
Metal Complexes for Dye-Sensitized Photoelectrochemical Cells (DSPECs)

Abstract
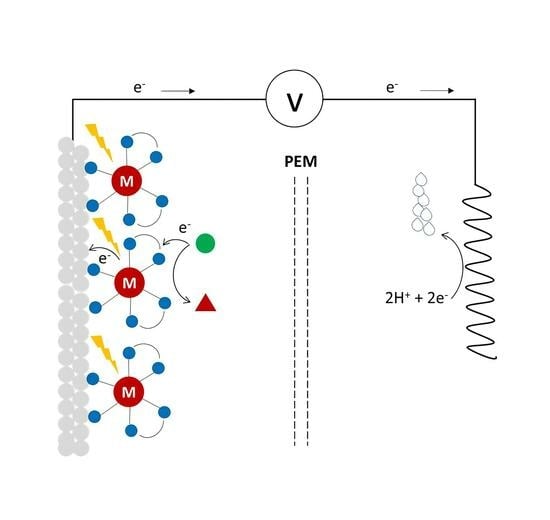
:
1. Introduction
2. Basic Concepts of DSPECs
3. Efficiency Parameters
4. Sensitizers and Surface Binding and Stabilization Strategies


















5. Water Splitting
5.1. Water Oxidation Catalyst (WOC)
5.2. Examples of Water splitting
Molecular Sensitizer–Catalyst Assembly










6. Hydrobromic Acid Oxidation (HBr)




7. Organic Reactions




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
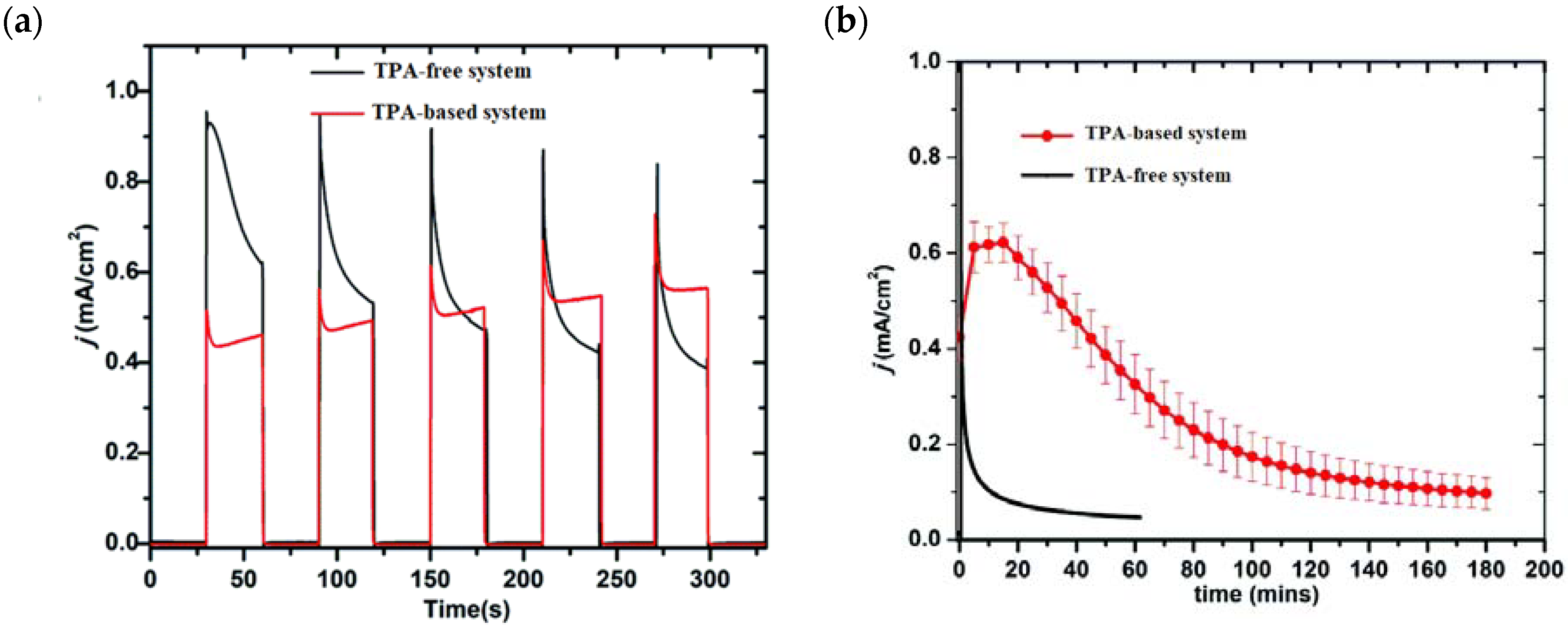{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
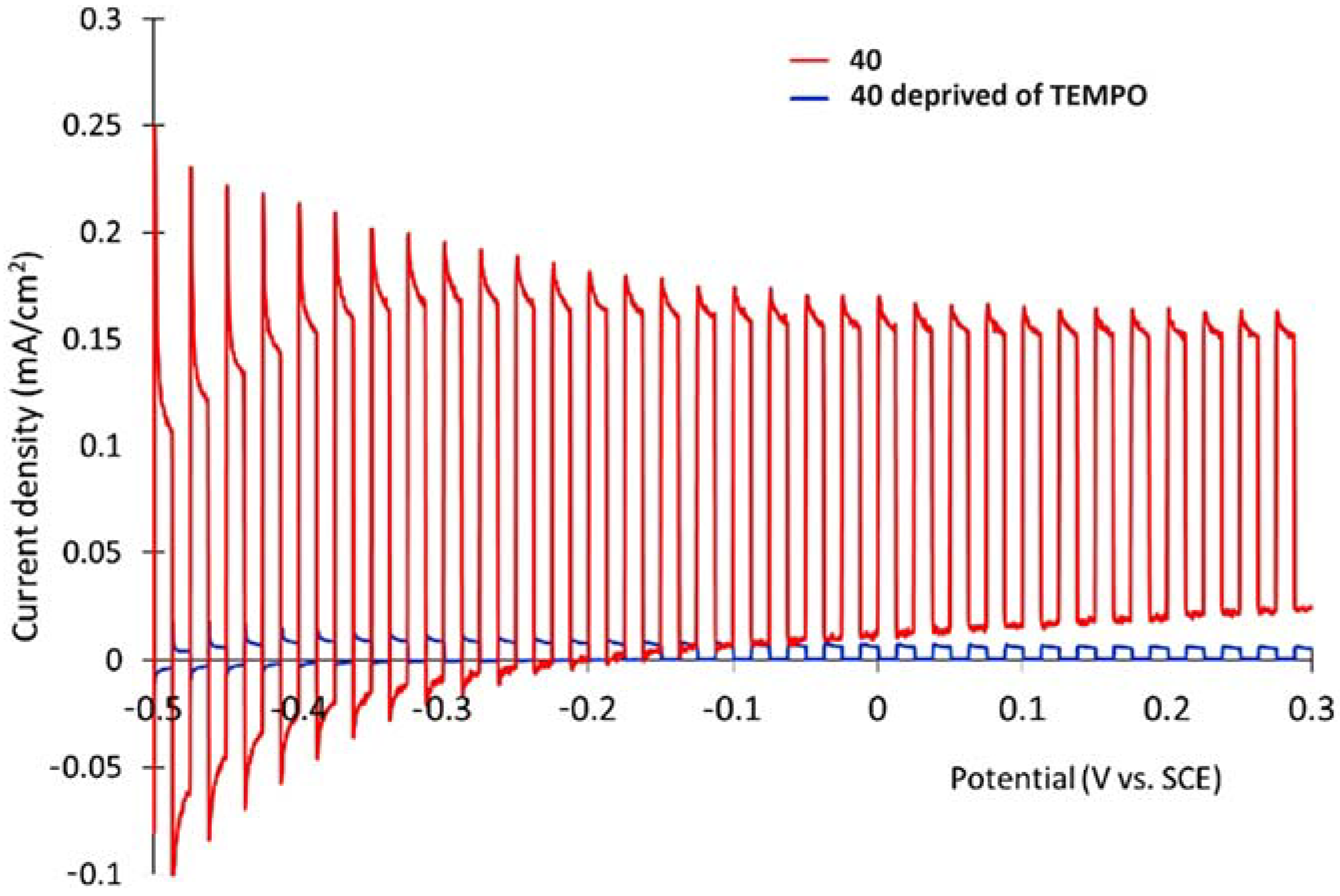{kind=link}
{kind=link}
{kind=link}
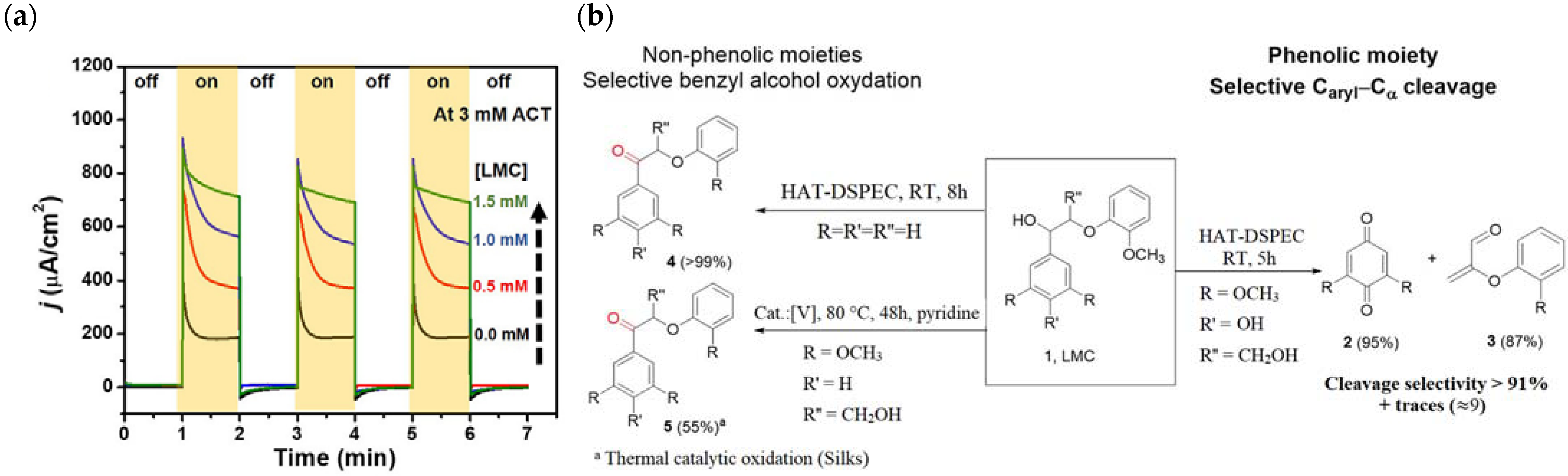{kind=link}
8. Conclusions and Future Outlooks
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Hammarstrom, L.; Hammes-Schiffer, S. Artificial photosynthesis and solar fuels. Acc. Chem. Res. 2009, 42, 1859–1860. [Google Scholar] [CrossRef] [PubMed]
- Maddah, H.A. Modeling the Relation between Carbon Dioxide Emissions and Sea Level Rise for the Determination of Future (2100) Sea Level. Am. J. Environ. Eng. 2016, 6, 52–61. [Google Scholar]
- Marques Lameirinhas, R.A.; Torres, J.P.N.; de Melo Cunha, J.P. A Photovoltaic Technology Review: History, Fundamentals and Applications. Energies 2022, 15, 1823. [Google Scholar] [CrossRef]
- Sudhagar, P.; Roy, N.; Vedarajan, R.; Devadoss, A.; Terashima, C.; Nakata, K.; Fujishima, A. Hydrogen and CO2 Reduction Reactions: Mechanisms and Catalysts. In Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices; Giménez, S., Bisquert, J., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 105–160. [Google Scholar]
- Newman, J.; Hoertz, P.G.; Bonino, C.A.; Trainham, J.A. Review: An Economic Perspective on Liquid Solar Fuels. J. Electrochem. Soc. 2012, 159, A1722–A1729. [Google Scholar] [CrossRef]
- Li, H.; Tu, W.; Zhou, Y.; Zou, Z. Z-Scheme Photocatalytic Systems for Promoting Photocatalytic Performance: Recent Progress and Future Challenges. Adv. Sci. 2016, 3, 1500389. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, K.; Grätzel, M. Cyclic Cleavage of Water into H2 and O2 by Visible Light with Coupled Redox Catalysts. Angew. Chem. Int. Ed. Engl. 1979, 18, 701–702. [Google Scholar] [CrossRef]
- Grätzel, M. Photochemical Methods for the Conversion of Light into Chemical Energy. Berichte Der Bunsenges. Für Phys. Chem. 1980, 84, 981–991. [Google Scholar] [CrossRef]
- Duonghong, D.; Borgarello, E.; Graetzel, M. Dynamics of light-induced water cleavage in colloidal systems. J. Am. Chem. Soc. 2002, 103, 4685–4690. [Google Scholar] [CrossRef]
- Cristino, V.; Caramori, S.; Argazzi, R.; Meda, L.; Marra, G.L.; Bignozzi, C.A. Efficient photoelectrochemical water splitting by anodically grown WO3 electrodes. Langmuir 2011, 27, 7276–7284. [Google Scholar] [CrossRef] [PubMed]
- Bignozzi, C.A.; Caramori, S.; Cristino, V.; Argazzi, R.; Meda, L.; Tacca, A. Nanostructured photoelectrodes based on WO3: Applications to photooxidation of aqueous electrolytes. Chem. Soc. Rev. 2013, 42, 2228–2246. [Google Scholar] [CrossRef] [PubMed]
- Berardi, S.; Cristino, V.; Bignozzi, C.A.; Grandi, S.; Caramori, S. Hematite-based photoelectrochemical interfaces for solar fuel production. Inorg. Chim. Acta 2022, 535, 120862. [Google Scholar] [CrossRef]
- Berardi, S.; Kopula Kesavan, J.; Amidani, L.; Meloni, E.M.; Marelli, M.; Boscherini, F.; Caramori, S.; Pasquini, L. Better Together: Ilmenite/Hematite Junctions for Photoelectrochemical Water Oxidation. ACS Appl. Mater. Interfaces 2020, 12, 47435–47446. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Pendlebury, S.R.; Reynal, A.; Le Formal, F.; Durrant, J.R. Dynamics of photogenerated holes in undoped BiVO4 photoanodes for solar water oxidation. Chem. Sci. 2014, 5, 2964–2973. [Google Scholar] [CrossRef]
- Vecchi, P.; Piccioni, A.; Mazzaro, R.; Mazzanti, M.; Cristino, V.; Caramori, S.; Pasquini, L. Charge Separation Efficiency in WO3/BiVO4 Photoanodes with CoFe Prussian Blue Catalyst Studied by Wavelength-Dependent Intensity-Modulated Photocurrent Spectroscopy. Sol. RRL 2022, 6, 2200108. [Google Scholar] [CrossRef]
- Lhermitte, C.R.; Bartlett, B.M. Advancing the Chemistry of CuWO4 for Photoelectrochemical Water Oxidation. Acc. Chem. Res. 2016, 49, 1121–1129. [Google Scholar] [CrossRef]
- Youngblood, W.J.; Lee, S.-H.A.; Kobayashi, Y.; Hernandez-Pagan, E.A.; Hoertz, P.G.; Moore, T.A.; Moore, A.L.; Gust, D.; Mallouk, T.E. Photoassisted Overall Water Splitting in a Visible Light-Absorbing Dye-Sensitized Photoelectrochemical Cell. J. Am. Chem. Soc. 2009, 131, 926–927. [Google Scholar] [CrossRef]
- Yu, Z.; Li, F.; Sun, L. Recent advances in dye-sensitized photoelectrochemical cells for solar hydrogen production based on molecular components. Energy Environ. Sci. 2015, 8, 760–775. [Google Scholar] [CrossRef]
- Liang, X.; Cao, X.; Sun, W.; Ding, Y. Recent Progress in Visible Light Driven Water Oxidation Using Semiconductors Coupled with Molecular Catalysts. ChemCatChem 2019, 11, 6190–6202. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, L. Artificial photosynthesis: Opportunities and challenges of molecular catalysts. Chem. Soc. Rev. 2019, 48, 2216–2264. [Google Scholar] [CrossRef]
- Nocera, D.G. Chemistry of personalized solar energy. Inorg. Chem. 2009, 48, 10001–10017. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Steeper, R.; Lutz, A. The hydrogen-fueled internal combustion engine: A technical review. Int. J. Hydrogen Energy 2006, 31, 1292–1305. [Google Scholar] [CrossRef]
- Livshits, V.; Ulus, A.; Peled, E. High-power H2/Br2 fuel cell. Electrochem. Commun. 2006, 8, 1358–1362. [Google Scholar] [CrossRef]
- Kreutzer, H.; Yarlagadda, V.; Van Nguyen, T. Performance Evaluation of a Regenerative Hydrogen-Bromine Fuel Cell. J. Electrochem. Soc. 2012, 159, F331–F337. [Google Scholar] [CrossRef]
- Cho, K.T.; Ridgway, P.; Weber, A.Z.; Haussener, S.; Battaglia, V.; Srinivasan, V. High Performance Hydrogen/Bromine Redox Flow Battery for Grid-Scale Energy Storage. J. Electrochem. Soc. 2012, 159, A1806–A1815. [Google Scholar] [CrossRef]
