
Aromatase Inhibitors as a Promising Direction for the Search for New Anticancer Drugs




Abstract
:1. Introduction
2. Materials and Methods
3. Aromatase Inhibitors
3.1. Steroidal Aromatase Inhibitors
3.2. Non-Steroidal Aromatase Inhibitors
3.2.1. Azoles
Imidazoles
Benzimidazoles
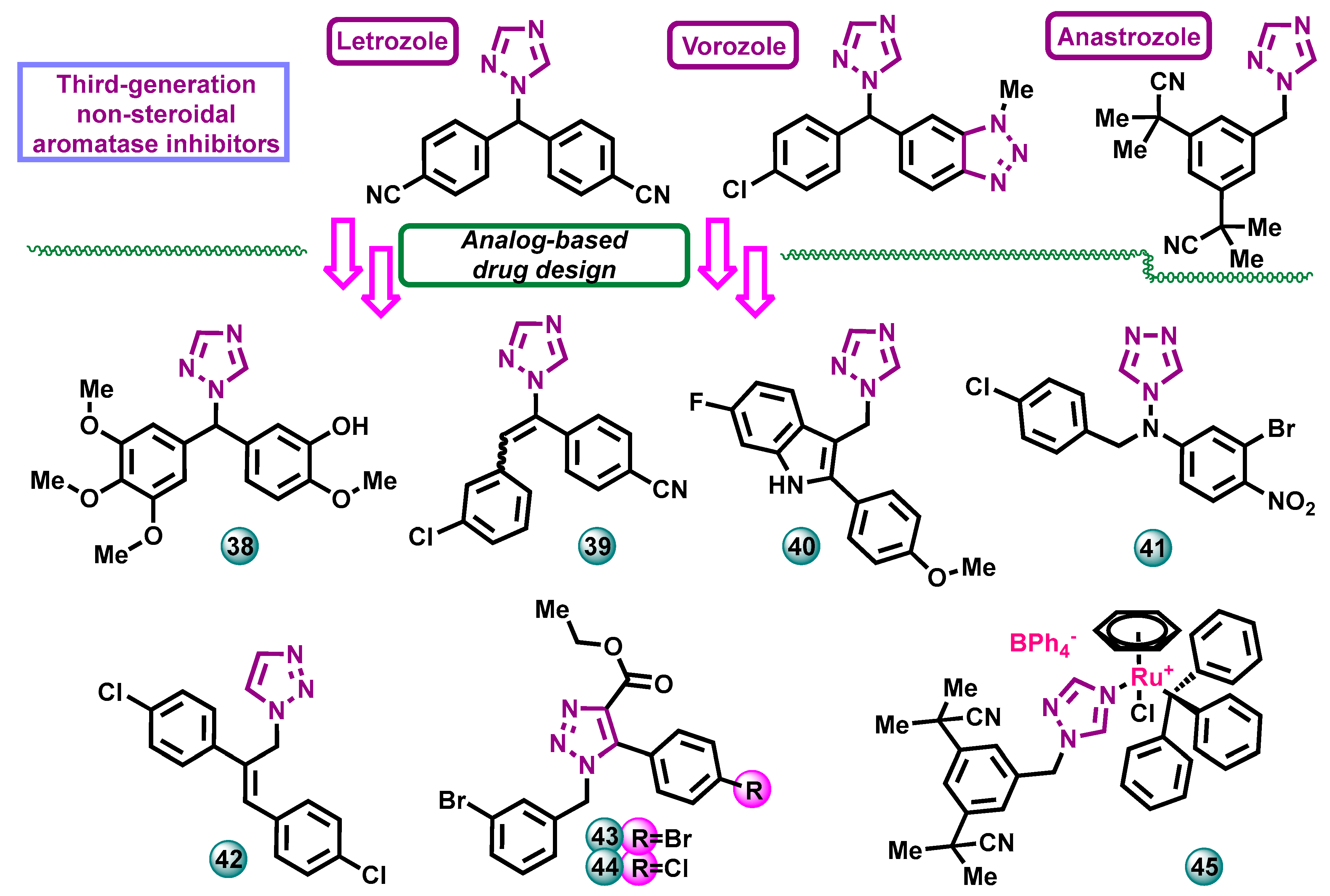
Triazoles
3.2.2. Coumarin-Based and Coumarin/Chromone/Heterocycle-Bearing Hybrid Molecules
3.2.3. Other Heterocycles
3.2.4. Aryl/Hetarylhydrazones
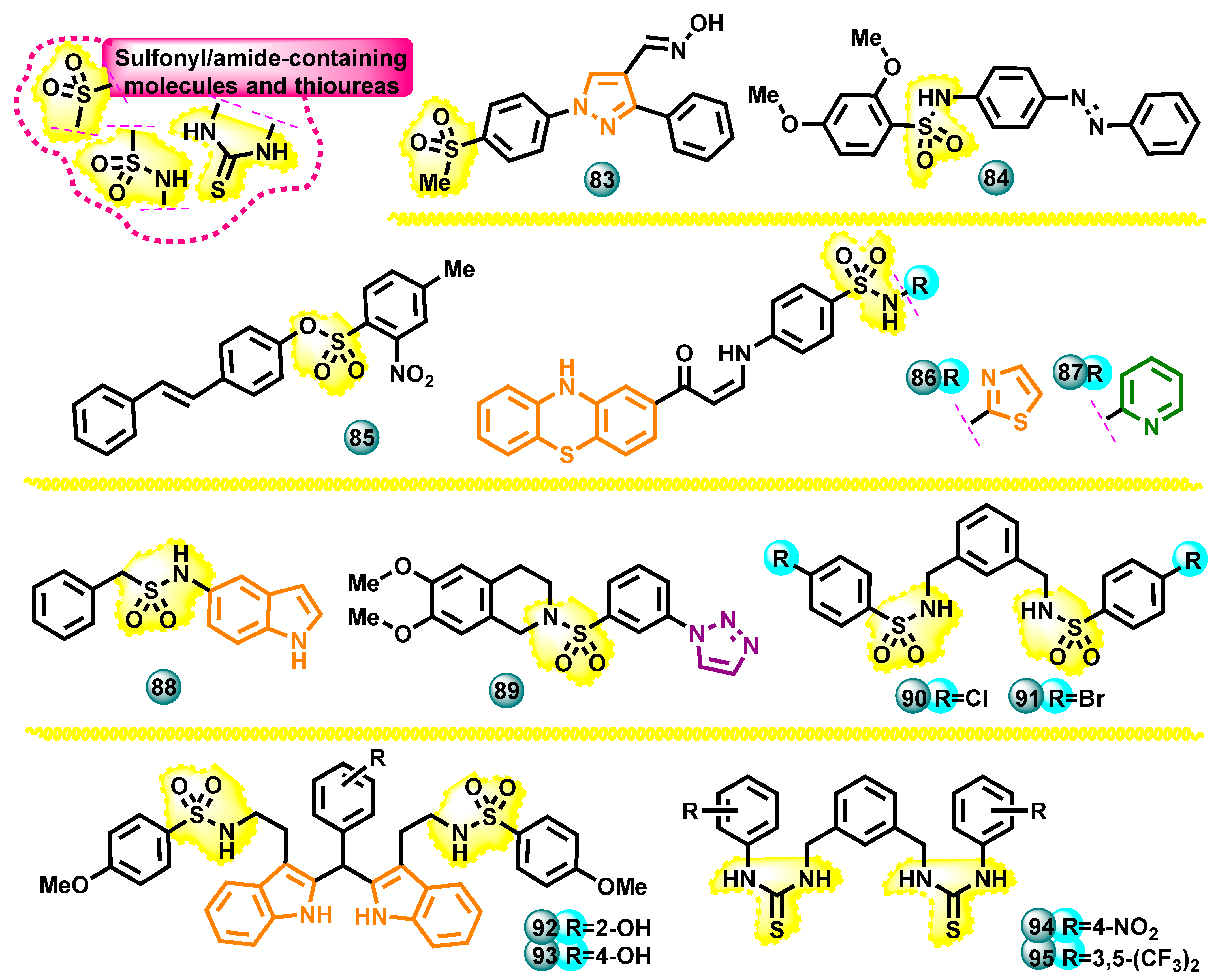
3.2.5. Sulfonyl/Amide Containing Molecules and Thioureas as Potential Aromatase Inhibitors
3.2.6. Triphenylethylene Derivatives
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sayyad, N.B.; Sabale, P.M.; Umare, M.D.; Bajaj, K.K. Aromatase Inhibitors: Development and Current Perspectives. Indian J. Pharm. Educ. Res. 2022, 56, 311–320. [Google Scholar] [CrossRef]
- Çetiner, G.; Acar Çevik, U.; Celik, I.; Bostancı, H.E.; Özkay, Y.; Kaplancıklı, Z.A. New Imidazole Derivatives as Aromatase Inhibitor: Design, Synthesis, Biological Activity, Molecular Docking, and Computational ADME-Tox Studies. J. Mol. Struct. 2023, 1278, 134920. [Google Scholar] [CrossRef]
- Ghosh, D.; Griswold, J.; Erman, M.; Pangborn, W. Structural Basis for Androgen Specificity and Oestrogen Synthesis in Human Aromatase. Nature 2009, 457, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Besman, M.J.; Sparkes, R.S.; Zollman, S.; Klisak, I.; Mohandas, T.; Hall, P.F.; Shively, J.E. Human Aromatase: CDNA Cloning, Southern Blot Analysis, and Assignment of the Gene to Chromosome 15. DNA 1988, 7, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.A.; Siiteri, P.K. Utilization of Oxygen and Reduced Nicotinamide Adenine Dinucleotide Phosphate by Human Placental Microsomes during Aromatization of Androstenedione. J. Biol. Chem. 1974, 249, 5364–5372. [Google Scholar] [CrossRef] [PubMed]
- Gilep, A.A.; Sushko, T.A.; Usanov, S.A. At the Crossroads of Steroid Hormone Biosynthesis: The Role, Substrate Specificity and Evolutionary Development of CYP17. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2011, 1814, 200–209. [Google Scholar] [CrossRef]
- Chumsri, S.; Howes, T.; Bao, T.; Sabnis, G.; Brodie, A. Aromatase, Aromatase Inhibitors, and Breast Cancer. J. Steroid Biochem. Mol. Biol. 2011, 125, 13–22. [Google Scholar] [CrossRef]
- Ghosh, D.; Griswold, J.; Erman, M.; Pangborn, W. X-ray Structure of Human Aromatase Reveals an Androgen-Specific Active Site. J. Steroid Biochem. Mol. Biol. 2010, 118, 197–202. [Google Scholar] [CrossRef]
- McDonnell, D.P. The Molecular Pharmacology of SERMs. Trends Endocrinol. Metab. 1999, 10, 301–311. [Google Scholar] [CrossRef]
- Pingaew, R.; Mandi, P.; Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis, Molecular Docking, and QSAR Study of Sulfonamide-Based Indoles as Aromatase Inhibitors. Eur. J. Med. Chem. 2018, 143, 1604–1615. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.; Iqbal, A.-W.; Zafar, H.; Aziz, A.; Shaikh, N. Naveed Atta-ur-Rahman Synthesis of a Potent Aromatase Inhibitor 17-α-Acetoxy-10-β,11-β-Dihydroxy-Progesterone for the Treatment of Estrogen Receptor Positive Breast Cancer. U.S. Patent Application 18/128,246, 3 August 2023. [Google Scholar]
