
Thioxanthone-Based Siloxane Photosensitizer for Cationic/Radical Photopolymerization and Photoinduced Sol–Gel Reactions
 and
and
Abstract
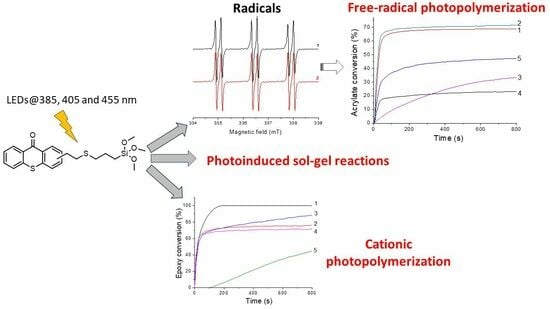
:
1. Introduction
2. Results and Discussion
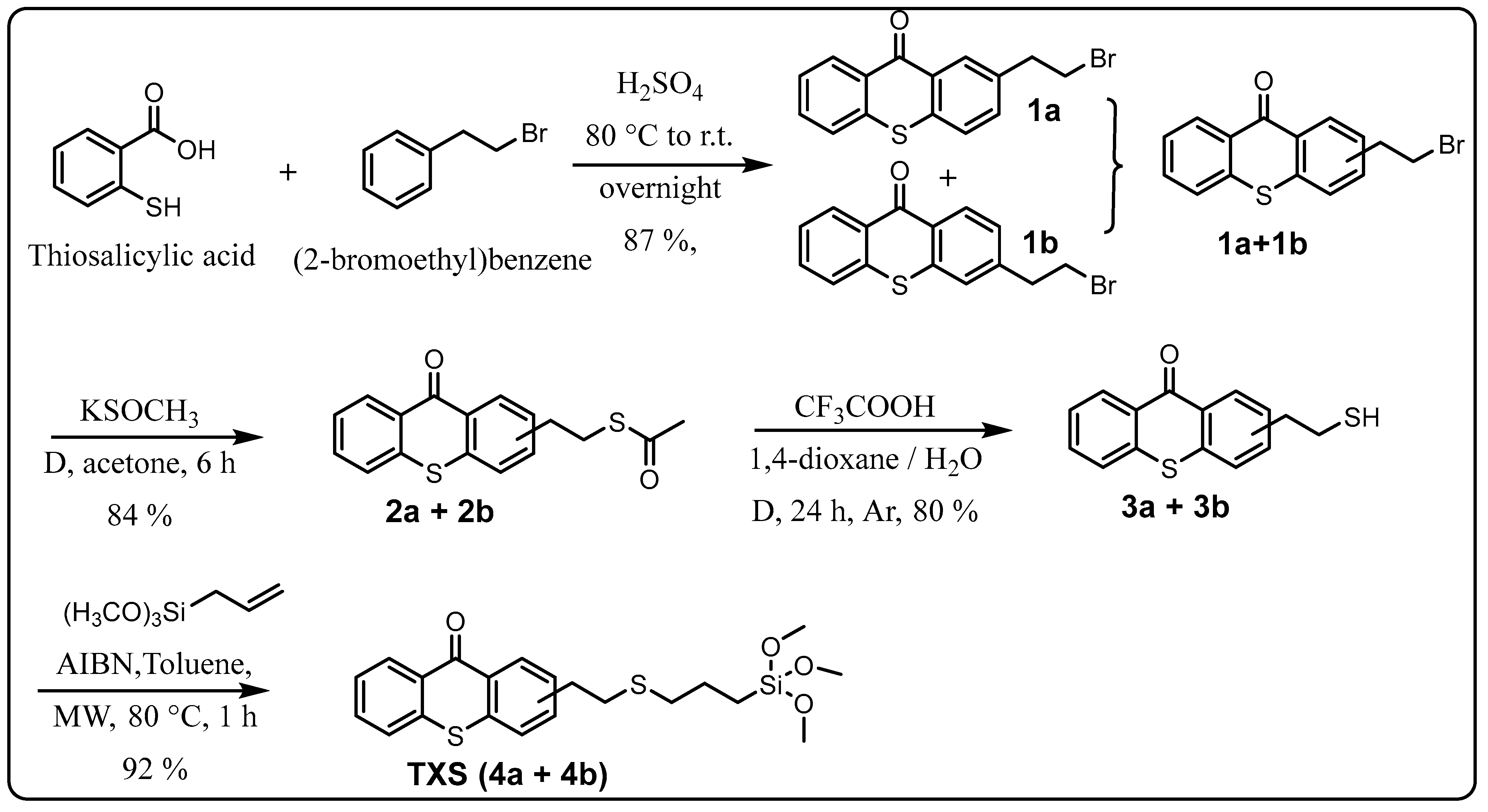
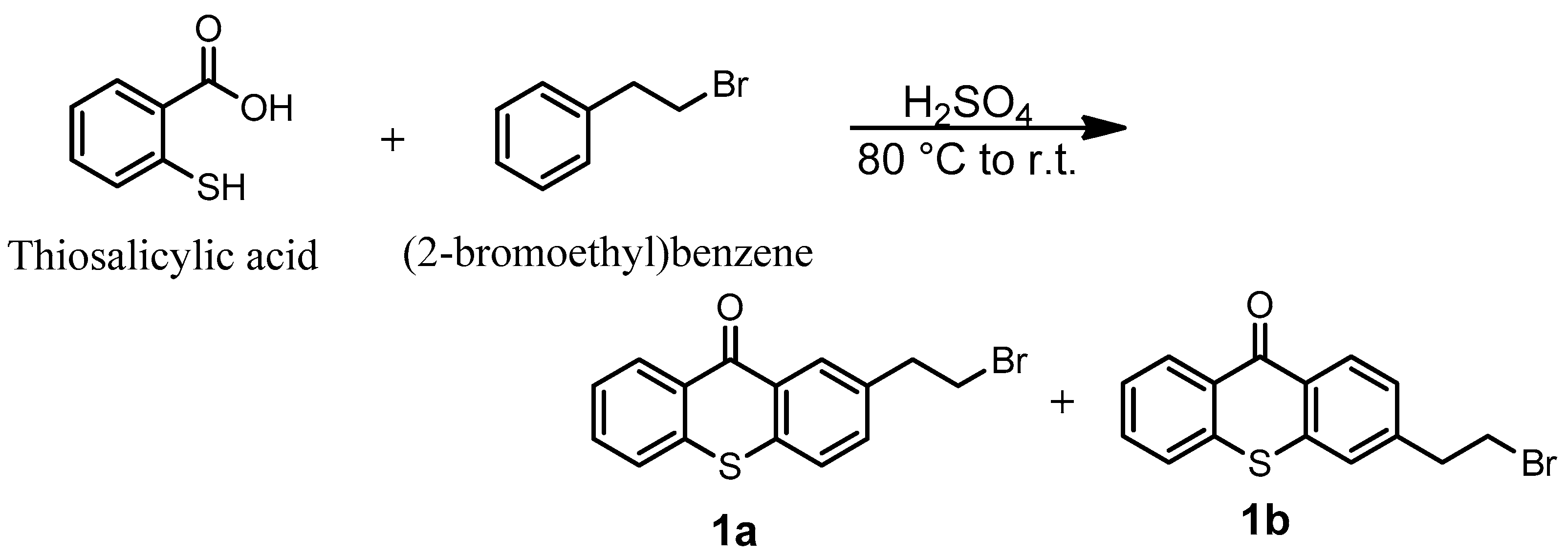