- Nikoloudakis, E.; Pati, P.B.; Charalambidis, G.; Budkina, D.S.; Diring, S.; Planchat, A.; Jacquemin, D.; Vauthey, E.; Coutsolelos, A.G.; Odobel, F. Dye-Sensitized Photoelectrosynthesis Cells for Benzyl Alcohol Oxidation Using a Zinc Porphyrin Sensitizer and TEMPO Catalyst. ACS Catal. 2021, 11, 12075–12086. [Google Scholar] [CrossRef]
- Rahimi, N.; Pax, R.A.; Gray, E.M. Review of functional titanium oxides. I: TiO2 and its modifications. Prog. Solid State Chem. 2016, 44, 86–105. [Google Scholar] [CrossRef]
- Sun, C.; Yang, J.; Xu, M.; Cui, Y.; Ren, W.; Zhang, J.; Zhao, H.; Liang, B. Recent intensification strategies of SnO2-based photocatalysts: A review. Chem. Eng. J. 2022, 427, 131564. [Google Scholar] [CrossRef]
- Ghaffar, S.; Abbas, A.; Naeem-Ul-Hassan, M.; Assad, N.; Sher, M.; Ullah, S.; Alhazmi, H.A.; Najmi, A.; Zoghebi, K.; Al Bratty, M.; et al. Improved Photocatalytic and Antioxidant Activity of Olive Fruit Extract-Mediated ZnO Nanoparticles. Antioxidants 2023, 12, 1201. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef]
- Bomben, P.G.; Robson, K.C.D.; Koivisto, B.D.; Berlinguette, C.P. Cyclometalated ruthenium chromophores for the dye-sensitized solar cell. Coord. Chem. Rev. 2012, 256, 1438–1450. [Google Scholar] [CrossRef]
- Kaneko, H.; Minegishi, T.; Nakabayashi, M.; Shibata, N.; Kuang, Y.; Yamada, T.; Domen, K. A Novel Photocathode Material for Sunlight-Driven Overall Water Splitting: Solid Solution of ZnSe and Cu(In,Ga)Se2. Adv. Funct. Mater. 2016, 26, 4570–4577. [Google Scholar] [CrossRef]
- Hammarstrom, L. Accumulative charge separation for solar fuels production: Coupling light-induced single electron transfer to multielectron catalysis. Acc. Chem. Res. 2015, 48, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Nozik, A.J. Photoelectrolysis of water using semiconducting TiO2 crystals. Nature 1975, 257, 383–386. [Google Scholar] [CrossRef]
- Nayak, P.K.; Garcia-Belmonte, G.; Kahn, A.; Bisquert, J.; Cahen, D. Photovoltaic efficiency limits and material disorder. Energy Environ. Sci. 2012, 5, 6022–6039. [Google Scholar] [CrossRef]
- Yun, S.; Vlachopoulos, N.; Qurashi, A.; Ahmad, S.; Hagfeldt, A. Dye sensitized photoelectrolysis cells. Chem. Soc. Rev. 2019, 48, 3705–3722. [Google Scholar] [CrossRef] [PubMed]
- Galoppini, E. Linkers for anchoring sensitizers to semiconductor nanoparticles. Coord. Chem. Rev. 2004, 248, 1283–1297. [Google Scholar] [CrossRef]
- Bignozzi, C.A.; Argazzi, R.; Boaretto, R.; Busatto, E.; Carli, S.; Ronconi, F.; Caramori, S. The role of transition metal complexes in dye sensitized solar devices. Coord. Chem. Rev. 2013, 257, 1472–1492. [Google Scholar] [CrossRef]
- Polo, A.S.; Itokazu, M.K.; Murakami Iha, N.Y. Metal complex sensitizers in dye-sensitized solar cells. Coord. Chem. Rev. 2004, 248, 1343–1361. [Google Scholar] [CrossRef]
- Park, H.; Bae, E.; Lee, J.J.; Park, J.; Choi, W. Effect of the anchoring group in Ru-bipyridyl sensitizers on the photoelectrochemical behavior of dye-sensitized TiO2 electrodes: Carboxylate versus phosphonate linkages. J. Phys. Chem. B 2006, 110, 8740–8749. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.; Choi, W. Effect of the anchoring group (carboxylate vs phosphonate) in Ru-complex-sensitized TiO2 on hydrogen production under visible light. J. Phys. Chem. B 2006, 110, 14792–14799. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.; Brennaman, M.K.; Luo, H.; Glasson, C.R.; Concepcion, J.J.; Song, W.; Meyer, T.J. Photostability of phosphonate-derivatized, Ru(II) polypyridyl complexes on metal oxide surfaces. ACS Appl. Mater. Interfaces 2012, 4, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.T.; Hanson, K.; Vannucci, A.K.; Lapides, A.M.; Alibabaei, L.; Norris, M.R.; Meyer, T.J.; Harrison, D.P. Electrochemical Instability of Phosphonate-Derivatized, Ruthenium(III) Polypyridyl Complexes on Metal Oxide Surfaces. ACS Appl. Mater. Interfaces 2015, 7, 9554–9562. [Google Scholar] [CrossRef] [PubMed]
- Raber, M.M.; Brady, M.D.; Troian-Gautier, L.; Dickenson, J.C.; Marquard, S.L.; Hyde, J.T.; Lopez, S.J.; Meyer, G.J.; Meyer, T.J.; Harrison, D.P. Fundamental Factors Impacting the Stability of Phosphonate-Derivatized Ruthenium Polypyridyl Sensitizers Adsorbed on Metal Oxide Surfaces. ACS Appl. Mater. Interfaces 2018, 10, 22821–22833. [Google Scholar] [CrossRef] [PubMed]
- Gillaizeau-Gauthier, I.; Odobel, F.; Alebbi, M.; Argazzi, R.; Costa, E.; Bignozzi, C.A.; Qu, P.; Meyer, G.J. Phosphonate-based bipyridine dyes for stable photovoltaic devices. Inorg. Chem. 2001, 40, 6073–6079. [Google Scholar] [CrossRef] [PubMed]
- Wee, K.R.; Brennaman, M.K.; Alibabaei, L.; Farnum, B.H.; Sherman, B.; Lapides, A.M.; Meyer, T.J. Stabilization of ruthenium(II) polypyridyl chromophores on nanoparticle metal-oxide electrodes in water by hydrophobic PMMA overlayers. J. Am. Chem. Soc. 2014, 136, 13514–13517. [Google Scholar] [CrossRef]
- Lapides, A.M.; Ashford, D.L.; Hanson, K.; Torelli, D.A.; Templeton, J.L.; Meyer, T.J. Stabilization of a ruthenium(II) polypyridyl dye on nanocrystalline TiO2 by an electropolymerized overlayer. J. Am. Chem. Soc. 2013, 135, 15450–15458. [Google Scholar] [CrossRef]
- Ashford, D.L.; Lapides, A.M.; Vannucci, A.K.; Hanson, K.; Torelli, D.A.; Harrison, D.P.; Templeton, J.L.; Meyer, T.J. Water oxidation by an electropolymerized catalyst on derivatized mesoporous metal oxide electrodes. J. Am. Chem. Soc. 2014, 136, 6578–6581. [Google Scholar] [CrossRef]
- Ashford, D.L.; Sherman, B.D.; Binstead, R.A.; Templeton, J.L.; Meyer, T.J. Electro-assembly of a chromophore-catalyst bilayer for water oxidation and photocatalytic water splitting. Angew. Chem. Int. Ed. Engl. 2015, 54, 4778–4781. [Google Scholar] [CrossRef]