- Cobos-Ontiveros, L.A.; Romero-Hernández, L.L.; Mastranzo-Sánchez, E.B.; Colín-Lozano, B.; Puerta, A.; Padrón, J.M.; Merino-Montiel, P.; Vega Baez, J.L.; Montiel-Smith, S. Synthesis, Antiproliferative Evaluation and in Silico Studies of a Novel Steroidal Spiro Morpholinone. Steroids 2023, 192, 109173. [Google Scholar] [CrossRef] [PubMed]
- Roleira, F.M.F.; Costa, S.C.; Gomes, A.R.; Varela, C.L.; Amaral, C.; Augusto, T.V.; Correia-da-Silva, G.; Romeo, I.; Costa, G.; Alcaro, S.; et al. Design, Synthesis, Biological Activity Evaluation and Structure-Activity Relationships of New Steroidal Aromatase Inhibitors. The Case of C-Ring and 7β Substituted Steroids. Bioorg. Chem. 2023, 131, 106286. [Google Scholar] [CrossRef]
- Amaral, C.; Correia-da-Silva, G.; Almeida, C.F.; Valente, M.J.; Varela, C.; Tavares-da-Silva, E.; Vinggaard, A.M.; Teixeira, N.; Roleira, F.M.F. An Exemestane Derivative, Oxymestane-D1, as a New Multi-Target Steroidal Aromatase Inhibitor for Estrogen Receptor-Positive (ER+) Breast Cancer: Effects on Sensitive and Resistant Cell Lines. Molecules 2023, 28, 789. [Google Scholar] [CrossRef] [PubMed]
- Wahab, A.-T.; Choudhary, M.I.; Farooq, R.; Szejk, N.N.; Rahman, A.-U. Bio-Catalyzed Synthesis of New Aromatase Inhibitors through Structural Modifications of Anticancer Drug Formestane. U.S. Patent Application 17/403,899, 9 December 2021. [Google Scholar]
- Banday, A.H.; Saeed, B.A.; Al-Masoudi, N.A. Synthesis, Aromatase Inhibitory, Antiproliferative and Molecular Modeling Studies of Functionally Diverse D-Ring Pregnenolone Pyrazoles. Anti-Cancer Agents Med. Chem. 2021, 21, 1671–1679. [Google Scholar] [CrossRef]
- Roleira, F.M.F.; Varela, C.; Amaral, C.; Costa, S.C.; Correia-da-Silva, G.; Moraca, F.; Costa, G.; Alcaro, S.; Teixeira, N.A.A.; Tavares da Silva, E.J. C-6α- vs C-7α-Substituted Steroidal Aromatase Inhibitors: Which Is Better? Synthesis, Biochemical Evaluation, Docking Studies, and Structure–Activity Relationships. J. Med. Chem. 2019, 62, 3636–3657. [Google Scholar] [CrossRef] [PubMed]
- Amaral, C.; Varela, C.L.; Maurício, J.; Sobral, A.F.; Costa, S.C.; Roleira, F.M.F.; Tavares-da-Silva, E.J.; Correia-da-Silva, G.; Teixeira, N. Anti-Tumor Efficacy of New 7α-Substituted Androstanes as Aromatase Inhibitors in Hormone-Sensitive and Resistant Breast Cancer Cells. J. Steroid Biochem. Mol. Biol. 2017, 171, 218–228. [Google Scholar] [CrossRef]
- Jójárt, R.; Traj, P.; Kovács, É.; Horváth, Á.; Schneider, G.; Szécsi, M.; Pál, A.; Paragi, G.; Mernyák, E. Synthesis, Biological Evaluation and Docking Studies of 13-Epimeric 10-Fluoro- and 10-Chloroestra-1,4-Dien-3-Ones as Potential Aromatase Inhibitors. Molecules 2019, 24, 1783. [Google Scholar] [CrossRef]
