
Photocatalytic Reduction of CO2 into CO with Cyclometalated Pt(II) Complexes of N^C^N Pincer Dipyridylbenzene Ligands: A DFT Study

Abstract
:1. Introduction
2. Results and Discussion
2.1. Photoexcitation/Reductive Quenching Step
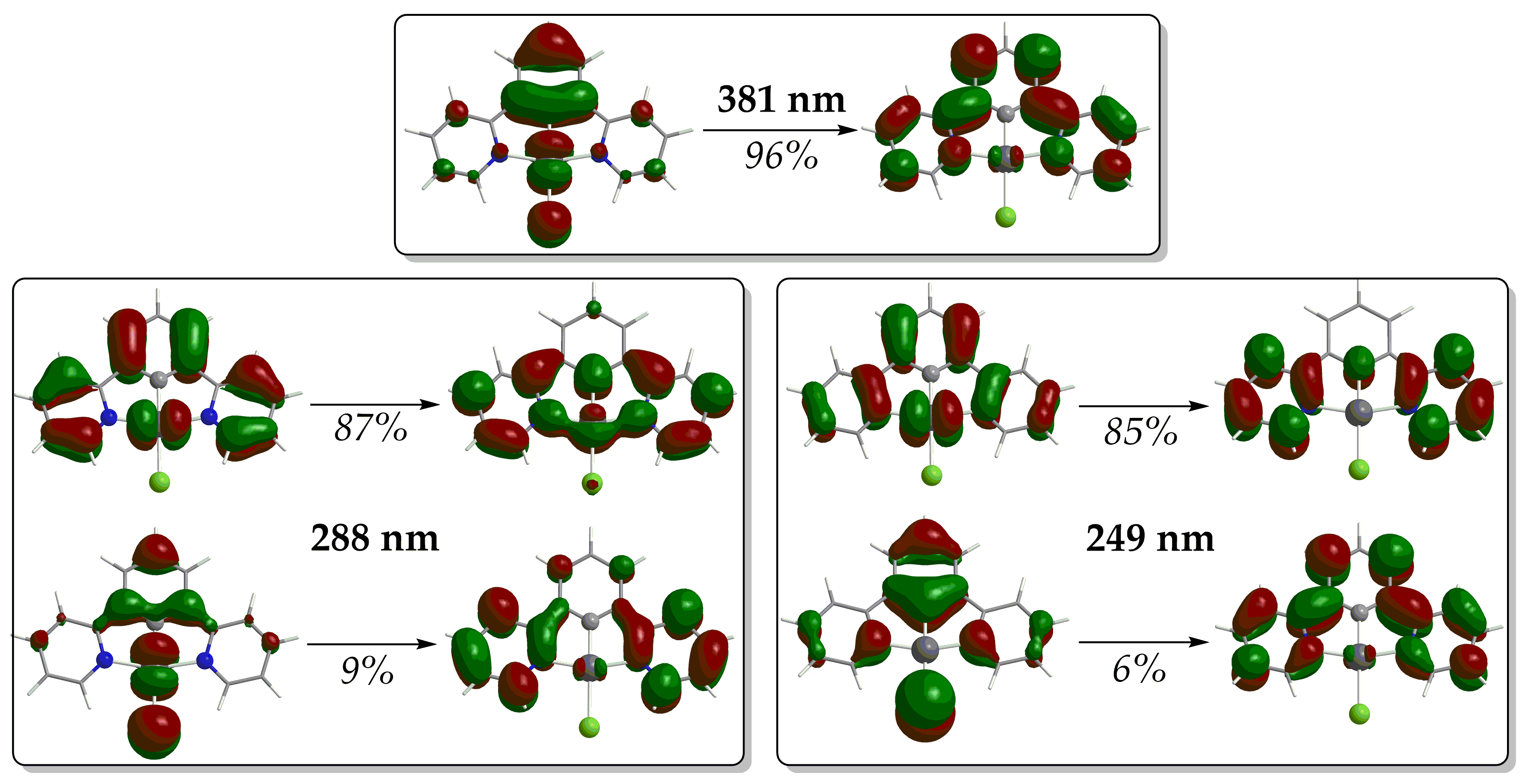
2.1.1. Absorption Spectra
2.1.2. Excited State Electrochemistry
2.2. Catalytic Cycle Step
2.2.1. The η1–CO2 Complex
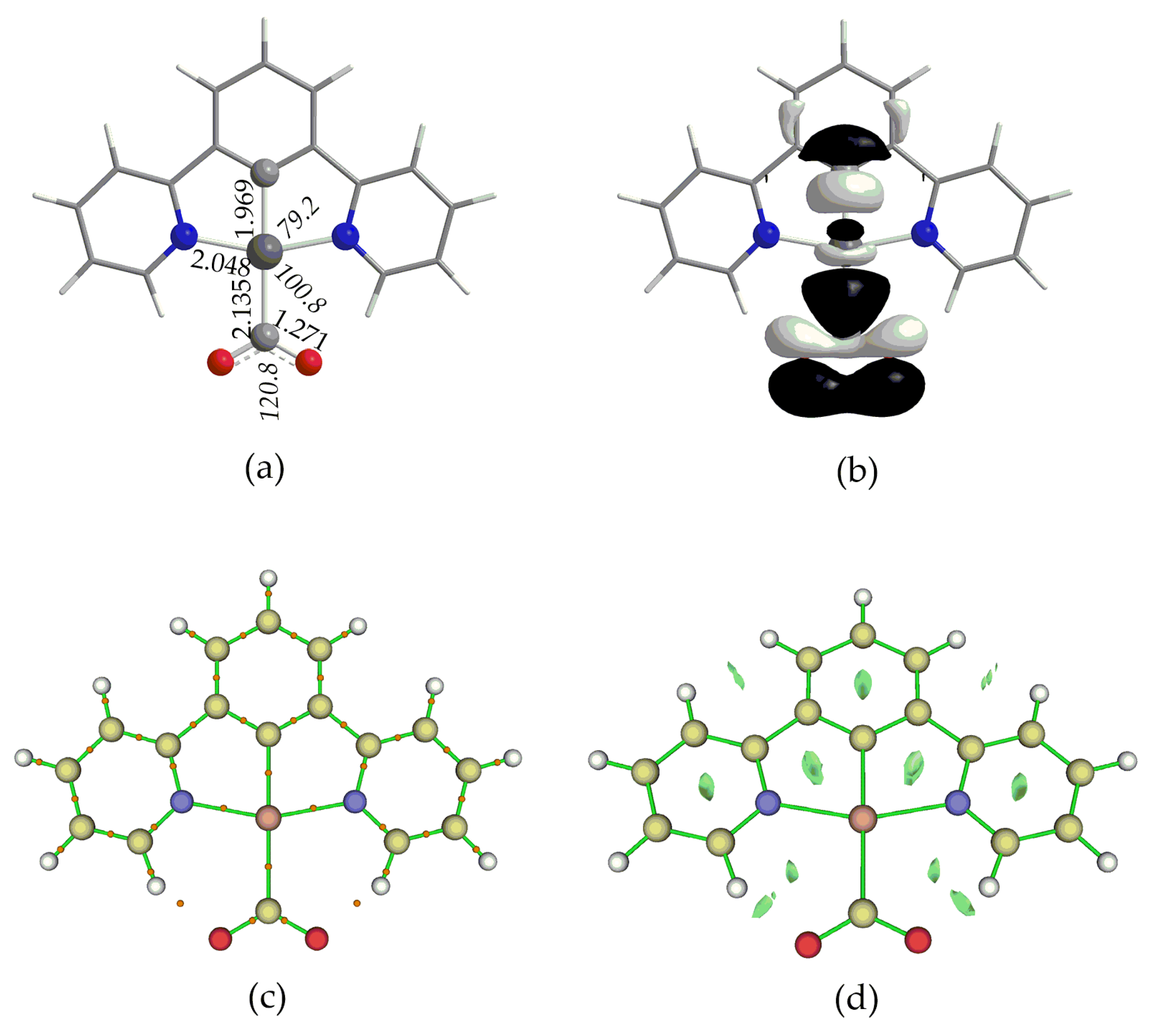
Structural Properties
Bonding and Electronic Properties
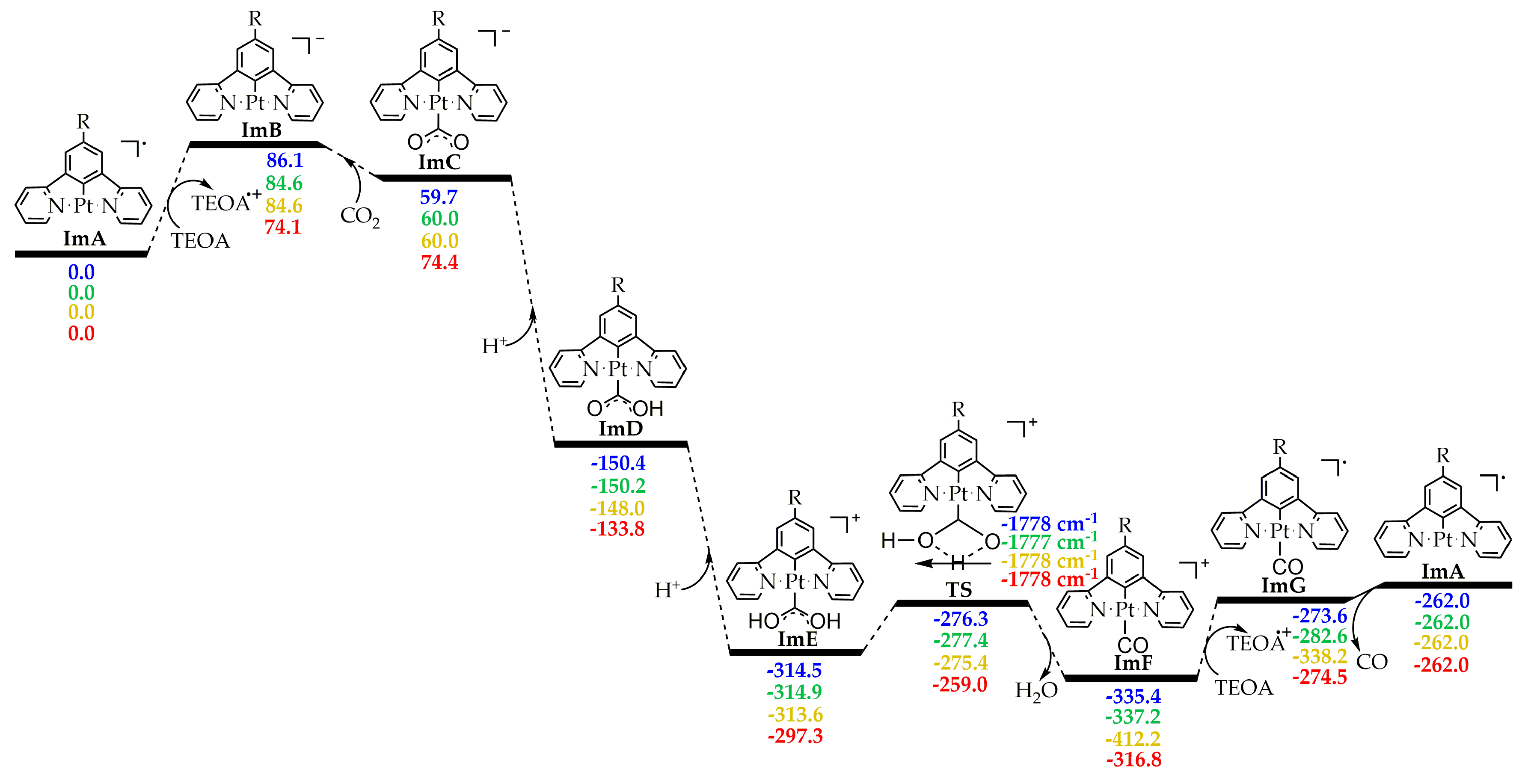
2.2.2. Energetic Reaction Profiles
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lahijani, P.; Zainal, Z.A.; Mohammadi, M.; Mohamed, A.R. Conversion of the greenhouse gas CO2 to the fuel gas CO via the Boudouard reaction: A review. Renew. Sustain. Energy Rev. 2015, 41, 615–632. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Ishitani, O.; Ishida, H. Reaction mechanisms of catalytic photochemical CO2 reduction using Re(I) and Ru(II) complexes. Coord. Chem. Rev. 2018, 373, 333–356. [Google Scholar] [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kühn, F.E. Transformation of carbon dioxide with homogeneous transition-metal catalysts: A molecular solution to a global challenge? Angew. Chemie Int. Ed. 2011, 50, 8510–8537. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, S.; Talbot, A.; Gotico, P.; Caillé, F.; Loreau, O.; Del Vecchio, A.; Malandain, A.; Sallustrau, A.; Leibl, W.; Aukauloo, A.; et al. Unlocking full and fast conversion in photocatalytic carbon dioxide reduction for applications in radio-carbonylation. Nat. Comm. 2023, 14, 4451–4461. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.J.; Meyer, G.J.; Fujita, E. Molecular approaches to the photocatalytic reduction of carbon dioxide for solar fuels. Acc. Chem. Res. 2009, 42, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Koike, K.; Morimoto, T.; Inumaru, H.; Ishitani, O. Photochemistry and photocatalysis of rhenium(I) diimine complexes. Adv. Inorg. Chem. 2011, 63, 137–186. [Google Scholar]
- Costentin, C.; Robert, M.; Savéant, J.M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef]
- Das, S.; Wan Daud, W.M.A. Photocatalytic CO2 transformation into fuel: A review on advances in photocatalyst and photoreactor. Renew. Sustain. Energy Rev. 2014, 39, 765–805. [Google Scholar] [CrossRef]