- Moss, J.A.; Yang, J.C.; Stipkala, J.M.; Wen, X.; Bignozzi, C.A.; Meyer, G.J.; Meyer, T.J. Sensitization and stabilization of TiO2 photoanodes with electropolymerized overlayer films of ruthenium and zinc polypyridyl complexes: A stable aqueous photoelectrochemical cell. Inorg. Chem. 2004, 43, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Schauer, P.A.; Borau-Garcia, J.; Fancy, B.R.; Berlinguette, C.P. Stabilization of ruthenium sensitizers to TiO2 surfaces through cooperative anchoring groups. J. Am. Chem. Soc. 2013, 135, 1692–1695. [Google Scholar] [CrossRef] [PubMed]
- Szpakolski, K.; Latham, K.; Rix, C.; Rani, R.A.; Kalantar-zadeh, K. Silane: A new linker for chromophores in dye-sensitised solar cells. Polyhedron 2013, 52, 719–732. [Google Scholar] [CrossRef]
- Materna, K.L.; Rudshteyn, B.; Brennan, B.J.; Kane, M.H.; Bloomfield, A.J.; Huang, D.L.; Shopov, D.Y.; Batista, V.S.; Crabtree, R.H.; Brudvig, G.W. Heterogenized Iridium Water-Oxidation Catalyst from a Silatrane Precursor. ACS Catal. 2016, 6, 5371–5377. [Google Scholar] [CrossRef]
- Materna, K.L.; Brennan, B.J.; Brudvig, G.W. Silatranes for binding inorganic complexes to metal oxide surfaces. Dalton Trans. 2015, 44, 20312–20315. [Google Scholar] [CrossRef]
- Brennan, B.J.; Llansola Portoles, M.J.; Liddell, P.A.; Moore, T.A.; Moore, A.L.; Gust, D. Comparison of silatrane, phosphonic acid, and carboxylic acid functional groups for attachment of porphyrin sensitizers to TiO2 in photoelectrochemical cells. Phys. Chem. Chem. Phys. 2013, 15, 16605–16614. [Google Scholar] [CrossRef] [PubMed]
- Zakeeruddin, S.M.; Nazeeruddin, M.K.; Humphry-Baker, R.; Péchy, P.; Quagliotto, P.; Barolo, C.; Viscardi, G.; Grätzel, M. Design, Synthesis, and Application of Amphiphilic Ruthenium Polypyridyl Photosensitizers in Solar Cells Based on Nanocrystalline TiO2 Films. Langmuir 2002, 18, 952–954. [Google Scholar] [CrossRef]
- Vannucci, A.K.; Alibabaei, L.; Losego, M.D.; Concepcion, J.J.; Kalanyan, B.; Parsons, G.N.; Meyer, T.J. Crossing the divide between homogeneous and heterogeneous catalysis in water oxidation. Proc. Natl. Acad. Sci. USA 2013, 110, 20918–20922. [Google Scholar] [CrossRef]
- Lapides, A.M.; Sherman, B.D.; Brennaman, M.K.; Dares, C.J.; Skinner, K.R.; Templeton, J.L.; Meyer, T.J. Synthesis, characterization, and water oxidation by a molecular chromophore-catalyst assembly prepared by atomic layer deposition. The “mummy” strategy. Chem. Sci. 2015, 6, 6398–6406. [Google Scholar] [CrossRef]
- Takijiri, K.; Morita, K.; Nakazono, T.; Sakai, K.; Ozawa, H. Highly stable chemisorption of dyes with pyridyl anchors over TiO2: Application in dye-sensitized photoelectrochemical water reduction in aqueous media. Chem. Commun. 2017, 53, 3042–3045. [Google Scholar] [CrossRef]
- Ooyama, Y.; Inoue, S.; Nagano, T.; Kushimoto, K.; Ohshita, J.; Imae, I.; Komaguchi, K.; Harima, Y. Dye-Sensitized Solar Cells Based On Donor-Acceptor π-Conjugated Fluorescent Dyes with a Pyridine Ring as an Electron-Withdrawing Anchoring Group. Angew. Chem. 2011, 123, 7567–7571. [Google Scholar] [CrossRef]
- Ooyama, Y.; Nagano, T.; Inoue, S.; Imae, I.; Komaguchi, K.; Ohshita, J.; Harima, Y. Dye-sensitized solar cells based on donor-pi-acceptor fluorescent dyes with a pyridine ring as an electron-withdrawing-injecting anchoring group. Chemistry 2011, 17, 14837–14843. [Google Scholar] [CrossRef]
- Shibayama, N.; Ozawa, H.; Abe, M.; Ooyama, Y.; Arakawa, H. A new cosensitization method using the Lewis acid sites of a TiO2 photoelectrode for dye-sensitized solar cells. Chem. Commun. 2014, 50, 6398–6401. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.L.; Xu, P.; Rothenberger, A.J.; Koehler, S.J.; Elacqua, E.; Milosavljevic, B.H.; Mallouk, T.E. Oligomeric Ruthenium Polypyridyl Dye for Improved Stability of Aqueous Photoelectrochemical Cells. J. Phys. Chem. C 2020, 124, 3542–3550. [Google Scholar] [CrossRef]
- Swierk, J.R.; McCool, N.S.; Saunders, T.P.; Barber, G.D.; Mallouk, T.E. Effects of electron trapping and protonation on the efficiency of water-splitting dye-sensitized solar cells. J. Am. Chem. Soc. 2014, 136, 10974–10982. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Ardo, S. Direct observation of sequential oxidations of a titania-bound molecular proxy catalyst generated through illumination of molecular sensitizers. Nat. Chem. 2018, 10, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ardo, S.; Meyer, G.J. Direct observation of photodriven intermolecular hole transfer across TiO2 nanocrystallites: Lateral self-exchange reactions and catalyst oxidation. J. Am. Chem. Soc. 2010, 132, 9283–9285. [Google Scholar] [CrossRef]
- Bangle, R.; Sampaio, R.N.; Troian-Gautier, L.; Meyer, G.J. Surface Grafting of Ru(II) Diazonium-Based Sensitizers on Metal Oxides Enhances Alkaline Stability for Solar Energy Conversion. ACS Appl. Mater. Interfaces 2018, 10, 3121–3132. [Google Scholar] [CrossRef]
- Linfoot, C.L.; Richardson, P.; McCall, K.L.; Durrant, J.R.; Morandeira, A.; Robertson, N. A nickel-complex sensitiser for dye-sensitised solar cells. Sol. Energy 2011, 85, 1195–1203. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, K.; Zhang, H.; Liu, H.; Tian, Y.; Wang, Y.; Zhong, C. Sensitizers of Metal Complexes with Sulfur Coordination Achieving a Power Conversion Efficiency of 12.89. ACS Appl. Mater. Interfaces 2023, 15, 35251–35260. [Google Scholar] [CrossRef]
- Conradie, J. Polypyridyl copper complexes as dye sensitizer and redox mediator for dye-sensitized solar cells. Electrochem. Commun. 2022, 134, 107182. [Google Scholar] [CrossRef]
- Marri, A.R.; Marchini, E.; Cabanes, V.D.; Argazzi, R.; Pastore, M.; Caramori, S.; Bignozzi, C.A.; Gros, P.C. A Series of Iron(II)-NHC Sensitizers with Remarkable Power Conversion Efficiency in Photoelectrochemical Cells. Chemistry 2021, 27, 16260–16269. [Google Scholar] [CrossRef]
- Tichnell, C.R.; Miller, J.N.; Liu, C.; Mukherjee, S.; Jakubikova, E.; McCusker, J.K. Influence of Electrolyte Composition on Ultrafast Interfacial Electron Transfer in Fe-Sensitized TiO2-Based Solar Cells. J. Phys. Chem. C 2020, 124, 1794–1811. [Google Scholar] [CrossRef]