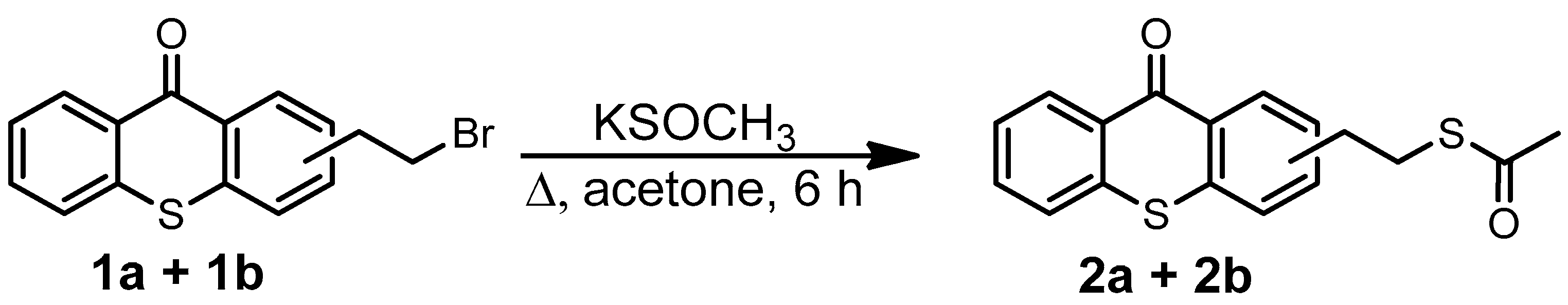
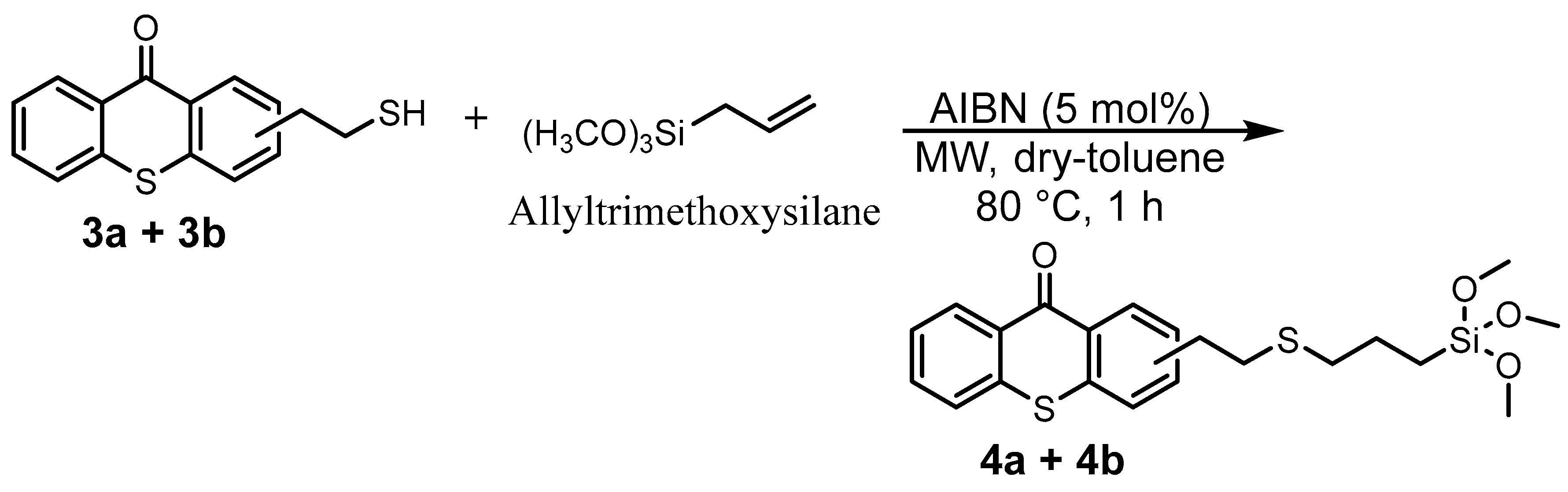