- Vandezande, J.E.; Schaefer, H.F. CO2 Reduction Pathways on MnBr(N-C)(CO)3 Electrocatalysts. Organometallics 2018, 37, 337–342. [Google Scholar] [CrossRef]
- Paquin, F.; Rivnay, J.; Salleo, A.; Stingelin, N.; Silva, C. Multi-phase semicrystalline microstructures drive exciton dissociation in neat plastic semiconductors. J. Mater. Chem. C 2015, 3, 10715–10722. [Google Scholar] [CrossRef]
- Yang, C.; Wang, Y.; Qian, L.; Al-Enizi, A.M.; Zhang, L.; Zheng, G. Heterogeneous Electrocatalysts for CO2 Reduction. Appl. Energy Mater. 2021, 4, 1034–1044. [Google Scholar] [CrossRef]
- Li, K.; Peng, B.; Peng, T. Recent Advances in Heterogeneous Photocatalytic CO2 Conversion to Solar Fuels. ACS Catal. 2016, 6, 7485–7527. [Google Scholar] [CrossRef]
- Ishida, H. Electrochemical/Photochemical CO2 Reduction Catalyzed by Transition Metal Complexes. In Carbon Dioxide Chemistry, Capture and Oil Recovery; Iyad, K., Shaya, J., Srour, H., Eds.; InTechOpen Ltd.: London, UK, 2018; pp. 17–40. [Google Scholar]
- Boutin, E.; Merakeb, L.; Ma, B.; Boudy, B.; Wang, M.; Bonin, J.; Anxolabèhère-Mallart, E.; Robert, M. Molecular catalysis of CO2 reduction: Recent advances and perspectives in electrochemical and light-driven processes with selected Fe, Ni and Co aza macrocyclic and polypyridine complexes. Chem. Soc. Rev. 2020, 49, 5772–5809. [Google Scholar] [CrossRef] [PubMed]
- Chapovetsky, A.; Do, T.H.; Haiges, R.; Takase, M.K.; Marinescu, S.C. Proton-Assisted Reduction of CO2 by Cobalt Aminopyridine Macrocycles. J. Am. Chem. Soc. 2016, 138, 5765–5768. [Google Scholar] [CrossRef] [PubMed]
- Chapovetsky, A.; Welborn, M.; Luna, J.M.; Haiges, R.; Miller, T.F.; Marinescu, S.C. Pendant Hydrogen-Bond Donors in Cobalt Catalysts Independently Enhance CO2 Reduction. ACS Cent. Sci. 2018, 4, 397–404. [Google Scholar]
- Costentin, C.; Robert, M.; Saveant, J.M.; Tatin, A. Efficient and selective molecular catalyst for the CO2-to-CO electrochemical conversion in water. Proc. Natl. Acad. Sci. USA 2015, 112, 6882–6886. [Google Scholar] [CrossRef] [PubMed]
- Furuya, N.; Matsui, K. Electroreduction of carbon dioxide on gas-diffusion electrodes modified by metal phthalocyanines. J. Electroanal. Chem. 1989, 271, 181–190. [Google Scholar] [CrossRef]
