
Magnetic Anisotropy of Homo- and Heteronuclear Terbium(III) and Dysprosium(III) Trisphthalocyaninates Derived from Paramagnetic 1H-NMR Investigation

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Synthesis and Characterization
2.2. Analysis of Lanthanide-Induced Shifts in 1H-NMR Spectra
2.2.1. Analysis of Lanthanide-Induced Shifts in 1H-NMR Spectra of Heteronuclear Complexes [B4]Dy[B4]Y[C4] and [B4]Y[B4]Dy[C4]
2.2.2. Analysis of Lanthanide-Induced Shifts in 1H-NMR Spectra of the Homonuclear Complex [B4]Dy[B4]Dy[C4]
2.2.3. Analysis of Lanthanide-Induced Shifts in 1H-NMR Spectra of Heteronuclear Complexes [B4]Dy[B4]Tb[C4] and [B4]Tb[B4]Dy[C4]
3. Discussion
- The trisphthalocyanine scaffold affords the synthesis of heteronuclear complexes with a precise arrangement of rare-earth ions due to its thermodynamical and kinetic stability. Complexes with different combinations of paramagnetic lanthanides can be efficiently obtained, which provides the basis for further investigation of intramolecular f-f interactions and the elaboration of molecular magnetic materials. In the present work, the combinations of two strongly paramagnetic Tb3+ and Dy3+ ions were used to obtain isomeric heteronuclear complexes, but, obviously, other combinations of middle and late lanthanides can also be used in this type of chemistry to obtain complexes with the required number of unpaired f-electrons.
- The comprehensive 1H-NMR spectroscopic characterization of strongly paramagnetic complexes is based on the appropriate structural model; therefore, this work provides algorithms for dealing with the spectra of complexes containing either one or two lanthanide ions, which are not necessarily equivalent. In this regard, the results presented demonstrate that the application of paramagnetic 1H-NMR spectroscopy should not be limited to routine identification but can be used to extract the magnetic properties of lanthanide ions [53]. In this context, our report follows the strategies applied by Enders and Yamashita, where Tb(III) and Dy(III) sandwich phthalocyaninates were comprehensively studied using NMR spectroscopy [26,35,54,57,58], and the influence of electronic and structural effects on their magnetic properties, especially , was revealed. In summary, it is expected that further magnetochemical studies of the newly synthesized lanthanide phthalocyaninates will provide more correlations between the term and the energetic properties of slow magnetic relaxation.
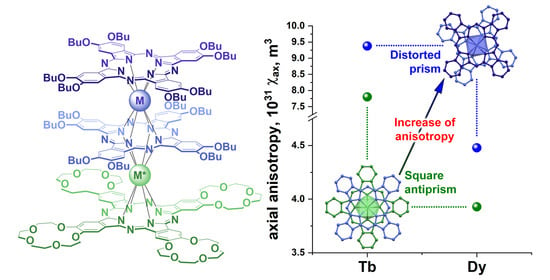
- The addition of controllable conformational flexibility gives one more degree of freedom to control the magnetic properties of sandwich lanthanide complexes. The previously discovered correlations between the symmetry of the coordination polyhedron of the Tb3+ ion and its magnetic properties are also valid for the Dy3+ ion, and the effect of the conformational switching can be studied for other lanthanides to find the capabilities and limitations of theoretical models.
- Importantly, our study evidences that the values for Dy3+ are nearly twice smaller than those for Tb3+; however, this observation does not match the expectations from Bleaney’s theory, where the largest anisotropy in the lanthanide series is expected for dysprosium [43]. Moreover, it contradicts the results of theoretical modeling obtained by Mironov et al. [32] for various polyhedra of lanthanide complexes, where the most pronounced influence of the surrounding coordination was anticipated for dysprosium complexes. The reason for this discrepancy may be a violation of the theory’s basic assumption that the thermal energy is larger than the ligand field splitting; thus, further theoretical modeling using ab initio methods might be particularly helpful [59,60].