2.1. Synthesis of TXS
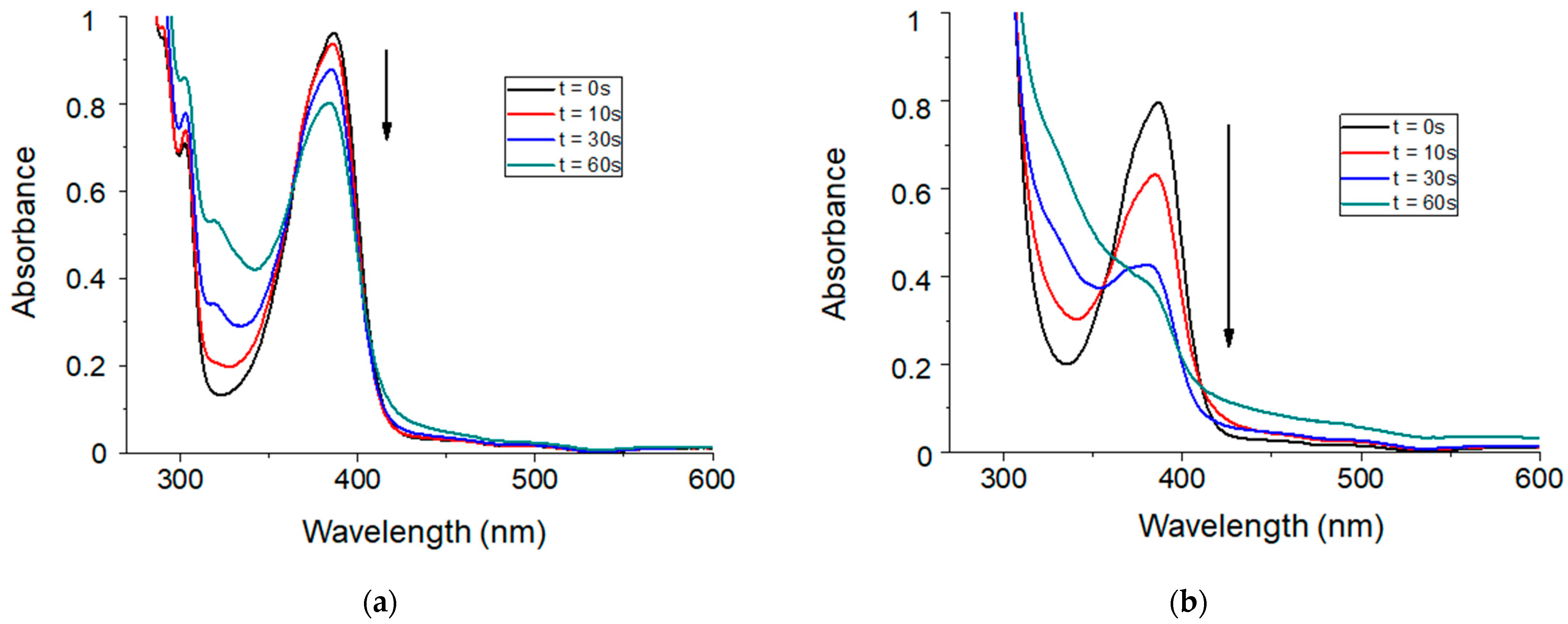
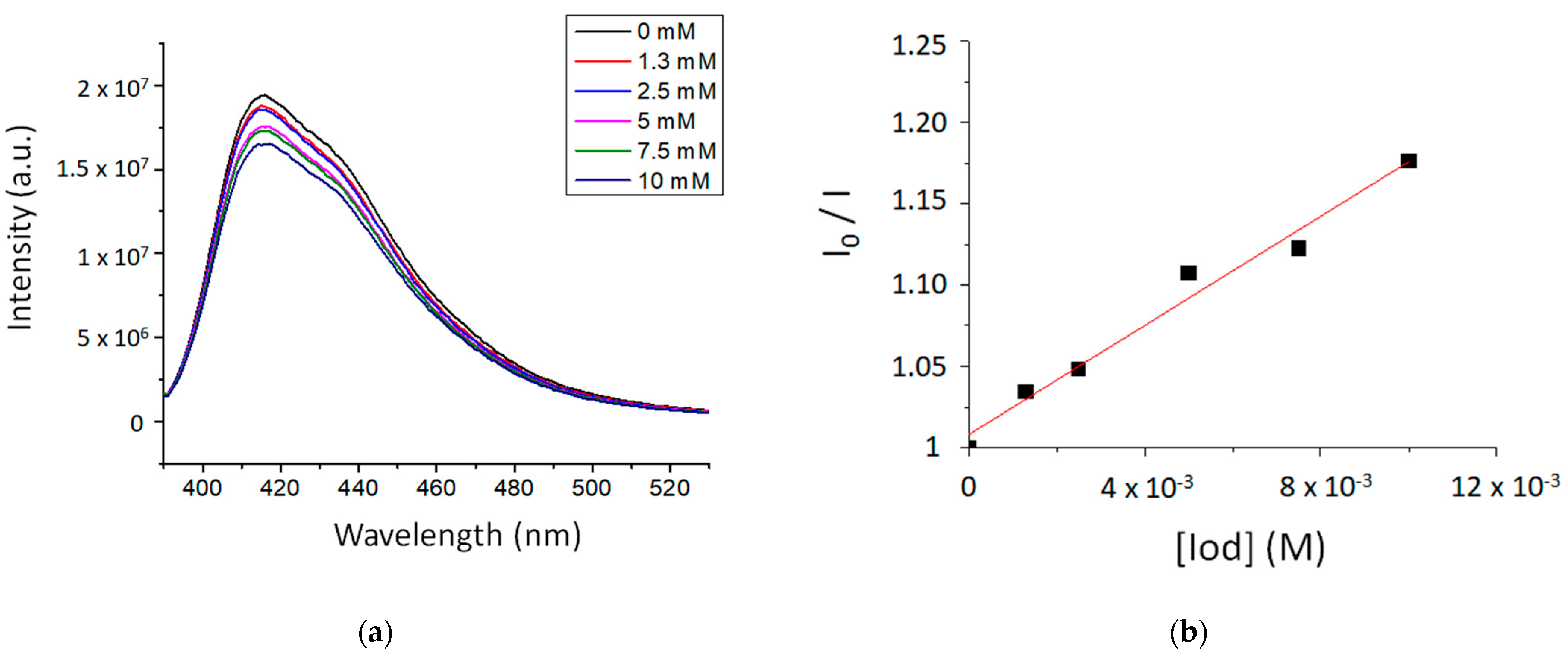