- Fernández, S.; Franco, F.; Casadevall, C.; Martin-Diaconescu, V.; Luis, J.M.; Lloret-Fillol, J. A Unified Electro- and Photocatalytic CO2 to CO Reduction Mechanism with Aminopyridine Cobalt Complexes. J. Am. Chem. Soc. 2020, 142, 120–133. [Google Scholar] [CrossRef]
- Guo, Z.; Cheng, S.; Cometto, C.; Anxolabéhère-Mallart, E.; Ng, S.-M.; Ko, C.C.; Liu, G.; Chen, L.; Robert, M.; Lau, T.-C. Highly Efficient and Selective Photocatalytic CO2 Reduction by Iron and Cobalt Quaterpyridine Complexes. J. Am. Chem. Soc. 2016, 138, 9413–9416. [Google Scholar] [CrossRef]
- Tinnemans, A.H.A.; Koster, T.P.M.; Thewissen, D.H.M.W.; Mackor, A. Tetraaza-macrocyclic cobalt(II) and nickel(II) complexes as electron-transfer agents in the photo(electro)chemical and electrochemical reduction of carbon dioxide. Recl. Trav. Chim. Pays-Bas 1984, 103, 288–295. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.-P.; Ruppert, R.; Sauvage, J.-P. Nickel (II)-cyclam: An extremely selective electrocatalyst for reduction of CO2 in water. J. Chem. Soc. Chem. Commun. 1984, 1315–1316. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.-P.; Ruppert, R.; Sauvage, J.-P. Electrocatalytic reduction of carbon dioxide by nickel cyclam2+ in water: Study of the factors affecting the efficiency and the selectivity of the process. J. Am. Chem. Soc. 1986, 108, 7461–7467. [Google Scholar] [CrossRef]
- Collin, J.-P.; Jouaiti, A.; Sauvage, J.-P. Electrocatalytic properties of (tetraazacyclotetradecane) nickel (2+) and Ni2(biscyclam)4+ with respect to carbon dioxide and water reduction. Inorg. Chem. 1988, 27, 1986–1990. [Google Scholar] [CrossRef]
- Nandal, N.; Jain, S.L. A review on progress and perspective of molecular catalysis in photoelectrochemical reduction of CO2. Coord. Chem. Rev. 2022, 451, 214271. [Google Scholar] [CrossRef]
- Lehn, J.-M.; Ziessel, R. Photochemical generation of carbon monoxide and hydrogen by reduction of carbon dioxide and water under visible light irradiation. Proc. Natl. Acad. Sci. USA 1982, 79, 701–704. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Efficient photochemical reduction of CO2 to CO by visible light irradiation of systems containing Re(bipy)(CO)3X or Ru(bipy)32+–Co2+ combinations as homogeneous catalysts. Chem. Commun. 1983, 536–538. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Photochemical and Electrochemical Reduction of Carbon Dioxide to Carbon Monoxide Mediated by (2,2′-Bipyridine)tricarbonylchlororhenium(I) and Related Complexes as Homogeneous Catalysts. Helv. Chim. Acta 1986, 69, 1990–2012. [Google Scholar] [CrossRef]