- Varela, C.L.; Amaral, C.; Correia-da-Silva, G.; Costa, S.C.; Carvalho, R.A.; Costa, G.; Alcaro, S.; Teixeira, N.A.A.; Tavares-da-Silva, E.J.; Roleira, F.M.F. Exploring New Chemical Functionalities to Improve Aromatase Inhibition of Steroids. Bioorg. Med. Chem. 2016, 24, 2823–2831. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; Gallorini, M.; Fantacuzzi, M.; Gambacorta, N.; De Filippis, B.; Giampietro, L.; Maccallini, C.; Nicolotti, O.; Cataldi, A.; Amoroso, R. Design, Synthesis and Biological Evaluation of Imidazole and Triazole-Based Carbamates as Novel Aromatase Inhibitors. Eur. J. Med. Chem. 2021, 211, 113115. [Google Scholar] [CrossRef]
- Maccallini, C.; Gallorini, M.; Sisto, F.; Akdemir, A.; Ammazzalorso, A.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Carradori, S.; Cataldi, A.; et al. New Azolyl-Derivatives as Multitargeting Agents against Breast Cancer and Fungal Infections: Synthesis, Biological Evaluation and Docking Study. J. Enzym. Inhib. Med. Chem. 2021, 36, 1631–1644. [Google Scholar] [CrossRef] [PubMed]
- Ghodsi, R.; Azizi, E.; Grazia Ferlin, M.; Pezzi, V.; Zarghi, A. Design, Synthesis and Biological Evaluation of 4-(Imidazolylmethyl)-2- Aryl-Quinoline Derivatives as Aromatase Inhibitors and Anti-Breast Cancer Agents. Lett. Drug. Des. Discov. 2015, 13, 89–97. [Google Scholar] [CrossRef]
- Di Matteo, M.; Ammazzalorso, A.; Andreoli, F.; Caffa, I.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Nencioni, A.; Parenti, M.D.; et al. Synthesis and Biological Characterization of 3-(Imidazol-1-Ylmethyl)Piperidine Sulfonamides as Aromatase Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3192–3194. [Google Scholar] [CrossRef] [PubMed]
- Kalalinia, F.; Jouya, M.; Komachali, A.K.; Aboutourabzadeh, S.M.; Karimi, G.; Behravan, J.; Abnous, K.; Etemad, L.; Kamali, H.; Hadizadeh, F. Design, Synthesis, and Biological Evaluation of New Azole Derivatives as Potent Aromatase Inhibitors with Potential Effects against Breast Cancer. Anti-Cancer Agents Med. Chem. 2018, 18, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Acar Çevik, U.; Celik, I.; Işık, A.; Ahmad, I.; Patel, H.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, Molecular Modeling, DFT, ADME and Biological Evaluation Studies of Some New 1,3,4-Oxadiazole Linked Benzimidazoles as Anticancer Agents and Aromatase Inhibitors. J. Biomol. Struct. Dyn. 2023, 41, 1944–1958. [Google Scholar] [CrossRef]
- Sayyad, N.B.; Sabale, P.M. Rational Drug Design and In Vitro Cell Line Studies of Some N-(4-(1Hbenzo[d]Imidazol-2-Yl)Phenyl)Arylamine Derivatives as Aromatase Inhibitors for the Treatment of Cancer. Curr. Enzym. Inhib. 2023, 19, 38–48. [Google Scholar] [CrossRef]
- Çevik, U.A.; Celik, I.; Mella, J.; Mellado, M.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, and Molecular Modeling Studies of a Novel Benzimidazole as an Aromatase Inhibitor. ACS Omega 2022, 7, 16152–16163. [Google Scholar] [CrossRef]
- Sağlık, B.N.; Şen, A.M.; Evren, A.E.; Çevik, U.A.; Osmaniye, D.; Kaya Çavuşoğlu, B.; Levent, S.; Karaduman, A.B.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis, Investigation of Biological Effects and In Silico Studies of New Benzimidazole Derivatives as Aromatase Inhibitors. Z. Naturforsch. C 2020, 75, 353–362. [Google Scholar] [CrossRef]