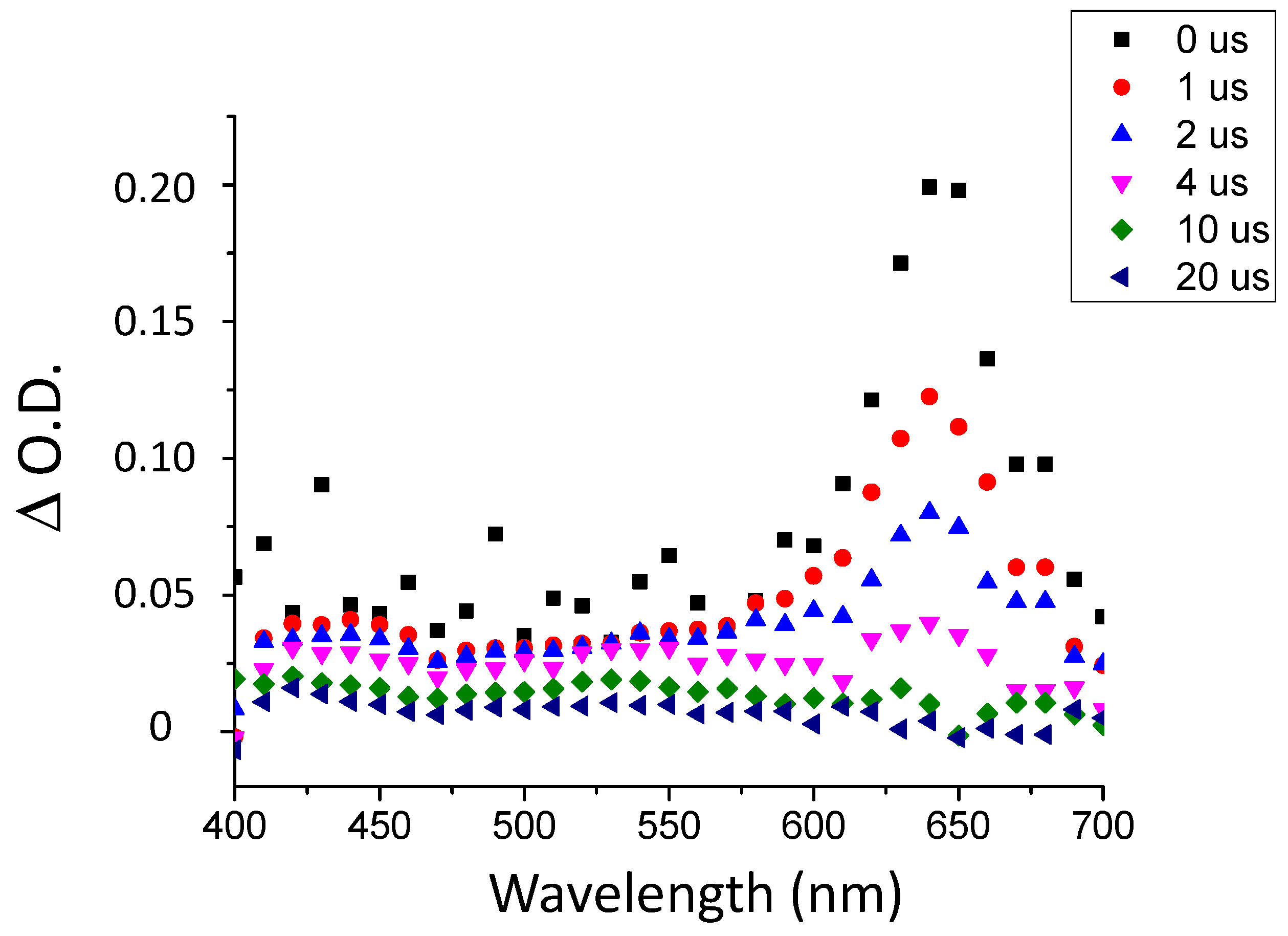
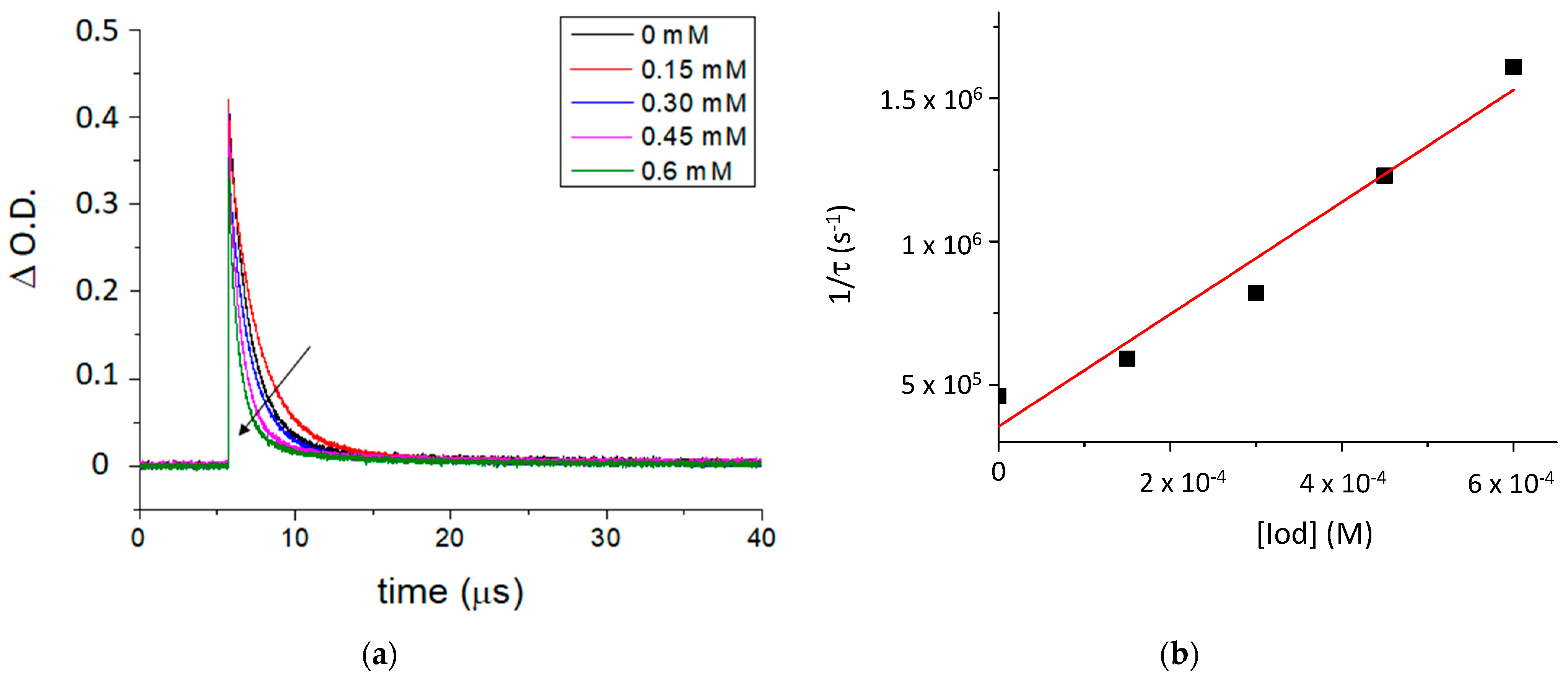