- Elgrishi, N.; Chambers, M.B.; Wang, X.; Fontecave, M. Molecular polypyridine-based metal complexes as catalysts for the reduction of CO2. Chem. Soc. Rev. 2017, 46, 761–796. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Takeda, H.; Ishitani, O. Photocatalytic reduction of CO2 using metal complexes. J. Photochem. Photobiol. C Photochem. Rev. 2015, 25, 106–137. [Google Scholar] [CrossRef]
- Voyame, P.; Toghill, K.E.; Méndez, M.A.; Girault, H.H. Photoreduction of CO2 Using [Ru(bpy)2(CO)L]n+ Catalysts in Biphasic Solution/Supercritical CO2 Systems. Inorg. Chem. 2013, 52, 10949–10957. [Google Scholar] [CrossRef]
- Morimoto, T.; Nakajima, T.; Sawa, S.; Nakanishi, R.; Imori, D.; Ishitani, O. CO2 Capture by a Rhenium(I) Complex with the Aid of Triethanolamine. J. Am. Chem. Soc. 2013, 135, 16825–16828. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Morimoto, T.; Koike, K.; Ishitani, O. Photocatalytic CO2 reduction with high turnover frequency and selectivity of formic acid formation using Ru(II) multinuclear complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 15673–15678. [Google Scholar] [CrossRef] [PubMed]
- Tsipis, A.C.; Sarantou, A.A. DFT insights into the photocatalytic reduction of CO2 to CO by Re(I) complexes: The crucial role of the triethanolamine “magic” sacrificial electron donor. Dalton Trans. 2021, 50, 14797–14809. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Kita, S.; Brunschwig, B.S.; Fujita, E. Involvement of a Binuclear Species with the Re-C(O)O-Re Moiety in CO2Reduction Catalyzed by Tricarbonyl Rhenium(I)Complexes with Diimine Ligands: Strikingly Slow Formation of the Re-Re and Re-C(O)O-Re Species from Re(dmb)(CO)3S (dmb = 4,4’-Dimethyl-2,2’-bipyridine, S = Solvent). J. Am. Chem. Soc. 2003, 125, 11976–11987. [Google Scholar] [PubMed]
- Sullivan, B.P.; Bolinger, C.M.; Conrad, D.; Vining, W.J.; Meyer, T.J. One- and Two-electron Pathways in the Electrocatalytic Reduction of CO2 by fac-Re(bpy)(CO)3Cl (bpy = 2,2’-bipyridine). J. Chem. Soc. Chem. Commun. 1985, 20, 1414–1416. [Google Scholar] [CrossRef]