- Orbelli Biroli, A.; Tessore, F.; Di Carlo, G.; Pizzotti, M.; Benazzi, E.; Gentile, F.; Berardi, S.; Bignozzi, C.A.; Argazzi, R.; Natali, M.; et al. Fluorinated Zn(II) Porphyrins for Dye-Sensitized Aqueous Photoelectrosynthetic Cells. ACS Appl. Mater. Interfaces 2019, 11, 32895–32908. [Google Scholar] [CrossRef] [PubMed]
- Berardi, S.; Caramori, S.; Benazzi, E.; Zabini, N.; Niorettini, A.; Orbelli Biroli, A.; Pizzotti, M.; Tessore, F.; Di Carlo, G. Electronic Properties of Electron-Deficient Zn(II) Porphyrins for HBr Splitting. Appl. Sci. 2019, 9, 2739. [Google Scholar] [CrossRef]
- Mussini, P.R.; Orbelli Biroli, A.; Tessore, F.; Pizzotti, M.; Biaggi, C.; Di Carlo, G.; Lobello, M.G.; De Angelis, F. Modulating the electronic properties of asymmetric push–pull and symmetric Zn(II)-diarylporphyrinates with para substituted phenylethynyl moieties in 5,15 meso positions: A combined electrochemical and spectroscopic investigation. Electrochim. Acta 2012, 85, 509–523. [Google Scholar] [CrossRef]
- Moore, G.F.; Blakemore, J.D.; Milot, R.L.; Hull, J.F.; Song, H.-e.; Cai, L.; Schmuttenmaer, C.A.; Crabtree, R.H.; Brudvig, G.W. A visible light water-splitting cell with a photoanode formed by codeposition of a high-potential porphyrin and an iridium water-oxidation catalyst. Energy Environ. Sci. 2011, 4, 2389–2392. [Google Scholar] [CrossRef]
- Nayak, A.; Roy, S.; Sherman, B.D.; Alibabaei, L.; Lapides, A.M.; Brennaman, M.K.; Wee, K.R.; Meyer, T.J. Phosphonate-Derivatized Porphyrins for Photoelectrochemical Applications. ACS Appl. Mater. Interfaces 2016, 8, 3853–3860. [Google Scholar] [CrossRef]
- Akamine, K.; Morita, K.; Sakai, K.; Ozawa, H. A Molecular-Based Water Electrolyzer Consisting of Two Mesoporous TiO2 Electrodes Modified with Metalloporphyrin Molecular Catalysts Showing a Quantitative Faradaic Efficiency. ACS Appl. Energy Mater. 2020, 3, 4860–4866. [Google Scholar] [CrossRef]
- Urbani, M.; Ragoussi, M.-E.; Nazeeruddin, M.K.; Torres, T. Phthalocyanines for dye-sensitized solar cells. Coord. Chem. Rev. 2019, 381, 1–64. [Google Scholar] [CrossRef]
- Atta, N.F.; Amin, H.M.; Khalil, M.W.; Galal, A. Nanotube arrays as photoanodes for dye sensitized solar cells using metal phthalocyanine dyes. Int. J. Electrochem. Sci. 2011, 6, 3. [Google Scholar] [CrossRef]
- Nada, F.; Galal, A.; Amin, H.M. Synthesis and photoelectrochemical behavior of a hybrid electrode composed of polyaniline encapsulated in highly ordered TiO2 nanotubes array. Int. J. Electrochem. Sci. 2012, 7, 3610–3626. [Google Scholar] [CrossRef]
- Orbelli Biroli, A.; Tessore, F.; Pizzotti, M.; Biaggi, C.; Ugo, R.; Caramori, S.; Aliprandi, A.; Bignozzi, C.A.; De Angelis, F.; Giorgi, G.; et al. A Multitechnique Physicochemical Investigation of Various Factors Controlling the Photoaction Spectra and of Some Aspects of the Electron Transfer for a Series of Push–Pull Zn(II) Porphyrins Acting as Dyes in DSSCs. J. Phys. Chem. C 2011, 115, 23170–23182. [Google Scholar] [CrossRef]
- Bessho, T.; Zakeeruddin, S.M.; Yeh, C.Y.; Diau, E.W.; Gratzel, M. Highly efficient mesoscopic dye-sensitized solar cells based on donor-acceptor-substituted porphyrins. Angew. Chem. Int. Ed. Engl. 2010, 49, 6646–6649. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Vlachopoulos, N.; Hao, Y.; Hagfeldt, A.; Boschloo, G. Efficient dye regeneration at low driving force achieved in triphenylamine dye LEG4 and TEMPO redox mediator based dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2015, 17, 15868–15875. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, K.; Xu, B.; Gabrielsson, E.; Daniel, Q.; Li, L.; Sun, L. Organic Dye-Sensitized Tandem Photoelectrochemical Cell for Light Driven Total Water Splitting. J. Am. Chem. Soc. 2015, 137, 9153–9159. [Google Scholar] [CrossRef]
- Loudet, A.; Burgess, K. BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef]
- Ulrich, G.; Ziessel, R.; Harriman, A. The chemistry of fluorescent bodipy dyes: Versatility unsurpassed. Angew. Chem. Int. Ed. Engl. 2008, 47, 1184–1201. [Google Scholar] [CrossRef]
- Kubo, Y.; Minowa, Y.; Shoda, T.; Takeshita, K. Synthesis of a new type of dibenzopyrromethene–boron complex with near-infrared absorption property. Tetrahedron Lett. 2010, 51, 1600–1602. [Google Scholar] [CrossRef]
- Kubo, Y.; Eguchi, D.; Matsumoto, A.; Nishiyabu, R.; Yakushiji, H.; Shigaki, K.; Kaneko, M. Boron–dibenzopyrromethene-based organic dyes for application in dye-sensitized solar cells. J. Mater. Chem. A 2014, 2, 5204–5211. [Google Scholar] [CrossRef]
- Kubo, Y.; Watanabe, K.; Nishiyabu, R.; Hata, R.; Murakami, A.; Shoda, T.; Ota, H. Near-infrared absorbing boron-dibenzopyrromethenes that serve as light-harvesting sensitizers for polymeric solar cells. Org. Lett. 2011, 13, 4574–4577. [Google Scholar] [CrossRef] [PubMed]
- Suryani, O.; Higashino, Y.; Mulyana, J.Y.; Kaneko, M.; Hoshi, T.; Shigaki, K.; Kubo, Y. A near-infrared organic photosensitizer for use in dye-sensitized photoelectrochemical water splitting. Chem. Commun. 2017, 53, 6784–6787. [Google Scholar] [CrossRef] [PubMed]
- Badgurjar, D.; Shan, B.; Nayak, A.; Wu, L.; Chitta, R.; Meyer, T.J. Electron-Withdrawing Boron Dipyrromethene Dyes As Visible Light Absorber/Sensitizers on Semiconductor Oxide Surfaces. ACS Appl. Mater. Interfaces 2020, 12, 7768–7776. [Google Scholar] [CrossRef] [PubMed]
- Brimblecombe, R.; Koo, A.; Dismukes, G.C.; Swiegers, G.F.; Spiccia, L. Solar driven water oxidation by a bioinspired manganese molecular catalyst. J. Am. Chem. Soc. 2010, 132, 2892–2894. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Duan, L.; Xu, Y.; Gorlov, M.; Hagfeldt, A.; Sun, L. A photoelectrochemical device for visible light driven water splitting by a molecular ruthenium catalyst assembled on dye-sensitized nanostructured TiO2. Chem. Commun. 2010, 46, 7307–7309. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Vannucci, A.K.; Farnum, B.H.; Lapides, A.M.; Brennaman, M.K.; Kalanyan, B.; Alibabaei, L.; Concepcion, J.J.; Losego, M.D.; Parsons, G.N.; et al. Visible light driven benzyl alcohol dehydrogenation in a dye-sensitized photoelectrosynthesis cell. J. Am. Chem. Soc. 2014, 136, 9773–9779. [Google Scholar] [CrossRef] [PubMed]