4. Materials and Methods
4.1. Materials
4.2. Methods
4.3. Synthesis and Characterization of the Triple-Decker Complexes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.-Y.; Kaizu, Y. Lanthanide Double-Decker Complexes Functioning as Magnets at the Single-Molecular Level. J. Am. Chem. Soc. 2003, 125, 8694–8695. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Isshiki, H.; Komeda, T.; Yamashita, M. Multiple-Decker Phthalocyaninato Tb(III) Single-Molecule Magnets and Y(III) Complexes for next Generation Devices. Coord. Chem. Rev. 2011, 255, 2124–2148. [Google Scholar] [CrossRef]
- Wang, H.; Wang, B.W.; Bian, Y.; Gao, S.; Jiang, J. Single-Molecule Magnetism of Tetrapyrrole Lanthanide Compounds with Sandwich Multiple-Decker Structures. Coord. Chem. Rev. 2016, 306, 195–216. [Google Scholar] [CrossRef]
- Martynov, A.G.; Horii, Y.; Katoh, K.; Bian, Y.; Jiang, J.; Yamashita, M.; Gorbunova, Y.G. Rare-Earth Based Tetrapyrrolic Sandwiches: Chemistry, Materials and Applications. Chem. Soc. Rev. 2022, 51, 9262–9339. [Google Scholar] [CrossRef] [PubMed]
- Gross, T.; Chevalier, F.; Lindsey, J.S. Investigation of Rational Syntheses of Heteroleptic Porphyrinic Lanthanide (Europium, Cerium) Triple-Decker Sandwich Complexes. Inorg. Chem. 2001, 40, 4762–4774. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ng, D.K.P. A Decade Journey in the Chemistry of Sandwich-Type Tetrapyrrolato−Rare Earth Complexes. Acc. Chem. Res. 2009, 42, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.L.; Xie, C.; Lo, W.S.; Bünzli, J.C.G.; Wong, W.K.; Wong, K.L. Lanthanide-Tetrapyrrole Complexes: Synthesis, Redox Chemistry, Photophysical Properties, and Photonic Applications. Chem. Soc. Rev. 2021, 50, 12189–12257. [Google Scholar] [CrossRef]
- Pushkarev, V.E.; Tomilova, L.G.; Nemykin, V.N. Historic Overview and New Developments in Synthetic Methods for Preparation of the Rare-Earth Tetrapyrrolic Complexes. Coord. Chem. Rev. 2016, 319, 110–179. [Google Scholar] [CrossRef]
- Ishikawa, N.; Iino, T.; Kaizu, Y. Interaction between F-Electronic Systems in Dinuclear Lanthanide Complexes with Phthalocyanines. J. Am. Chem. Soc. 2002, 124, 11440–11447. [Google Scholar] [CrossRef]
- Ishikawa, N.; Iino, T.; Kaizu, Y. Determination of Ligand-Field Parameters and f-Electronic Structures of Hetero-Dinuclear Phthalocyanine Complexes with a Diamagnetic Yttrium(III) and a Paramagnetic Trivalent Lanthanide Ion. J. Phys. Chem. A 2002, 106, 9543–9550. [Google Scholar] [CrossRef]
- Katoh, K.; Aizawa, Y.; Morita, T.; Breedlove, B.K.; Yamashita, M. Elucidation of Dual Magnetic Relaxation Processes in Dinuclear Dysprosium(III) Phthalocyaninato Triple-Decker Single-Molecule Magnets Depending on the Octacoordination Geometry. Chem. Eur. J. 2017, 23, 15377–15386. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Asano, R.; Miura, A.; Horii, Y.; Morita, T.; Breedlove, B.K.; Yamashita, M. Effect of f-f Interactions on Quantum Tunnelling of the Magnetization: Mono- and Dinuclear Dy(III) Phthalocyaninato Triple-Decker Single-Molecule Magnets with the Same Octacoordination Environment. Dalton Trans. 2014, 43, 7716–7725. [Google Scholar] [CrossRef] [PubMed]