- Agarwal, J.; Fujita, E.; Schaefer, H.F., III; Muckerman, J.T. Mechanisms for CO Production from CO2 Using Reduced Rhenium Tricarbonyl Catalysts. J. Am. Chem. Soc. 2012, 134, 5180–5186. [Google Scholar] [CrossRef]
- Ceballos, B.M.; Yang, J.Y. Directing the reactivity of metal hydrides for selective CO2 reduction. Proc. Natl. Acad. Sci. USA 2018, 115, 12686–12691. [Google Scholar] [CrossRef]
- Zhang, Q.H.; Han, W.D.; Hong, Y.J.; Yu, J.G. Photocatalytic reduction of CO2 with H2O on Pt-loaded TiO2 catalyst. Catal. Today 2009, 148, 335–340. [Google Scholar] [CrossRef]
- Wang, W.; An, W.; Ramalingam, B.; Mukherjee, S.; Niedzwiedzki, D.M.; Gangopadhyay, S.; Biswas, P. Size and Structure Matter: Enhanced CO2 Photoreduction Efficiency by Size-Resolved Ultrafine Pt Nanoparticles on TiO2 Single Crystals. J. Am. Chem. Soc. 2012, 134, 11276–11281. [Google Scholar] [CrossRef]
- Katsumata, K.I.; Sakai, K.; Ikeda, K.; Carja, G.; Matsushita, N.; Okada, K. Preparation and photocatalytic reduction of CO2 on noble metal (Pt, Pd, Au) loaded Zn-Cr layered double hydroxides. Mater. Lett. 2013, 107, 138–140. [Google Scholar] [CrossRef]
- Xie, S.; Wang, Y.; Zhang, Q.; Fan, W.; Denga, W.; Wang, Y. Photocatalytic reduction of CO2 with H2O: Significant enhancement of the activity of Pt–TiO2 in CH4 formation by addition of MgO. Chem. Commun. 2013, 49, 2451–2453. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Lei, Z.; Kuang, C.C.; Chen, X.; Gong, B.; Zhao, Y.; Zhang, J.; Zheng, C.; Wu, J.C.S. Selective photocatalytic reduction of CO2 into CH4 over Pt-Cu2O TiO2 nanocrystals: The interaction between Pt and Cu2O cocatalysts. Appl. Catal. B Environ. 2017, 202, 695–703. [Google Scholar] [CrossRef]
- Kočí, K.; Dang Van, H.; Edelmannová, M.; Reli, M.; Wu, J.C.S. Photocatalytic reduction of CO2 using Pt/C3N4 photocatalyts. Appl. Surf. Sci. 2020, 503, 144426. [Google Scholar] [CrossRef]
- Tasbihi, M.; Kočí, K.; Edelmannová, M.; Troppová, I.; Reli, M.; Schomäcker, R. Pt/TiO2 photocatalysts deposited on commercial support for photocatalytic reduction of CO2. J. Photochem. Photobiol. A Chem. 2018, 366, 72–80. [Google Scholar] [CrossRef]
- Xu, J.; Liu, X.; Zhou, Z.; Deng, L.; Liu, L.; Xu, M. Platinum Nanoparticles with Low Content and High Dispersion over Exfoliated Layered Double Hydroxide for Photocatalytic CO2 Reduction. Energy Fuels 2021, 35, 10820–10831. [Google Scholar] [CrossRef]