- Michaux, K.E.; Gambardella, A.A.; Alibabaei, L.; Ashford, D.L.; Sherman, B.D.; Binstead, R.A.; Meyer, T.J.; Murray, R.W. Visible Photoelectrochemical Water Splitting Based on a Ru(II) Polypyridyl Chromophore and Iridium Oxide Nanoparticle Catalyst. J. Phys. Chem. C 2015, 119, 17023–17027. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, D.; Huang, Q.; Du, J.; Sun, L.; Li, F.; Meyer, T.J. Stabilization of a molecular water oxidation catalyst on a dye-sensitized photoanode by a pyridyl anchor. Nat. Commun. 2020, 11, 4610. [Google Scholar] [CrossRef]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar water splitting cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef]
- Cook, T.R.; Dogutan, D.K.; Reece, S.Y.; Surendranath, Y.; Teets, T.S.; Nocera, D.G. Solar energy supply and storage for the legacy and nonlegacy worlds. Chem. Rev. 2010, 110, 6474–6502. [Google Scholar] [CrossRef]
- Kirner, J.T.; Finke, R.G. Water-oxidation photoanodes using organic light-harvesting materials: A review. J. Mater. Chem. A 2017, 5, 19560–19592. [Google Scholar] [CrossRef]
- Murphy, A.; Barnes, P.; Randeniya, L.; Plumb, I.; Grey, I.; Horne, M.; Glasscock, J. Efficiency of solar water splitting using semiconductor electrodes. Int. J. Hydrogen Energy 2006, 31, 1999–2017. [Google Scholar] [CrossRef]
- Bolton, J.R.; Strickler, S.J.; Connolly, J.S. Limiting and realizable efficiencies of solar photolysis of water. Nature 1985, 316, 495–500. [Google Scholar] [CrossRef]
- Gersten, S.W.; Samuels, G.J.; Meyer, T.J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer. J. Am. Chem. Soc. 1982, 104, 4029–4030. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Eggleston, D.S.; Murphy, W.R.; Geselowitz, D.A.; Gersten, S.W.; Hodgson, D.J.; Meyer, T.J. Structure and redox properties of the water-oxidation catalyst [(bpy)2(OH2)RuORu(OH2)(bpy)2]4+. J. Am. Chem. Soc. 1985, 107, 3855–3864. [Google Scholar] [CrossRef]
- Karkas, M.D.; Verho, O.; Johnston, E.V.; Akermark, B. Artificial photosynthesis: Molecular systems for catalytic water oxidation. Chem. Rev. 2014, 114, 11863–12001. [Google Scholar] [CrossRef]
- Sens, C.; Romero, I.; Rodriguez, M.; Llobet, A.; Parella, T.; Benet-Buchholz, J. A new Ru complex capable of catalytically oxidizing water to molecular dioxygen. J. Am. Chem. Soc. 2004, 126, 7798–7799. [Google Scholar] [CrossRef] [PubMed]
- Zong, R.; Thummel, R.P. A new family of Ru complexes for water oxidation. J. Am. Chem. Soc. 2005, 127, 12802–12803. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Jurss, J.W.; Templeton, J.L.; Meyer, T.J. One site is enough. Catalytic water oxidation by [Ru(tpy)(bpm)(OH2)]2+ and [Ru(tpy)(bpz)(OH2)]2+. J. Am. Chem. Soc. 2008, 130, 16462–16463. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Jurss, J.W.; Brennaman, M.K.; Hoertz, P.G.; Patrocinio, A.O.; Murakami Iha, N.Y.; Templeton, J.L.; Meyer, T.J. Making oxygen with ruthenium complexes. Acc. Chem. Res. 2009, 42, 1954–1965. [Google Scholar] [CrossRef]
- Xu, Y.; Akermark, T.; Gyollai, V.; Zou, D.; Eriksson, L.; Duan, L.; Zhang, R.; Akermark, B.; Sun, L. A new dinuclear ruthenium complex as an efficient water oxidation catalyst. Inorg. Chem. 2009, 48, 2717–2719. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fischer, A.; Duan, L.; Tong, L.; Gabrielsson, E.; Åkermark, B.; Sun, L. Chemical and Light-Driven Oxidation of Water Catalyzed by an Efficient Dinuclear Ruthenium Complex. Angew. Chem. 2010, 122, 9118–9121. [Google Scholar] [CrossRef]
- Duan, L.; Fischer, A.; Xu, Y.; Sun, L. Isolated seven-coordinate Ru(IV) dimer complex with [HOHOH](-) bridging ligand as an intermediate for catalytic water oxidation. J. Am. Chem. Soc. 2009, 131, 10397–10399. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W. Molecular Catalysts for Water Oxidation. Chem. Rev. 2015, 115, 12974–13005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Sun, L. Ru-bda: Unique Molecular Water-Oxidation Catalysts with Distortion Induced Open Site and Negatively Charged Ligands. J. Am. Chem. Soc. 2019, 141, 5565–5580. [Google Scholar] [CrossRef] [PubMed]
- Harriman, A.; Pickering, I.J.; Thomas, J.M.; Christensen, P.A. Metal oxides as heterogeneous catalysts for oxygen evolution under photochemical conditions. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1988, 84, 2795–2806. [Google Scholar] [CrossRef]
- Ruttinger, W.; Dismukes, G.C. Synthetic Water-Oxidation Catalysts for Artificial Photosynthetic Water Oxidation. Chem. Rev. 1997, 97, 1–24. [Google Scholar] [CrossRef]
- Ruettinger, W.F.; Campana, C.; Dismukes, G.C. Synthesis and Characterization of Mn4O4L6Complexes with Cubane-like Core Structure: A New Class of Models of the Active Site of the Photosynthetic Water Oxidase. J. Am. Chem. Soc. 1997, 119, 6670–6671. [Google Scholar] [CrossRef]
- Brimblecombe, R.; Swiegers, G.F.; Dismukes, G.C.; Spiccia, L. Sustained water oxidation photocatalysis by a bioinspired manganese cluster. Angew. Chem. Int. Ed. Engl. 2008, 47, 7335–7338. [Google Scholar] [CrossRef]
- Duan, L.; Xu, Y.; Zhang, P.; Wang, M.; Sun, L. Visible light-driven water oxidation by a molecular ruthenium catalyst in homogeneous system. Inorg. Chem. 2010, 49, 209–215. [Google Scholar] [CrossRef]
- Hoogeveen, D.A.; Fournier, M.; Bonke, S.A.; Nattestad, A.; Mishra, A.; Bäuerle, P.; Spiccia, L.; Mozer, A.J.; Simonov, A.N. Origin of Photoelectrochemical Generation of Dihydrogen by a Dye-Sensitized Photocathode without an Intentionally Introduced Catalyst. J. Phys. Chem. C 2017, 121, 25836–25846. [Google Scholar] [CrossRef]
- Kamire, R.J.; Materna, K.L.; Hoffeditz, W.L.; Phelan, B.T.; Thomsen, J.M.; Farha, O.K.; Hupp, J.T.; Brudvig, G.W.; Wasielewski, M.R. Photodriven Oxidation of Surface-Bound Iridium-Based Molecular Water-Oxidation Catalysts on Perylene-3,4-dicarboximide-Sensitized TiO2 Electrodes Protected by an Al2O3 Layer. J. Phys. Chem. C 2017, 121, 3752–3764. [Google Scholar] [CrossRef]