- Horii, Y.; Katoh, K.; Cosquer, G.; Breedlove, B.K.; Yamashita, M. Weak DyIII-DyIII Interactions in DyIII-Phthalocyaninato Multiple-Decker Single-Molecule Magnets Effectively Suppress Magnetic Relaxation. Inorg. Chem. 2016, 55, 11782–11790. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Otsuka, S.; Kaizu, Y. The Effect of the f-f Interaction on the Dynamic Magnetism of a Coupled 4f8 System in a Dinuclear Terbium Complex with Phthalocyanines. Angew. Chem. Int. Ed. 2005, 44, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, R.J.; Polovkova, M.A.; Martynov, A.G.; Gorbunova, Y.G.; Murugesu, M. Impact of the Coordination Environment on the Magnetic Properties of Single-Molecule Magnets Based on Homo- and Hetero-Dinuclear Terbium(III) Heteroleptic Tris(Crownphthalocyaninate). Dalton Trans. 2016, 45, 9320–9327. [Google Scholar] [CrossRef]
- Pushkarev, V.E.; Shulishov, E.V.; Tomilov, Y.V.; Tomilova, L.G. The Development of Highly Selective Approaches to Sandwich-Type Heteroleptic Double- and Triple-Decker Lutetium(III) and Europium(III) Phthalocyanine Complexes. Tetrahedron Lett. 2007, 48, 5269–5273. [Google Scholar] [CrossRef]
- Nefedova, I.V.; Gorbunova, Y.G.; Sakharov, S.G.G.; Tsivadze, A.Y. Synthesis and Structure of Homo- and Heteronuclear Rare Earth Element Complexes with Tetra-15-Crown-5-Phthalocyanine. Mendeleev Commun. 2006, 16, 67–69. [Google Scholar] [CrossRef]
- Zhu, P.; Pan, N.; Li, R.; Dou, J.; Zhang, Y.; Cheng, D.Y.Y.; Wang, D.; Ng, D.K.P.; Jiang, J. Electron-Donating Alkoxy-Group-Driven Synthesis of Heteroleptic Tris(Phthalocyaninato) Lanthanide(III) Triple-Deckers with Symmetrical Molecular Structure. Chem. Eur. J. 2005, 11, 1425–1432. [Google Scholar] [CrossRef]
- Polovkova, M.A.; Martynov, A.G.; Birin, K.P.; Nefedov, S.E.; Gorbunova, Y.G.; Tsivadze, A.Y. Determination of the Structural Parameters of Heteronuclear (Phthalocyaninato)Bis(Crownphthalocyaninato)Lanthanide(III) Triple-Deckers in Solution by Simultaneous Analysis of NMR and Single-Crystal X-Ray Data. Inorg. Chem. 2016, 55, 9258–9269. [Google Scholar] [CrossRef]
- Lan, Y.; Klyatskaya, S.; Ruben, M.; Fuhr, O.; Wernsdorfer, W.; Candini, A.; Corradini, V.; Lodi Rizzini, A.; del Pennino, U.; Troiani, F.; et al. Magnetic Interplay between Two Different Lanthanides in a Tris-Phthalocyaninato Complex: A Viable Synthetic Route and Detailed Investigation in the Bulk and on the Surface. J. Mater. Chem. C 2015, 3, 9794–9801. [Google Scholar] [CrossRef]
- Katoh, K.; Yasuda, N.; Damjanović, M.; Wernsdorfer, W.; Breedlove, B.K.; Yamashita, M. Manipulation of the Coordination Geometry along the C4 Rotation Axis in a Dinuclear Tb3+ Triple-Decker Complex via a Supramolecular Approach. Chem. Eur. J. 2020, 26, 4805–4815. [Google Scholar] [CrossRef]
- Martynov, A.G.; Polovkova, M.A.; Berezhnoy, G.S.; Sinelshchikova, A.A.; Khrustalev, V.N.; Birin, K.P.; Kirakosyan, G.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Heteroleptic Crown-Substituted Tris(Phthalocyaninates) as Dynamic Supramolecular Scaffolds with Switchable Rotational States and Tunable Magnetic Properties. Inorg. Chem. 2021, 60, 9110–9121. [Google Scholar] [CrossRef] [PubMed]
- Martynov, A.G.; Sinelshchikova, A.A.; Dorovatovskii, P.V.; Polovkova, M.A.; Ovchenkova, A.E.; Birin, K.P.; Kirakosyan, G.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Solvation-Induced Conformational Switching of Trisphthalocyanates for Control of Their Magnetic Properties. Inorg. Chem. 2023, 62, 10329–10342. [Google Scholar] [CrossRef]
- Martynov, A.G.; Birin, K.P.; Kirakosyan, G.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Site-Selective Solvation-Induced Conformational Switching of Heteroleptic Heteronuclear Tb(III) and Y(III) Trisphthalocyaninates for the Control of Their Magnetic Anisotropy. Molecules 2023, 28, 4474. [Google Scholar] [CrossRef] [PubMed]