- Acar Çevik, U.; Sağlık, B.N.; Osmaniye, D.; Levent, S.; Kaya Çavuşoğlu, B.; Karaduman, A.B.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis and Docking Study of Benzimidazole–Triazolothiadiazine Hybrids as Aromatase Inhibitors. Arch. Pharm. 2020, 353, e2000008. [Google Scholar] [CrossRef]
- Acar Çevik, U.; Kaya Çavuşoğlu, B.; Sağlık, B.N.; Osmaniye, D.; Levent, S.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis, Docking Studies and Biological Activity of New Benzimidazole- Triazolothiadiazine Derivatives as Aromatase Inhibitor. Molecules 2020, 25, 1642. [Google Scholar] [CrossRef]
- Osmaniye, D.; Hıdır, A.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of New Pyrimidine-Triazole Derivatives and Investigation of Their Anticancer Activities. Chem. Biodivers. 2022, 19, e202200216. [Google Scholar] [CrossRef] [PubMed]
- Osmaniye, D.; Sağlık, B.N.; Levent, S.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, in Vitro and in Silico Studies of Some Novel Triazoles as Anticancer Agents for Breast Cancer. J. Mol. Struct. 2021, 1246, 131198. [Google Scholar] [CrossRef]
- Xi, Y.; Liu, J.; Wang, H.; Li, S.; Yi, Y.; Du, Y. New Small-molecule Compound Hu-17 Inhibits Estrogen Biosynthesis by Aromatase in Human Ovarian Granulosa Cancer Cells. Cancer Med. 2020, 9, 9081–9095. [Google Scholar] [CrossRef] [PubMed]
- Ana, G.; Kelly, P.M.; Malebari, A.M.; Noorani, S.; Nathwani, S.M.; Twamley, B.; Fayne, D.; O’Boyle, N.M.; Zisterer, D.M.; Pimentel, E.F.; et al. Synthesis and Biological Evaluation of 1-(Diarylmethyl)-1H-1,2,4-Triazoles and 1-(Diarylmethyl)-1H-Imidazoles as a Novel Class of Anti-Mitotic Agent for Activity in Breast Cancer. Pharmaceuticals 2021, 14, 169. [Google Scholar] [CrossRef] [PubMed]
- Vosooghi, M.; Firoozpour, L.; Rodaki, A.; Pordeli, M.; Safavi, M.; Ardestani, S.K.; Dadgar, A.; Asadipour, A.; Moshafi, M.H.; Foroumadi, A. Design, Synthesis, Docking Study and Cytotoxic Activity Evaluation of Some Novel Letrozole Analogs. DARU J. Pharm. Sci. 2014, 22, 83. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Xiao, X.; Huang, C.; Yuan, Y.; Tang, D.; Dai, X.; Zeng, X. Potent Aromatase Inhibitors and Molecular Mechanism of Inhibitory Action. Eur. J. Med. Chem. 2018, 143, 426–437. [Google Scholar] [CrossRef]
- Song, Z.; Liu, Y.; Dai, Z.; Liu, W.; Zhao, K.; Zhang, T.; Hu, Y.; Zhang, X.; Dai, Y. Synthesis and Aromatase Inhibitory Evaluation of 4-N-Nitrophenyl Substituted Amino-4H-1,2,4-Triazole Derivatives. Bioorg. Med. Chem. 2016, 24, 4723–4730. [Google Scholar] [CrossRef]
- Nielsen, A.J.; Raez-Villanueva, S.; Crankshaw, D.J.; Holloway, A.C.; McNulty, J. Synthesis of α-Methylstilbenes Using an Aqueous Wittig Methodology and Application toward the Development of Potent Human Aromatase Inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 1395–1398. [Google Scholar] [CrossRef]