2.2. Photoreactivity of the TXS/Iod Photoinitiating System
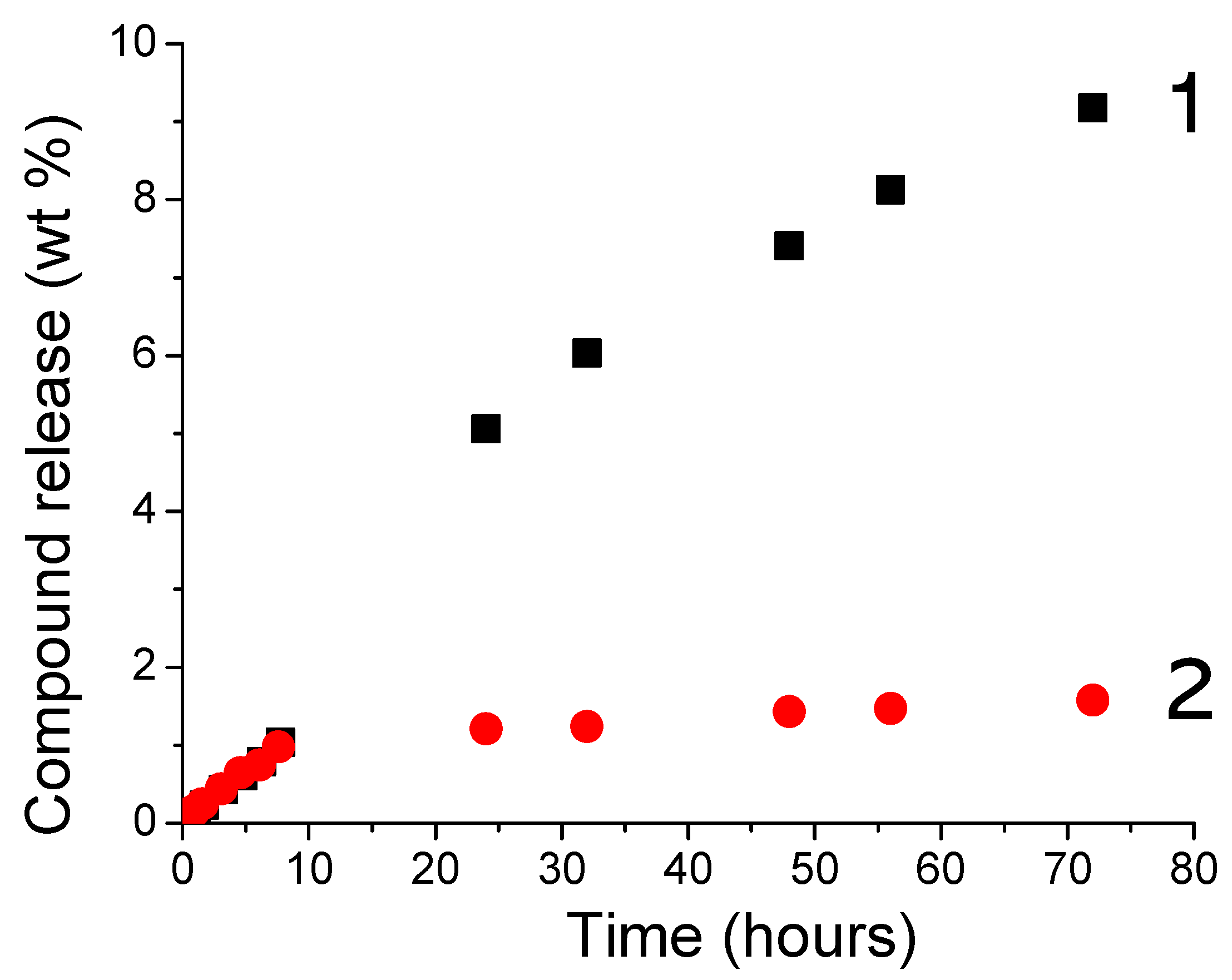