- Martynov, A.G.; Polovkova, M.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Redox-Triggered Switching of Conformational State in Triple-Decker Lanthanide Phthalocyaninates. Molecules 2022, 27, 6498. [Google Scholar] [CrossRef] [PubMed]
- Horii, Y.; Damjanović, M.; Ajayakumar, M.R.; Katoh, K.; Kitagawa, Y.; Chibotaru, L.; Ungur, L.; Mas-Torrent, M.; Wernsdorfer, W.; Breedlove, B.K.; et al. Highly Oxidized States of Phthalocyaninato Terbium(III) Multiple-Decker Complexes Showing Structural Deformations, Biradical Properties and Decreases in Magnetic Anisotropy. Chem. Eur. J. 2020, 26, 8621–8630. [Google Scholar] [CrossRef] [PubMed]
- Mironov, V.S.; Galyametdinov, Y.G.; Ceulemans, A.; Görller-Walrand, C.; Binnemans, K. Influence of Crystal-Field Perturbations on the Room-Temperature Magnetic Anisotropy of Lanthanide Complexes. Chem. Phys. Lett. 2001, 345, 132–140. [Google Scholar] [CrossRef]
- Sakaue, S.; Fuyuhiro, A.; Fukuda, T.; Ishikawa, N. Dinuclear Single-Molecule Magnets with Porphyrin–Phthalocyanine Mixed Triple-Decker Ligand Systems Giving SAP and SP Coordination Polyhedra. Chem. Commun. 2012, 48, 5337. [Google Scholar] [CrossRef]
- Katoh, K.; Breedlove, B.K.; Yamashita, M. Symmetry of Octa-Coordination Environment Has a Substantial Influence on Dinuclear TbIII Triple-Decker Single-Molecule Magnets. Chem. Sci. 2016, 7, 4329–4340. [Google Scholar] [CrossRef]
- Görller-Walran, C.; Binnemans, K. Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K.A., Eyring, L., Eds.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 23, ISBN 9780444825070. [Google Scholar]
- Hiller, M.; Krieg, S.; Ishikawa, N.; Enders, M. Ligand-Field Energy Splitting in Lanthanide-Based Single-Molecule Magnets by NMR Spectroscopy. Inorg. Chem. 2017, 56, 15285–15294. [Google Scholar] [CrossRef] [PubMed]
- Mironov, V.S.; Galyametdinov, Y.G.; Ceulemans, A.; Görller-Walrand, C.; Binnemans, K. Room-Temperature Magnetic Anisotropy of Lanthanide Complexes: A Model Study for Various Coordination Polyhedra. J. Chem. Phys. 2002, 116, 4673–4685. [Google Scholar] [CrossRef]
- Blackburn, O.A.; Edkins, R.M.; Faulkner, S.; Kenwright, A.M.; Parker, D.; Rogers, N.J.; Shuvaev, S. Electromagnetic Susceptibility Anisotropy and Its Importance for Paramagnetic NMR and Optical Spectroscopy in Lanthanide Coordination Chemistry. Dalton Trans. 2016, 45, 6782–6800. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.; Suturina, E.A.; Kuprov, I.; Chilton, N.F. How the Ligand Field in Lanthanide Coordination Complexes Determines Magnetic Susceptibility Anisotropy, Paramagnetic NMR Shift, and Relaxation Behavior. Acc. Chem. Res. 2020, 53, 1520–1534. [Google Scholar] [CrossRef]
- Morita, T.; Damjanović, M.; Katoh, K.; Kitagawa, Y.; Yasuda, N.; Lan, Y.; Wernsdorfer, W.; Breedlove, B.K.; Enders, M.; Yamashita, M. Comparison of the Magnetic Anisotropy and Spin Relaxation Phenomenon of Dinuclear Terbium(III) Phthalocyaninato Single-Molecule Magnets Using the Geometric Spin Arrangement. J. Am. Chem. Soc. 2018, 140, 2995–3007. [Google Scholar] [CrossRef]
- Gigli, L.; Di Grande, S.; Ravera, E.; Parigi, G.; Luchinat, C. Nmr for Single Ion Magnets. Magnetochemistry 2021, 7, 96. [Google Scholar] [CrossRef]
- Ishikawa, N.; Kaizu, Y. Excited States of the Lutetium Phthalocyanine Trimer: Semiempirical Molecular Orbital and Localized Orbital Study. J. Phys. Chem. 1996, 100, 8722–8730. [Google Scholar] [CrossRef]