- McNulty, J.; Keskar, K.; Crankshaw, D.J.; Holloway, A.C. Discovery of a New Class of Cinnamyl-Triazole as Potent and Selective Inhibitors of Aromatase (Cytochrome P450 19A1). Bioorg. Med. Chem. Lett. 2014, 24, 4586–4589. [Google Scholar] [CrossRef]
- Golbaghi, G.; Haghdoost, M.M.; Yancu, D.; López de los Santos, Y.; Doucet, N.; Patten, S.A.; Sanderson, J.T.; Castonguay, A. Organoruthenium(II) Complexes Bearing an Aromatase Inhibitor: Synthesis, Characterization, In Vitro Biological Activity and In Vivo Toxicity in Zebrafish Embryos. Organometallics 2019, 38, 702–711. [Google Scholar] [CrossRef]
- Takla, F.N.; Bayoumi, W.A.; El-Messery, S.M.; Nasr, M.N.A. Developing Multitarget Coumarin Based Anti-Breast Cancer Agents: Synthesis and Molecular Modeling Study. Sci. Rep. 2023, 13, 13370. [Google Scholar] [CrossRef]
- Pingaew, R.; Prachayasittikul, V.; Mandi, P.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis and Molecular Docking of 1,2,3-Triazole-Based Sulfonamides as Aromatase Inhibitors. Bioorg. Med. Chem. 2015, 23, 3472–3480. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Alsaid, M.S.; Al-Ansary, G.H.; Abdel-Latif, G.A.; Abou El Ella, D.A. Analogue Based Drug Design, Synthesis, Molecular Docking and Anticancer Evaluation of Novel Chromene Sulfonamide Hybrids as Aromatase Inhibitors and Apoptosis Enhancers. Eur. J. Med. Chem. 2016, 124, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.; Patel, S.; Patel, M.; Jain, N.; Pandey, N.; Chauhan, A.; Patel, A.; Patel, S. In Vitro Cytotoxicity and Aromatase Inhibitory Activity of Flavonoids: Synthesis, Molecular Docking and In Silico ADME Prediction. Anti-Cancer Agents Med. Chem. 2022, 22, 1370–1385. [Google Scholar] [CrossRef] [PubMed]
- Sable, P.M.; Potey, L.C. Synthesis and Antiproliferative Activity of Imidazole and Triazole Derivatives of Flavonoids. Pharm. Chem. J. 2018, 52, 438–443. [Google Scholar] [CrossRef]
- Eissa, A.G.; Powell, L.E.; Gee, J.; Foster, P.A.; Simons, C. Pyridine Based Dual Binding Site Aromatase (CYP19A1) Inhibitors. RSC Med. Chem. 2023, 14, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Eissa, A.G.; Barrow, D.; Gee, J.; Powell, L.E.; Foster, P.A.; Simons, C. 4th Generation Nonsteroidal Aromatase Inhibitors: An Iterative SAR-Guided Design, Synthesis, and Biological Evaluation towards Picomolar Dual Binding Inhibitors. Eur. J. Med. Chem. 2022, 240, 114569. [Google Scholar] [CrossRef]
- Sobh, E.A.; Khalil, N.A.; Faggal, S.I.; Hassan, M.S.A. New Benzothienopyrimidine Derivatives as Dual EGFR/ARO Inhibitors: Design, Synthesis, and Their Cytotoxic Effect on MCF-7 Breast Cancer Cell Line. Drug. Dev. Res. 2022, 83, 1075–1096. [Google Scholar] [CrossRef]
- Golani, P.; Shah, S.K.; Tyagi, C.K. Novel 5-(4-bromophenyl)-1,3-oxazole derivatives: Synthesis, Characterization, Docking and Evaluation of in vitro Anticancer Activity. Int. J. Pharm. Res. 2021, 13, 328. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Mad Nasir, N.; Stanslas, J.; Imran Faisal Hamdi, A.; Alif Mohammad Latif, M.; Farhana Baharuddin, F. One-Pot Two-Component Synthesis of Halogenated Xanthone, 3-o Substituted Xanthone, and Prenylated Xanthone Derivatives as Aromatase Inhibitors. Results Chem. 2023, 5, 100789. [Google Scholar] [CrossRef]