- Li, X.; Bi, W.; Zhang, Q.; Tao, S.; Chu, W.; Zhang, Q.; Luo, Y.; Wu, C.; Xie, Y. Single-atom Pt as co-catalyst for enhanced photocatalytic H2 evolution. Adv. Mater. 2016, 28, 2427–2431. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, D.; Lin, Y.; Liu, W.; Cao, L.; Liu, X.; Zhang, W.; Mou, X.; Fang, S.; Shen, X.; et al. Single Pt atom with highly vacant d-orbital for accelerating photocatalytic H2 evolution. ACS Appl. Energy Mater. 2018, 1, 6082–6088. [Google Scholar] [CrossRef]
- Williams, J.A.G.; Beeby, A.; Davies, S.; Weinstein, J.A.; Wilson, C. An Alternative Route to Highly Luminescent Platinum(II) Complexes: Cyclometalation with N^C^N-Coordinating Dipyridylbenzene Ligands. Inorg. Chem. 2003, 42, 8609–8611. [Google Scholar] [CrossRef]
- Cárdenas, D.J.; Echavarren, A.M.; Ramírez de Arellano, M.C. Divergent Behavior of Palladium(II) and Platinum(II) in the Metalation of 1,3-Di(2-pyridyl)benzene. Organometallics 1999, 18, 3337–3341. [Google Scholar] [CrossRef]
- Demissie, T.B.; Ruud, K.; Hansen, J.H. DFT as a Powerful Predictive Tool in Photoredox Catalysis: Redox Potentials and Mechanistic Analysis. Organometallics 2015, 34, 4218–4228. [Google Scholar] [CrossRef]
- Grice, K.A. Carbon dioxide reduction with homogenous early transition metal complexes: Opportunities and challenges for developing CO2 catalysis. Coord. Chem. Rev. 2017, 336, 78–95. [Google Scholar] [CrossRef]
- Kinzel, N.W.; Werl, C.; Leitner, W. Transition Metal Complexes as Catalysts for the Electroconversion of CO2: An Organometallic Perspective. Angew. Chem. Int. Ed. 2021, 60, 11628–11686. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Macchi, P.; Sironi, A. Chemical bonding in transition metal carbonyl clusters: Complementary analysis of theoretical and experimental electron densities. Coord. Chem. Rev. 2003, 383, 238–239. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Johnson, E.R.; Shahar Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Yin, X.; Moss, J.R. Recent developments in the activation of carbon dioxide by metal complexes. Coord. Chem. Rev. 1999, 181, 27–59. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Dapprich, S.; Frenking, G. Investigation of Donor-Acceptor Interactions: A Charge Decomposition Analysis Using Fragment Molecular Orbitals. J. Phys. Chem. 1995, 99, 9352–9362. [Google Scholar] [CrossRef]