- Rousseau, R.; Aroca, R.; Rodríguez-Méndez, M.L. Extended Hückel Molecular Orbital Model for Lanthanide Bisphthalocyanine Complexes. J. Mol. Struct. 1995, 356, 49–62. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Cryst. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Utochnikova, V.V. The Use of Luminescent Spectroscopy to Obtain Information about the Composition and the Structure of Lanthanide Coordination Compounds. Coord. Chem. Rev. 2019, 398, 113006. [Google Scholar] [CrossRef]
- Bulach, V.; Sguerra, F.; Hosseini, M.W. Porphyrin Lanthanide Complexes for NIR Emission. Coord. Chem. Rev. 2012, 256, 1468–1478. [Google Scholar] [CrossRef]
- Golding, R.; Halton, M. A Theoretical Study of the 14N and 17O N.M.R. Shifts in Lanthanide Complexes. Aust. J. Chem. 1972, 25, 2577–2581. [Google Scholar] [CrossRef]
- Bleaney, B. Nuclear Magnetic Resonance Shifts in Solution Due to Lanthanide Ions. J. Magn. Reson. 1972, 8, 91–100. [Google Scholar] [CrossRef]
- Gorbunova, Y.G.; Martynov, A.G.; Birin, K.P.; Tsivadze, A.Y. NMR Spectroscopy—A Versatile Tool for Studying the Structure and Magnetic Properties of Paramagnetic Lanthanide Complexes in Solutions (Review). Russ. J. Inorg. Chem. 2021, 66, 202–216. [Google Scholar] [CrossRef]
- Vogel, R.; Müntener, T.; Häussinger, D. Intrinsic Anisotropy Parameters of a Series of Lanthanoid Complexes Deliver New Insights into the Structure-Magnetism Relationship. Chem 2021, 7, 3144–3156. [Google Scholar] [CrossRef]
- Piguet, C.; Geraldes, C.F.G.C. Paramagnetic NMR Lanthanide Induced Shifts for Extracting Solution Structures. In Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K.A., Bünzli, J.-C.G., Pecharsky, V.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 33, pp. 353–463. ISBN 9780444513236. [Google Scholar]
- Zapolotsky, E.N.; Qu, Y.; Babailov, S.P. Lanthanide Complexes with Polyaminopolycarboxylates as Prospective NMR/MRI Diagnostic Probes: Peculiarities of Molecular Structure, Dynamics and Paramagnetic Properties. J. Incl. Phenom. Macrocycl. Chem. 2021, 102, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Ravera, E.; Gigli, L.; Fiorucci, L.; Luchinat, C.; Parigi, G. The Evolution of Paramagnetic NMR as a Tool in Structural Biology. Phys. Chem. Chem. Phys. 2022, 24, 17397–17416. [Google Scholar] [CrossRef] [PubMed]
- Miao, Q.; Nitsche, C.; Orton, H.; Overhand, M.; Otting, G.; Ubbink, M. Paramagnetic Chemical Probes for Studying Biological Macromolecules. Chem. Rev. 2022, 122, 9571–9642. [Google Scholar] [CrossRef]
- Santria, A.; Fuyuhiro, A.; Fukuda, T.; Ishikawa, N. Determination of Ligand Field Splitting in Lanthanide(III) Monoporphyrinato Complexes. Dalton Trans. 2019, 48, 7685–7692. [Google Scholar] [CrossRef]
- Hiller, M.; Sittel, T.; Wadepohl, H.; Enders, M. A New Class of Lanthanide Complexes with Three Ligand Centered Radicals: NMR Evaluation of Ligand Field Energy Splitting and Magnetic Coupling. Chem. Eur. J. 2019, 25, 10668–10677. [Google Scholar] [CrossRef]
- Hiller, M.; Maier, M.; Wadepohl, H.; Enders, M. Paramagnetic NMR Analysis of Substituted Biscyclooctatetraene Lanthanide Complexes. Organometallics 2016, 35, 1916–1922. [Google Scholar] [CrossRef]
- Ince, R.; Doudouh, A.; Claiser, N.; Furet, É.; Guizouarn, T.; Le Pollès, L.; Kervern, G. Determining Local Magnetic Susceptibility Tensors in Paramagnetic Lanthanide Crystalline Powders from Solid-State NMR Chemical Shift Anisotropies. J. Phys. Chem. A 2023, 127, 1547–1554. [Google Scholar] [CrossRef]