- Lekgau, K.; Raphoko, L.A.; Lebepe, C.M.; Mongokoana, D.F.; Leboho, T.C.; Matsebatlela, T.M.; Gumede, N.J.; Nxumalo, W. Design and Synthesis of 6-Amino-Quinoxaline-Alkynyl as Potential Aromatase (CYP19A1) Inhibitors. J. Mol. Struct. 2022, 1255, 132473. [Google Scholar] [CrossRef]
- Burdzhiev, N.T.; Baramov, T.I.; Stanoeva, E.R.; Yanev, S.G.; Stoyanova, T.D.; Dimitrova, D.H.; Kostadinova, K.A. Synthesis of Novel Trans-4-(Phthalimidomethyl)- and 4-(Imidazol-1-Ylmethyl)-3-Indolyl-Tetrahydroisoquinolinones as Possible Aromatase Inhibitors. Chem. Pap. 2019, 73, 1263–1277. [Google Scholar] [CrossRef]
- Yi, X.; El-Idreesy, T.T.; Eldebss, T.M.A.; Farag, A.M.; Abdulla, M.M.; Hassan, S.A.; Mabkhot, Y.N. Synthesis, Biological Evaluation, and Molecular Docking Studies of New Pyrazol-3-one Derivatives with Aromatase Inhibition Activities. Chem. Biol. Drug Des. 2016, 88, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Ertas, M.; Sahin, Z.; Berk, B.; Yurttas, L.; Biltekin, S.N.; Demirayak, S. Pyridine-substituted Thiazolylphenol Derivatives: Synthesis, Modeling Studies, Aromatase Inhibition, and Antiproliferative Activity Evaluation. Arch. Pharm. 2018, 351, 1700272. [Google Scholar] [CrossRef] [PubMed]
- Gomha, S.M.; Abdelrazek, F.M.; Abdulla, M.M. Synthesis of New Functionalised Derivatives of [1,2,4]Triazolo[4,3-a]Pyrimidine and Pyrimido[2,1-b][1,3,5]Thiadiazine as Aromatase Inhibitors. J. Chem. Res. 2015, 39, 425–429. [Google Scholar] [CrossRef]
- Osmaniye, D.; Karaca, Ş.; Kurban, B.; Baysal, M.; Ahmad, I.; Patel, H.; Özkay, Y.; Asım Kaplancıklı, Z. Design, Synthesis, Molecular Docking and Molecular Dynamics Studies of Novel Triazolothiadiazine Derivatives Containing Furan or Thiophene Rings as Anticancer Agents. Bioorg. Chem. 2022, 122, 105709. [Google Scholar] [CrossRef]
- El-Naggar, M.; Abd El-All, A.S.; El-Naem, S.I.A.; Abdalla, M.M.; Rashdan, H.R.M. New Potent 5α- Reductase and Aromatase Inhibitors Derived from 1,2,3-Triazole Derivative. Molecules 2020, 25, 672. [Google Scholar] [CrossRef]
- Evren, A.E.; Nuha, D.; Dawbaa, S.; Sağlık, B.N.; Yurttaş, L. Synthesis of Novel Thiazolyl Hydrazone Derivatives as Potent Dual Monoamine Oxidase-Aromatase Inhibitors. Eur. J. Med. Chem. 2022, 229, 114097. [Google Scholar] [CrossRef]
- Osmaniye, D.; Görgülü, Ş.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, in Vitro and in Silico Studies of Some Novel Thiazole-Dihydrofuran Derivatives as Aromatase Inhibitors. Bioorg. Chem. 2021, 114, 105123. [Google Scholar] [CrossRef]
- Ozcan-Sezer, S.; Ince, E.; Akdemir, A.; Ceylan, Ö.Ö.; Suzen, S.; Gurer-Orhan, H. Aromatase Inhibition by 2-Methyl Indole Hydrazone Derivatives Evaluated via Molecular Docking and In Vitro Activity Studies. Xenobiotica 2019, 49, 549–556. [Google Scholar] [CrossRef]