- Xu, P.; Gray, C.L.; Xiao, L.; Mallouk, T.E. Charge Recombination with Fractional Reaction Orders in Water-Splitting Dye-Sensitized Photoelectrochemical Cells. J. Am. Chem. Soc. 2018, 140, 11647–11654. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, T.; Kubota, J.; Domen, K. Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem. Soc. Rev. 2014, 43, 7520–7535. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zhang, P.; Qin, P.; Sun, D.; Zhang, S.; Guo, X.; Zhao, W.; Zhao, D.; Huang, F. Novel Black BiVO4/TiO2−x Photoanode with Enhanced Photon Absorption and Charge Separation for Efficient and Stable Solar Water Splitting. Adv. Energy Mater. 2019, 9, 1901287. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Zhong, D.K.; Szalda, D.J.; Muckerman, J.T.; Fujita, E. Mechanism of water oxidation by [Ru(bda)(L)2]: The return of the “blue dimer”. Chem. Commun. 2015, 51, 4105–4108. [Google Scholar] [CrossRef]
- Wang, D.; Sampaio, R.N.; Troian-Gautier, L.; Marquard, S.L.; Farnum, B.H.; Sherman, B.D.; Sheridan, M.V.; Dares, C.J.; Meyer, G.J.; Meyer, T.J. Molecular Photoelectrode for Water Oxidation Inspired by Photosystem II. J. Am. Chem. Soc. 2019, 141, 7926–7933. [Google Scholar] [CrossRef] [PubMed]
- Ashford, D.L.; Gish, M.K.; Vannucci, A.K.; Brennaman, M.K.; Templeton, J.L.; Papanikolas, J.M.; Meyer, T.J. Molecular Chromophore-Catalyst Assemblies for Solar Fuel Applications. Chem. Rev. 2015, 115, 13006–13049. [Google Scholar] [CrossRef]
- Alibabaei, L.; Brennaman, M.K.; Norris, M.R.; Kalanyan, B.; Song, W.; Losego, M.D.; Concepcion, J.J.; Binstead, R.A.; Parsons, G.N.; Meyer, T.J. Solar water splitting in a molecular photoelectrochemical cell. Proc. Natl. Acad. Sci. USA 2013, 110, 20008–20013. [Google Scholar] [CrossRef]
- Alibabaei, L.; Sherman, B.D.; Norris, M.R.; Brennaman, M.K.; Meyer, T.J. Visible photoelectrochemical water splitting into H2 and O2 in a dye-sensitized photoelectrosynthesis cell. Proc. Natl. Acad. Sci. USA 2015, 112, 5899–5902. [Google Scholar] [CrossRef]
- Lee, H.; Kepley, L.J.; Hong, H.G.; Akhter, S.; Mallouk, T.E. Adsorption of ordered zirconium phosphonate multilayer films on silicon and gold surfaces. J. Phys. Chem. 2002, 92, 2597–2601. [Google Scholar] [CrossRef]
- Lee, H.; Kepley, L.J.; Hong, H.G.; Mallouk, T.E. Inorganic analogs of Langmuir-Blodgett films: Adsorption of ordered zirconium 1,10-decanebisphosphonate multilayers on silicon surfaces. J. Am. Chem. Soc. 2002, 110, 618–620. [Google Scholar] [CrossRef]
- Ishida, T.; Terada, K.-i.; Hasegawa, K.; Kuwahata, H.; Kusama, K.; Sato, R.; Nakano, M.; Naitoh, Y.; Haga, M.-a. Self-assembled monolayer and multilayer formation using redox-active Ru complex with phosphonic acids on silicon oxide surface. Appl. Surf. Sci. 2009, 255, 8824–8830. [Google Scholar] [CrossRef]
- Terada, K.; Kobayashi, K.; Hikita, J.; Haga, M.-a. Electric Conduction Properties of Self-assembled Monolayer Films of Ru Complexes with Disulfide/Phosphonate Anchors in a Au–(Molecular Ensemble)–(Au Nanoparticle) Junction. Chem. Lett. 2009, 38, 416–417. [Google Scholar] [CrossRef]
- Hanson, K.; Torelli, D.A.; Vannucci, A.K.; Brennaman, M.K.; Luo, H.; Alibabaei, L.; Song, W.; Ashford, D.L.; Norris, M.R.; Glasson, C.R.K.; et al. Self-Assembled Bilayer Films of Ruthenium(II)/Polypyridyl Complexes through Layer-by-Layer Deposition on Nanostructured Metal Oxides. Angew. Chem. 2012, 124, 12954–12957. [Google Scholar] [CrossRef]
- Ding, X.; Gao, Y.; Zhang, L.; Yu, Z.; Liu, J.; Sun, L. Visible Light-Driven Water Splitting in Photoelectrochemical Cells with Supramolecular Catalysts on Photoanodes. ACS Catal. 2014, 4, 2347–2350. [Google Scholar] [CrossRef]
- Sheridan, M.V.; Sherman, B.D.; Coppo, R.L.; Wang, D.; Marquard, S.L.; Wee, K.-R.; Murakami Iha, N.Y.; Meyer, T.J. Evaluation of Chromophore and Assembly Design in Light-Driven Water Splitting with a Molecular Water Oxidation Catalyst. ACS Energy Lett. 2016, 1, 231–236. [Google Scholar] [CrossRef]
- Wang, D.; Xu, Z.; Sheridan, M.V.; Concepcion, J.J.; Li, F.; Lian, T.; Meyer, T.J. Photodriven water oxidation initiated by a surface bound chromophore-donor-catalyst assembly. Chem. Sci. 2021, 12, 14441–14450. [Google Scholar] [CrossRef]
- Yeo, R.S.; Chin, D.T. A Hydrogen-Bromine Cell for Energy Storage Applications. J. Electrochem. Soc. 2019, 127, 549–555. [Google Scholar] [CrossRef]
- Savinell, R.F.; Fritts, S.D. Theoretical performance of a hydrogen-bromine rechargeable SPE fuel cell. J. Power Sources 1988, 22, 423–440. [Google Scholar] [CrossRef]
- Park, J.W.; Wycisk, R.; Pintauro, P.N.; Yarlagadda, V.; Van Nguyen, T. Electrospun Nafion((R))/Polyphenylsulfone Composite Membranes for Regenerative Hydrogen Bromine Fuel Cells. Materials 2016, 9, 143. [Google Scholar] [CrossRef] [PubMed]
- Michalowski, T. Calculation of pH and Potential E for Bromine Aqueous Solution. J. Chem. Educ. 1994, 71, 560–561. [Google Scholar] [CrossRef]
- Sheridan, M.V.; Wang, Y.; Wang, D.; Troian-Gautier, L.; Dares, C.J.; Sherman, B.D.; Meyer, T.J. Light-Driven Water Splitting Mediated by Photogenerated Bromine. Angew. Chem. 2018, 130, 3507–3511. [Google Scholar] [CrossRef]
- Cettou, P.; Robertson, P.M.; Ibl, N. On the electrolysis of aqueous bromide solutions to bromate. Electrochim. Acta 1984, 29, 875–885. [Google Scholar] [CrossRef]
- Mastragostino, M.; Gramellini, C. Kinetic study of the electrochemical processes of the bromine/bromine aqueous system on vitreous carbon electrodes. Electrochim. Acta 1985, 30, 373–380. [Google Scholar] [CrossRef]
- Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 2012, 4, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Concepcion, J.J.; Binstead, R.A.; Rudd, J.A.; Vannucci, A.K.; Dares, C.J.; Coggins, M.K.; Meyer, T.J. Base-enhanced catalytic water oxidation by a carboxylate-bipyridine Ru(II) complex. Proc. Natl. Acad. Sci. USA 2015, 112, 4935–4940. [Google Scholar] [CrossRef]
- Li, G.; Ward, W.M.; Meyer, G.J. Visible Light Driven Nanosecond Bromide Oxidation by a Ru Complex with Subsequent Br-Br Bond Formation. J. Am. Chem. Soc. 2015, 137, 8321–8323. [Google Scholar] [CrossRef]