- Damjanovic, M.; Katoh, K.; Yamashita, M.; Enders, M. Combined NMR Analysis of Huge Residual Dipolar Couplings and Pseudocontact Shifts in Terbium(III)-Phthalocyaninato Single Molecule Magnets. J. Am. Chem. Soc. 2013, 135, 14349–14358. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.P.; Jiang, J. Distinction between Light and Heavy Lanthanide(III) Ions Based on the 1H NMR Spectra of Heteroleptic Triple-Decker Phthalocyaninato Sandwich Complexes. J. Phys. Chem. A 2001, 105, 7525–7533. [Google Scholar] [CrossRef]
- Ishikawa, N.; Iino, T.; Kaizu, Y. Study of 1H NMR Spectra of Dinuclear Complexes of Heavy Lanthanides with Phthalocyanines Based on Separation of the Effects of Two Paramagnetic Centers. J. Phys. Chem. A 2003, 107, 7879–7884. [Google Scholar] [CrossRef]
- Damjanović, M.; Horie, Y.; Morita, T.; Horii, Y.; Katoh, K.; Yamashita, M.; Enders, M. α-Substituted Bis(Octabutoxyphthalocyaninato)Terbium(III) Double-Decker Complexes: Preparation and Study of Protonation by NMR and DFT. Inorg. Chem. 2015, 54, 11986–11992. [Google Scholar] [CrossRef] [PubMed]
- Damjanović, M.; Morita, T.; Katoh, K.; Yamashita, M.; Enders, M. Ligand π-Radical Interaction with f-Shell Unpaired Electrons in Phthalocyaninato-Lanthanoid Single-Molecule Magnets: A Solution NMR Spectroscopic and DFT Study. Chem. Eur. J. 2015, 21, 14421–14432. [Google Scholar] [CrossRef]
- Funk, A.M.; Finney, K.L.N.A.; Harvey, P.; Kenwright, A.M.; Neil, E.R.; Rogers, N.J.; Kanthi Senanayake, P.; Parker, D. Critical Analysis of the Limitations of Bleaney’s Theory of Magnetic Anisotropy in Paramagnetic Lanthanide Coordination Complexes. Chem. Sci. 2015, 6, 1655–1662. [Google Scholar] [CrossRef]
- Suturina, E.A.; Mason, K.; Geraldes, C.F.G.C.; Kuprov, I.; Parker, D. Beyond Bleaney’s Theory: Experimental and Theoretical Analysis of Periodic Trends in Lanthanide-Induced Chemical Shift. Angew. Chem. 2017, 129, 12383–12386. [Google Scholar] [CrossRef]
- Takahashi, K.; Tomita, Y.; Hada, Y.; Tsubota, K.; Handa, M.; Kasuga, K.; Sogabe, K.; Tokii, T. Preparation and Electrochemical Properties of the Green Ytterbium(III) and Lutetium(III) Sandwich Complexes of Octabutoxy-Substituted Phthalocyanine. Chem. Lett. 1992, 21, 759–762. [Google Scholar] [CrossRef]
- Katoh, K.; Horii, Y.; Yasuda, N.; Wernsdorfer, W.; Toriumi, K.; Breedlove, B.K.; Yamashita, M. Multiple-Decker Phthalocyaninato Dinuclear Lanthanoid(III) Single-Molecule Magnets with Dual-Magnetic Relaxation Processes. Dalton Trans. 2012, 41, 13582–13600. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kormschikov, I.D.; Polovkova, M.A.; Kirakosyan, G.A.; Martynov, A.G.; Gorbunova, Y.G.; Tsivadze, A.Y. Magnetic Anisotropy of Homo- and Heteronuclear Terbium(III) and Dysprosium(III) Trisphthalocyaninates Derived from Paramagnetic 1H-NMR Investigation. Molecules 2024, 29, 510. https://doi.org/10.3390/molecules29020510
Kormschikov ID, Polovkova MA, Kirakosyan GA, Martynov AG, Gorbunova YG, Tsivadze AY. Magnetic Anisotropy of Homo- and Heteronuclear Terbium(III) and Dysprosium(III) Trisphthalocyaninates Derived from Paramagnetic 1H-NMR Investigation. Molecules. 2024; 29(2):510. https://doi.org/10.3390/molecules29020510
Chicago/Turabian StyleKormschikov, Ilya D., Marina A. Polovkova, Gayane A. Kirakosyan, Alexander G. Martynov, Yulia G. Gorbunova, and Aslan Yu. Tsivadze. 2024. "Magnetic Anisotropy of Homo- and Heteronuclear Terbium(III) and Dysprosium(III) Trisphthalocyaninates Derived from Paramagnetic 1H-NMR Investigation" Molecules 29, no. 2: 510. https://doi.org/10.3390/molecules29020510