- Farghaly, A.M.; AboulWafa, O.M.; Baghdadi, H.H.; Abd El Razik, H.A.; Sedra, S.M.Y.; Shamaa, M.M. New Thieno[3,2-d]Pyrimidine-Based Derivatives: Design, Synthesis and Biological Evaluation as Antiproliferative Agents, EGFR and ARO Inhibitors Inducing Apoptosis in Breast Cancer Cells. Bioorg. Chem. 2021, 115, 105208. [Google Scholar] [CrossRef] [PubMed]
- Osmaniye, D.; Levent, S.; Sağlık, B.N.; Karaduman, A.B.; Özkay, Y.; Kaplancıklı, Z.A. Novel Imidazole Derivatives as Potential Aromatase and Monoamine Oxidase-B Inhibitors against Breast Cancer. New J. Chem. 2022, 46, 7442–7451. [Google Scholar] [CrossRef]
- Fadaly, W.A.; Elshaier, Y.A.; Nemr, M.T.; Abdellatif, K.R. Design, Synthesis, Modeling Studies and Biological Evaluation of Pyrazole Derivatives Linked to Oxime and Nitrate Moieties as Nitric Oxide Donor Selective COX-2 and Aromatase Inhibitors with Dual Anti-Inflammatory and Anti-Neoplastic Activities. Bioorg. Chem. 2023, 134, 106428. [Google Scholar] [CrossRef] [PubMed]
- Giampietro, L.; Gallorini, M.; Gambacorta, N.; Ammazzalorso, A.; De Filippis, B.; Della Valle, A.; Fantacuzzi, M.; Maccallini, C.; Mollica, A.; Cataldi, A.; et al. Synthesis, Structure-Activity Relationships and Molecular Docking Studies of Phenyldiazenyl Sulfonamides as Aromatase Inhibitors. Eur. J. Med. Chem. 2021, 224, 113737. [Google Scholar] [CrossRef] [PubMed]
- Fantacuzzi, M.; Gallorini, M.; Gambacorta, N.; Ammazzalorso, A.; Aturki, Z.; Balaha, M.; Carradori, S.; Giampietro, L.; Maccallini, C.; Cataldi, A.; et al. Design, Synthesis and Biological Evaluation of Aromatase Inhibitors Based on Sulfonates and Sulfonamides of Resveratrol. Pharmaceuticals 2021, 14, 984. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Alsaid, M.S.; Samir, N.; Abdel-Latif, G.A.; Soliman, A.M.; Ragab, F.A.; Abou El Ella, D.A. Aromatase Inhibitors and Apoptotic Inducers: Design, Synthesis, Anticancer Activity and Molecular Modeling Studies of Novel Phenothiazine Derivatives Carrying Sulfonamide Moiety as Hybrid Molecules. Eur. J. Med. Chem. 2017, 134, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Fantacuzzi, M.; De Filippis, B.; Gallorini, M.; Ammazzalorso, A.; Giampietro, L.; Maccallini, C.; Aturki, Z.; Donati, E.; Ibrahim, R.S.; Shawky, E.; et al. Synthesis, Biological Evaluation, and Docking Study of Indole Aryl Sulfonamides as Aromatase Inhibitors. Eur. J. Med. Chem. 2020, 185, 111815. [Google Scholar] [CrossRef]
- Chamduang, C.; Pingaew, R.; Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Novel Triazole-Tetrahydroisoquinoline Hybrids as Human Aromatase Inhibitors. Bioorg. Chem. 2019, 93, 103327. [Google Scholar] [CrossRef]
- Leechaisit, R.; Pingaew, R.; Prachayasittikul, V.; Worachartcheewan, A.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis, Molecular Docking, and QSAR Study of Bis-Sulfonamide Derivatives as Potential Aromatase Inhibitors. Bioorg. Med. Chem. 2019, 27, 115040. [Google Scholar] [CrossRef]
- Pingaew, R.; Prachayasittikul, V.; Anuwongcharoen, N.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis and Molecular Docking of N,N′-Disubstituted Thiourea Derivatives as Novel Aromatase Inhibitors. Bioorg. Chem. 2018, 79, 171–178. [Google Scholar] [CrossRef]
