11.0 | 118.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Embo-Ibouanga, A.W.; Nguyen, M.; Paloque, L.; Coustets, M.; Joly, J.-P.; Augereau, J.-M.; Vanthuyne, N.; Bikanga, R.; Coquin, N.; Robert, A.; et al. Hybrid Peptide-Alkoxyamine Drugs: A Strategy for the Development of a New Family of Antiplasmodial Drugs. Molecules 2024, 29, 1397. https://doi.org/10.3390/molecules29061397
Embo-Ibouanga AW, Nguyen M, Paloque L, Coustets M, Joly J-P, Augereau J-M, Vanthuyne N, Bikanga R, Coquin N, Robert A, et al. Hybrid Peptide-Alkoxyamine Drugs: A Strategy for the Development of a New Family of Antiplasmodial Drugs. Molecules. 2024; 29(6):1397. https://doi.org/10.3390/molecules29061397
Chicago/Turabian StyleEmbo-Ibouanga, Ange W., Michel Nguyen, Lucie Paloque, Mathilde Coustets, Jean-Patrick Joly, Jean-Michel Augereau, Nicolas Vanthuyne, Raphaël Bikanga, Naomie Coquin, Anne Robert, and et al. 2024. "Hybrid Peptide-Alkoxyamine Drugs: A Strategy for the Development of a New Family of Antiplasmodial Drugs" Molecules 29, no. 6: 1397. https://doi.org/10.3390/molecules29061397