- Sheng, H.; Frei, H. Direct Observation by Rapid-Scan FT-IR Spectroscopy of Two-Electron-Reduced Intermediate of Tetraaza Catalyst [CoIIN4H(MeCN)]2+ Converting CO2 to CO. J. Am. Chem. Soc. 2016, 138, 9959–9967. [Google Scholar] [CrossRef] [PubMed]
- Call, A.; Cibian, M.; Yamamoto, K.; Nakazono, T.; Yamauchi, K.; Sakai, K. Highly Efficient and Selective Photocatalytic CO2 Reduction to CO in Water by a Cobalt Porphyrin Molecular Catalyst. ACS Catal. 2019, 9, 4867–4874. [Google Scholar] [CrossRef]
- Vetere, V.; Adamo, C.; Maldivi, P. Performance of the ‘parameter free’ PBE0 functional for the modeling of molecular properties of heavy metals. Chem. Phys. Lett. 2000, 325, 99–105. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Inexpensive and accurate predictions of optical excitations in transition-metal complexes: The TDDFT/PBE0 route. Theor. Chem. Acc. 2000, 105, 169–172. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Adamo, C.; Scuseria, G.E.; Barone, V. Accurate excitation energies from time-dependent density functional theory: Assessing the PBE0 model. J. Chem. Phys. 1999, 111, 2889–2899. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable adiabatic connection models free from adjustable parameters. Chem. Phys. Lett. 1997, 274, 242–250. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16W, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Wu, P.; Chaudret, R.; Hu, X.; Yang, W. Noncovalent Interaction Analysis in Fluctuating Environments. J. Chem. Theory Comput. 2013, 9, 2226–2234. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyser. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Van Gisbergen, S.J.A.; Kootstra, F.; Schipper, P.R.T.; Gritsenko, O.V.; Snijders, J.G.; Baerends, E.J. Density-functional-theory response-property calculations with accurate exchange-correlation potentials. Phys. Rev. A At. Mol. Opt. Phys. 1998, 57, 2556–2571. [Google Scholar] [CrossRef]
- Jamorski, C.; Casida, M.E.; Salahub, D.R. Dynamic polarizabilities and excitation spectra from a molecular implementation of time-dependent density-functional response theory: N2 as a case study. J. Chem. Phys. 1996, 104, 5134–5147. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Laurent, A.D.; Jacquemine, D. TD-DFT Benchmarks: A review. Int. J. Quant. Chem. 2013, 113, 2019–2039. [Google Scholar] [CrossRef]