- Li, G.; Swords, W.B.; Meyer, G.J. Bromide Photo-oxidation Sensitized to Visible Light in Consecutive Ion Pairs. J. Am. Chem. Soc. 2017, 139, 14983–14991. [Google Scholar] [CrossRef]
- Li, G.; Brady, M.D.; Meyer, G.J. Visible Light Driven Bromide Oxidation and Ligand Substitution Photochemistry of a Ru Diimine Complex. J. Am. Chem. Soc. 2018, 140, 5447–5456. [Google Scholar] [CrossRef]
- Tsai, K.Y.; Chang, I.J. Photocatalytic Oxidation of Bromide to Bromine. Inorg. Chem. 2017, 56, 693–696. [Google Scholar] [CrossRef]
- Tsai, K.Y.; Chang, I.J. Oxidation of Bromide to Bromine by Ruthenium(II) Bipyridine-Type Complexes Using the Flash-Quench Technique. Inorg. Chem. 2017, 56, 8497–8503. [Google Scholar] [CrossRef] [PubMed]
- Brady, M.D.; Sampaio, R.N.; Wang, D.; Meyer, T.J.; Meyer, G.J. Dye-Sensitized Hydrobromic Acid Splitting for Hydrogen Solar Fuel Production. J. Am. Chem. Soc. 2017, 139, 15612–15615. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.V.; de Oliveira, K.T.; Meyer, G.J.; Polo, A.S. Inhibiting Charge Recombination in cis-Ru(NCS)(2) Diimine Sensitizers with Aromatic Substituents. ACS Appl. Mater. Interfaces 2019, 11, 43223–43234. [Google Scholar] [CrossRef] [PubMed]
- Reddy-Marri, A.; Marchini, E.; Cabanes, V.D.; Argazzi, R.; Pastore, M.; Caramori, S.; Gros, P.C. Panchromatic light harvesting and record power conversion efficiency for carboxylic/cyanoacrylic Fe(ii) NHC co-sensitized FeSSCs. Chem. Sci. 2023, 14, 4288–4301. [Google Scholar] [CrossRef] [PubMed]
- Slama-Schwok, A.; Gershuni, S.; Rabani, J.; Cohen, H.; Meyerstein, D. An iridium-bipyridine complex as a photosensitizer for the bromide oxidation to bromine by oxygen. J. Phys. Chem. 2002, 89, 2460–2464. [Google Scholar] [CrossRef]
- Brady, M.D.; Troian-Gautier, L.; Sampaio, R.N.; Motley, T.C.; Meyer, G.J. Optimization of Photocatalyst Excited- and Ground-State Reduction Potentials for Dye-Sensitized HBr Splitting. ACS Appl. Mater. Interfaces 2018, 10, 31312–31323. [Google Scholar] [CrossRef]
- Wilkinson, G.; Gillard, R.D.; McCleverty, J.A. Comprehensive Coordination Chemistry. The Synthesis, Reactions, Properties and Applications of Coordination Compounds. V. 3. Main Group and Early Transition Elements; Pergamon Press: Oxford, UK, 1987. [Google Scholar]
- Bergeron, B.V.; Marton, A.; Oskam, G.; Meyer, G.J. Dye-sensitized SnO2 electrodes with iodide and pseudohalide redox mediators. J. Phys. Chem. B 2005, 109, 937–943. [Google Scholar] [CrossRef]
- Treadway, J.A.; Moss, J.A.; Meyer, T.J. Visible Region Photooxidation on TiO2 with a Chromophore-Catalyst Molecular Assembly. Inorg. Chem. 1999, 38, 4386–4387. [Google Scholar] [CrossRef]
- Ruberu, T.P.A.; Nelson, N.C.; Slowing, I.I.; Vela, J. Selective Alcohol Dehydrogenation and Hydrogenolysis with Semiconductor-Metal Photocatalysts: Toward Solar-to-Chemical Energy Conversion of Biomass-Relevant Substrates. J. Phys. Chem. Lett. 2012, 3, 2798–2802. [Google Scholar] [CrossRef]
- Shimizu, K.; Sugino, K.; Sawabe, K.; Satsuma, A. Oxidant-free dehydrogenation of alcohols heterogeneously catalyzed by cooperation of silver clusters and acid-base sites on alumina. Chemistry 2009, 15, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Angelici, R.J. Hydrogen Storage and Energy Recovery Using Aldehydes and Ketones: A Key Role for Catalysis. ACS Catal. 2011, 1, 772–776. [Google Scholar] [CrossRef]
- Song, W.; Ito, A.; Binstead, R.A.; Hanson, K.; Luo, H.; Brennaman, M.K.; Concepcion, J.J.; Meyer, T.J. Accumulation of multiple oxidative equivalents at a single site by cross-surface electron transfer on TiO2. J. Am. Chem. Soc. 2013, 135, 11587–11594. [Google Scholar] [CrossRef] [PubMed]
- Natali, M.; Sartorel, A.; Ruggi, A. Beyond Water Oxidation: Hybrid, Molecular-Based Photoanodes for the Production of Value-Added Organics. Front. Chem. 2022, 10, 907510. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Stahl, S.S. Electrochemical Oxidation of Organic Molecules at Lower Overpotential: Accessing Broader Functional Group Compatibility with Electron-Proton Transfer Mediators. Acc. Chem. Res. 2020, 53, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Nutting, J.E.; Rafiee, M.; Stahl, S.S. Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev. 2018, 118, 4834–4885. [Google Scholar] [CrossRef]
- Ciriminna, R.; Palmisano, G.; Pagliaro, M. Electrodes Functionalized with the 2,2,6,6-Tetramethylpiperidinyloxy Radical for the Waste-Free Oxidation of Alcohols. ChemCatChem 2015, 7, 552–558. [Google Scholar] [CrossRef]
- Ciriminna, R.; Ghahremani, M.; Karimi, B.; Pagliaro, M. Electrochemical Alcohol Oxidation Mediated by TEMPO-like Nitroxyl Radicals. ChemistryOpen 2017, 6, 5–10. [Google Scholar] [CrossRef]
- Heeres, A.; van Doren, H.A.; Bleeker, I.P.; Gotlieb, K.F. Method for Recovering or Recirculating Stable Nitroxide Radicals. WO Patent 9636621, 14 May 1996. [Google Scholar]
- Schnatbaum, K.; Schäfer, H.J. Electroorganic Synthesis 66: Selective Anodic Oxidation of Carbohydrates Mediated by TEMPO. Synthesis 1999, 1999, 864–872. [Google Scholar] [CrossRef]
- Cha, H.G.; Choi, K.S. Combined biomass valorization and hydrogen production in a photoelectrochemical cell. Nat. Chem. 2015, 7, 328–333. [Google Scholar] [CrossRef]
- Li, T.; Kasahara, T.; He, J.; Dettelbach, K.E.; Sammis, G.M.; Berlinguette, C.P. Photoelectrochemical oxidation of organic substrates in organic media. Nat. Commun. 2017, 8, 390. [Google Scholar] [CrossRef] [PubMed]
- Kato, F.; Kikuchi, A.; Okuyama, T.; Oyaizu, K.; Nishide, H. Nitroxide Radicals as Highly Reactive Redox Mediators in Dye-Sensitized Solar Cells. Angew. Chem. 2012, 124, 10324–10327. [Google Scholar] [CrossRef]
- Pati, P.B.; Abdellah, M.; Diring, S.; Hammarstrom, L.; Odobel, F. Molecular Triad Containing a TEMPO Catalyst Grafted on Mesoporous Indium Tin Oxide as a Photoelectrocatalytic Anode for Visible Light-Driven Alcohol Oxidation. ChemSusChem 2021, 14, 2902–2913. [Google Scholar] [CrossRef] [PubMed]