- Caciolla, J.; Martini, S.; Spinello, A.; Pavlin, M.; Turrini, E.; Simonelli, F.; Belluti, F.; Rampa, A.; Bisi, A.; Fimognari, C.; et al. Balanced Dual Acting Compounds Targeting Aromatase and Estrogen Receptor α as an Emerging Therapeutic Opportunity to Counteract Estrogen Responsive Breast Cancer. Eur. J. Med. Chem. 2021, 224, 113733. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Liu, J.; Skaar, T.C.; O’Neill, E.; Yu, G.; Flockhart, D.A.; Cushman, M. Synthesis of Triphenylethylene Bisphenols as Aromatase Inhibitors That Also Modulate Estrogen Receptors. J. Med. Chem. 2016, 59, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-M.; Jin, H.-S.; Liu, J.; Skaar, T.C.; Ipe, J.; Lv, W.; Flockhart, D.A.; Cushman, M. A New Suzuki Synthesis of Triphenylethylenes That Inhibit Aromatase and Bind to Estrogen Receptors α and β. Bioorg. Med. Chem. 2016, 24, 5400–5409. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Liu, J.; Skaar, T.C.; Flockhart, D.A.; Cushman, M. Design and Synthesis of Norendoxifen Analogues with Dual Aromatase Inhibitory and Estrogen Receptor Modulatory Activities. J. Med. Chem. 2015, 58, 2623–2648. [Google Scholar] [CrossRef]
- Neves, M.A.C.; Dinis, T.C.P.; Colombo, G.; Sá e Melo, M.L. Fast Three Dimensional Pharmacophore Virtual Screening of New Potent Non-Steroid Aromatase Inhibitors. J. Med. Chem. 2009, 52, 143–150. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name and Chemical Structure of Kinase Inhibitor | Aromatase Inhibitors Used in Clinical Trials/and Other Drug | ClinicalTrials.gov ID and the Phase of Clinical Trials |
|---|---|---|
Abemaciclib | NSAI/FUL ANA, LET ANA ANA | NCT04707196; 4 NCT0224621; 3 NCT02763566; 3 NCT02441946; 2 |
Afatinib | LET | NCT02115048; 2 |
Alpelisib | LET | NCT01923168; 3 |
Everolimus | EXE EXE EXE, ANA EXE, ANA EXE LET | NCT01743560; 4 NCT03176238; 3 NCT02137837; 3 NCT01674140; 3 NCT01783444; 2 NCT02520063; 2, 1 |
MK-2206 | ANA | NCT01776008; 2 |
Palbociclib | LET LE/FUL AI LET LET ANA LET | NCT02297438; 3 NCT02692755; 3, 2 NCT02040857; 2 NCT02764541; 2 NCT02296801; 2 NCT01723774; 2 NCT02499146; 1 |
Ribociclib | LET LET LET AI EXE | NCT02278120; 3 NCT02941926; 3 NCT03050398; 3 NCT03477396; 2 NCT02732119; 2,1 |
Sapanisertib | EXE | NCT02049957; 2 |
Taselisib | LET | NCT02273973; 2 |
Tucatinib | LET | NCT03054363; 2,1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janowska, S.; Holota, S.; Lesyk, R.; Wujec, M. Aromatase Inhibitors as a Promising Direction for the Search for New Anticancer Drugs. Molecules 2024, 29, 346. https://doi.org/10.3390/molecules29020346
Janowska S, Holota S, Lesyk R, Wujec M. Aromatase Inhibitors as a Promising Direction for the Search for New Anticancer Drugs. Molecules. 2024; 29(2):346. https://doi.org/10.3390/molecules29020346
Chicago/Turabian StyleJanowska, Sara, Serhii Holota, Roman Lesyk, and Monika Wujec. 2024. "Aromatase Inhibitors as a Promising Direction for the Search for New Anticancer Drugs" Molecules 29, no. 2: 346. https://doi.org/10.3390/molecules29020346