- Latouche, C.; Skouteris, D.; Palazzetti, F.; Barone, V. TD-DFT Benchmark on Inorganic Pt(II) and Ir(III) Complexes. J. Chem. Theory Comput. 2015, 11, 3281–3289. [Google Scholar] [CrossRef]
- Martin, R.L. Natural Transition Orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | E0red | E0*red | E0red vs. SHE | E0*red vs. SHE 1 | E0*redox 2 | |
|---|---|---|---|---|---|---|
| vs. TEOA | vs. TEA | |||||
| 1 | −2.48 | 1.80 | 0.32 | 4.60 | 7.43 | 7.19 |
| 2 | −2.46 | 1.82 | −0.36 | 3.92 | 7.45 | 7.21 |
| 3 | −2.54 | 1.74 | −0.34 | 3.94 | 7.37 | 7.13 |
| 4 | −3.48 | 0.80 | −1.88 | 2.40 | 6.43 | 6.19 |
| TEOA | −5.63 | −1.35 | ||||
| TEA | −5.39 | −1.11 | ||||
| Species | ρBCP 1 | ∇ρBCP 2 | GBCP 3 | VBCP 3 | |VBCP|/GBCP | HBCP 3 | GBCP/ρBCP 4 |
|---|---|---|---|---|---|---|---|
| 1_ImC | 0.124 | 0.137 | 0.089 | −0.145 | 1.629 | −0.056 | 0.718 |
| 2_ImC | 0.125 | 0.138 | 0.090 | −0.147 | 1.633 | −0.057 | 0.720 |
| 3_ImC | 0.125 | 0.135 | 0.090 | −0.147 | 1.633 | −0.057 | 0.720 |
| 4_ImC | 0.125 | 0.130 | 0.088 | −0.145 | 1.648 | −0.057 | 0.704 |
| Species | QPt | QC 1 | BD[σ(Pt-CO2)] | ΔE(2) | |
|---|---|---|---|---|---|
| σ(Pt-CN^C^N) → σ*(Pt-CO2) | σ(Pt-CO2) → σ*(Pt-CN^C^N) | ||||
| 1_ImC | 0.235 | 0.495 | 0.558hPt + 0.830hC | 58.3 | 27.8 |
| 2_ImC | 0.237 | 0.494 | 0.540hPt + 0.842hC | 58.1 | 26.4 |
| 3_ImC | 0.244 | 0.495 | 0.538hPt + 0.843hC | 58.3 | 27.2 |
| 4_ImC | 0.260 | 0.497 | 0.535hPt + 0.845hC | 56.9 | 23.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarantou, A.; Tsipis, A. Photocatalytic Reduction of CO2 into CO with Cyclometalated Pt(II) Complexes of N^C^N Pincer Dipyridylbenzene Ligands: A DFT Study. Molecules 2024, 29, 403. https://doi.org/10.3390/molecules29020403
Sarantou A, Tsipis A. Photocatalytic Reduction of CO2 into CO with Cyclometalated Pt(II) Complexes of N^C^N Pincer Dipyridylbenzene Ligands: A DFT Study. Molecules. 2024; 29(2):403. https://doi.org/10.3390/molecules29020403
Chicago/Turabian StyleSarantou, Antonia, and Athanassios Tsipis. 2024. "Photocatalytic Reduction of CO2 into CO with Cyclometalated Pt(II) Complexes of N^C^N Pincer Dipyridylbenzene Ligands: A DFT Study" Molecules 29, no. 2: 403. https://doi.org/10.3390/molecules29020403