- Skaisgirski, M.; Guo, X.; Wenger, O.S. Electron Accumulation on Naphthalene Diimide Photosensitized by [Ru(2,2′-Bipyridine)3]2+. Inorg. Chem. 2017, 56, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Llorente, M.; Froehlich, J.; Dang, T.; Sathrum, A.; Kubiak, C.P. Photochemical and photoelectrochemical reduction of CO2. Annu. Rev. Phys. Chem. 2012, 63, 541–569. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, J.C.; Cai, W.; Ma, J.; Yang, H.B.; Xiao, H.; Li, J.; Xiong, Y.; Huang, Y.; Liu, B. Selective photoelectrochemical oxidation of glycerol to high value-added dihydroxyacetone. Nat. Commun. 2019, 10, 1779. [Google Scholar] [CrossRef]
- Katryniok, B.; Kimura, H.; Skrzyńska, E.; Girardon, J.-S.; Fongarland, P.; Capron, M.; Ducoulombier, R.; Mimura, N.; Paul, S.; Dumeignil, F. Selective catalytic oxidation of glycerol: Perspectives for high value chemicals. Green Chem. 2011, 13, 1960–1979. [Google Scholar] [CrossRef]
- Bobbitt, J.M.; BrüCkner, C.; Merbouh, N. Oxoammonium- and Nitroxide-Catalyzed Oxidations of Alcohols. Organomet. React. 2010, 103–424. [Google Scholar] [CrossRef]
- Bruggeman, D.F.; Laporte, A.A.H.; Detz, R.J.; Mathew, S.; Reek, J.N.H. Aqueous Biphasic Dye-Sensitized Photosynthesis Cells for TEMPO-Based Oxidation of Glycerol. Angew. Chem. Int. Ed. Engl. 2022, 61, e202200175. [Google Scholar] [CrossRef]
- Yoo, C.G.; Yang, Y.; Pu, Y.; Meng, X.; Muchero, W.; Yee, K.L.; Thompson, O.A.; Rodriguez, M.; Bali, G.; Engle, N.L.; et al. Insights of biomass recalcitrance in natural Populus trichocarpa variants for biomass conversion. Green Chem. 2017, 19, 5467–5478. [Google Scholar] [CrossRef]
- Galkin, M.V.; Samec, J.S. Lignin Valorization through Catalytic Lignocellulose Fractionation: A Fundamental Platform for the Future Biorefinery. ChemSusChem 2016, 9, 1544–1558. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kim, S.; Davis, A.H.; Zhuang, J.; Shuler, E.W.; Willinger, D.; Lee, J.-J.; Zheng, W.; Sherman, B.D.; Yoo, C.G.; et al. Photocatalytic Chemoselective C–C Bond Cleavage at Room Temperature in Dye-Sensitized Photoelectrochemical Cells. ACS Catal. 2021, 11, 3771–3781. [Google Scholar] [CrossRef]

































| Meyer 1982 | Liobet 2004 | Thummel 2005 | Meyer 2008 |
|---|---|---|---|
 |  |  |  |
| I | II | III | IV |
| Sun 2009 | Sun 2010 | Sun 2009 | |
 |  |  | |
| V | VI | VII | |
| Sensitizer | Catalyst | Photocurrent | Details | Ref |
|---|---|---|---|---|
| TiO2@22 | IrO2·nH2O | 30 µA (>0.2 V vs. Ag/AgCl) | JV in 30 mM Na2SiF6/500 mM Na2SO4, pH 5.75 | [19] |
| TiO2@1 | 23 | 15 µA (0 V) | Cronoamperometry in 0.1 M Na2SO4 pH 6.5 | [95] |
| TiO2@4 | 24 | 50 µA (0 V) | Cronoamperometry in 0.1 M Na2SO4 | [96] |
| nanoITO/TiO2@5 | IrOx | 110 µA cm−2 (0.3 V vs. Ag/AgCl) | Cronoamperometry in NaSiF6 pH 5.8 | [98] |
| TiO2@8 | / | 1.1 mA cm−2 (−0.17 V vs. SCE) | Cronoamperometry in 0.1 M acetate buffer/30 mM EDTA/0.1 NaClO4 pH 5.0. Stable over 100 min | [61] |
| TiO2@4 | 25 26 27 | 1.5 mA cm−2 (0.2 V vs. NHE) 140 µA cm−2 (0.2 V vs. NHE) 0.8 mA cm−2 (0.2 V vs. NHE) | Chopped photocurrent density–time in a 0.1 M acetate buffer/0.5 M NaClO4 | [99] |
| nanoITO–TiO2@28 (assembly) | 100 μA cm−2 (0.2 V vs. NHE) | Cronoamperometry in 20 mM acetate buffer pH 4.6 | [130] | |
| SnO2/TiO2@28 (assembly) | 100 μA cm−2 (0.6 V vs. NHE) | Cronoamperometry in 0.1 M phosphate buffer at pH 7/0.5 M NaClO4 | [131] | |
| TiO2@30 (assembly) | 500 μA cm−2 (0.2 V vs. NHE) | Chopped cronoamperometry in 0.1 M Na2SO4 pH 6.4 | [137] | |
| SnO2/TiO2@5 SnO2/TiO2@6 | 31 | 0.97 mA cm−2 (0.1 V) 1.45 mA cm−2 | Cronoamperometry in, 0.1 M acetate buffer/1.0 M in NaClO4 pH 5.7 | [138] |
| SnO2/TiO2@32 | 600 μA cm−2 (0.6 V vs. NHE) | Cronoamperometry in 0.1 M phosphonate buffer/0.4 M NaClO4 pH 7.0 | [139] | |
| Sensitizer | Photocurrent | Details | Ref |
| SnO2/TiO2@33 | 1.5 mA cm−2 (>0.7 V vs. NHE) | JV in 1 M HBr. FE% (4) = 72% | [154] |
| SnO2/TiO2@4 | 0.9 mA cm−2 (>0.7 V vs. NHE) | JV in 1 M HBr. FE% (4) = 51%, FE% (33) = 81%, FE% (34) = 70%, FE% (35) = 54%, | [158] |
| SnO2/TiO2@33 | 1.6 mA cm−2 (>0.7 V vs. NHE) | ||
| SnO2/TiO2@34 | 1.0 mA cm−2 (>0.7 V vs. NHE) | ||
| SnO2/TiO2@35 | 4.0 µA cm−2 (>0.7 V vs. NHE) | ||
| SnO2/TiO2@15 | 100 μA cm−2 (>0.4 V vs. SCE) | JV in 0.3 M NaBr/0.1 M HBr | [75] |
| SnO2/TiO2@16 | 400 μA cm−2 (>0.4 V vs. SCE) | ||
| SnO2/TiO2@17 | 300 μA cm−2 (>0.4 V vs. SCE) | ||
| SnO2/TiO2@36 | 300 μA cm−2 (>0.4 V vs. SCE) | JV 0.1 M HBr | [76] |
| SnO2/TiO2@37 | 250 μA cm−2 (>0.4 V vs. SCE) |
| Sensitizer | Catalyst | Reaction | Photocurrent | Details | Ref |
|---|---|---|---|---|---|
| nanoITO/TiO2@4 | 7 | BZ | 200 µA cm−2 (0.2 V vs. NHE) | Cronoamperometry in 20 mM acetate/acetic acid buffer pH 4.5/0.1 M LiClO4/0.1 M Benzyl alcohol | [97] |
| ITO(NP)@38 (Assembly-TEMPO catalyst) | BZ | 180 µA cm−2 (>0.4 V vs. SCE) | JV in 50 mM MeO-BA/carbonate buffer pH 10 | [176] | |
| TiO2@39 (Assembly-TEMPO catalyst) | BZ | 200 µA cm−2 (>0.4 V vs. SCE) | JV in 0.1 M borate buffer/50 mM [MeO-Ph-CH2OH]/0.1 M [NaClO4] pH 8. FE% = 80% | [28] | |
| TiO2@41 | TEMPO | Glycerol | 400 µA cm−2 (0.1 V vs. Ag/AgCl) | Cronoamperometry in 1.0 M TEMPO 3 mm redox-gel (10% wt. PVDF-HFP, 1.2 M LiTFSI in ACN)/0.1 M glycerol aqueous solution (sat. NaCl, NaHCO3 pH 8.3. Stability >20 h | [182] |
| TiO2 NRAs@1 | ACT | Lignin | 690 μA cm−2 (0.1 V vs. Ag/AgCl) | Chopped cronoamperometry in 3 mM ACT/1.5 mM LMC/ACN | [185] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchini, E.; Caramori, S.; Carli, S. Metal Complexes for Dye-Sensitized Photoelectrochemical Cells (DSPECs). Molecules 2024, 29, 293. https://doi.org/10.3390/molecules29020293
Marchini E, Caramori S, Carli S. Metal Complexes for Dye-Sensitized Photoelectrochemical Cells (DSPECs). Molecules. 2024; 29(2):293. https://doi.org/10.3390/molecules29020293
Chicago/Turabian StyleMarchini, Edoardo, Stefano Caramori, and Stefano Carli. 2024. "Metal Complexes for Dye-Sensitized Photoelectrochemical Cells (DSPECs)" Molecules 29, no. 2: 293. https://doi.org/10.3390/molecules29020293