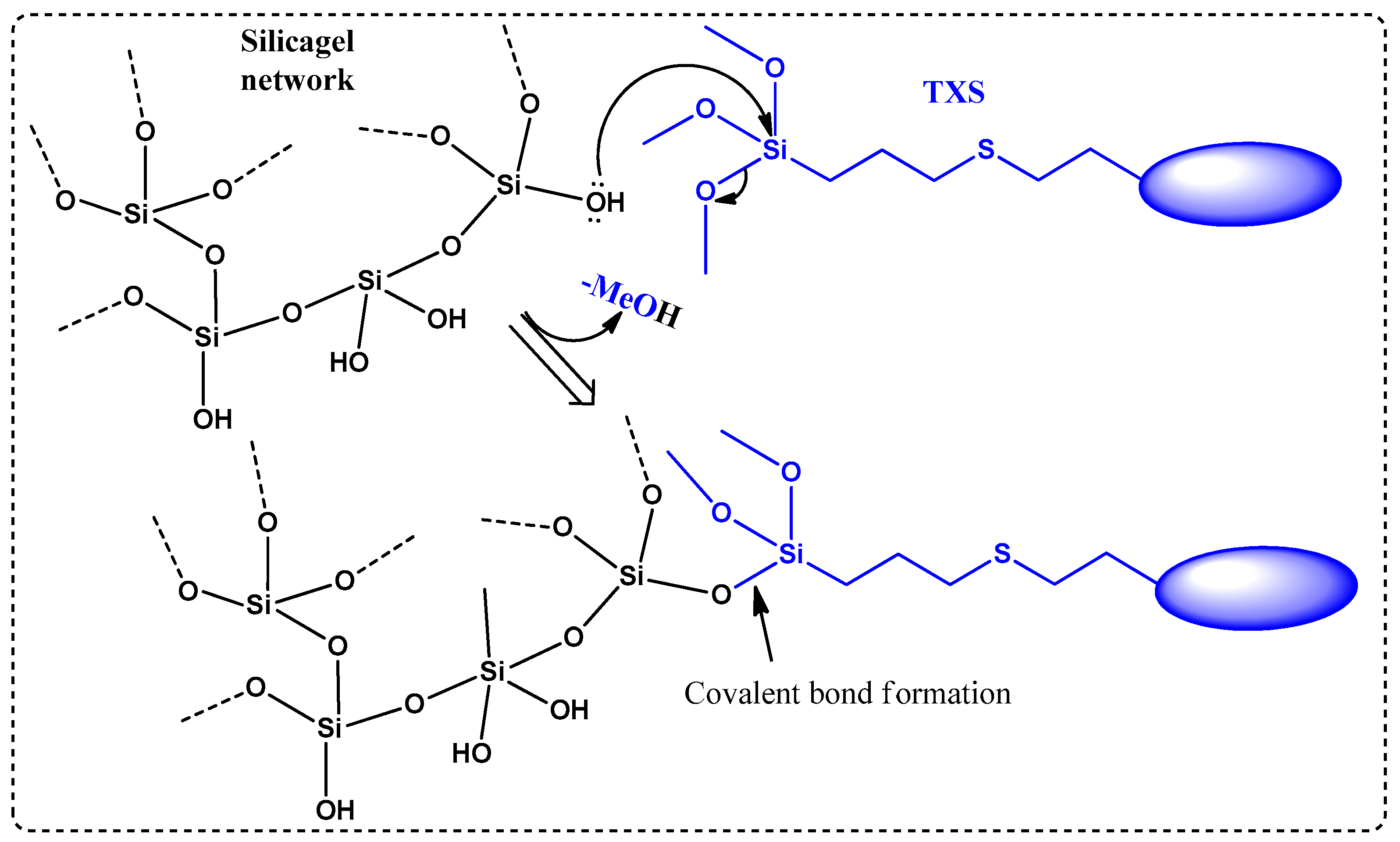
2.3. Photoinduced Sol–Gel Reaction with TXS/Iod System
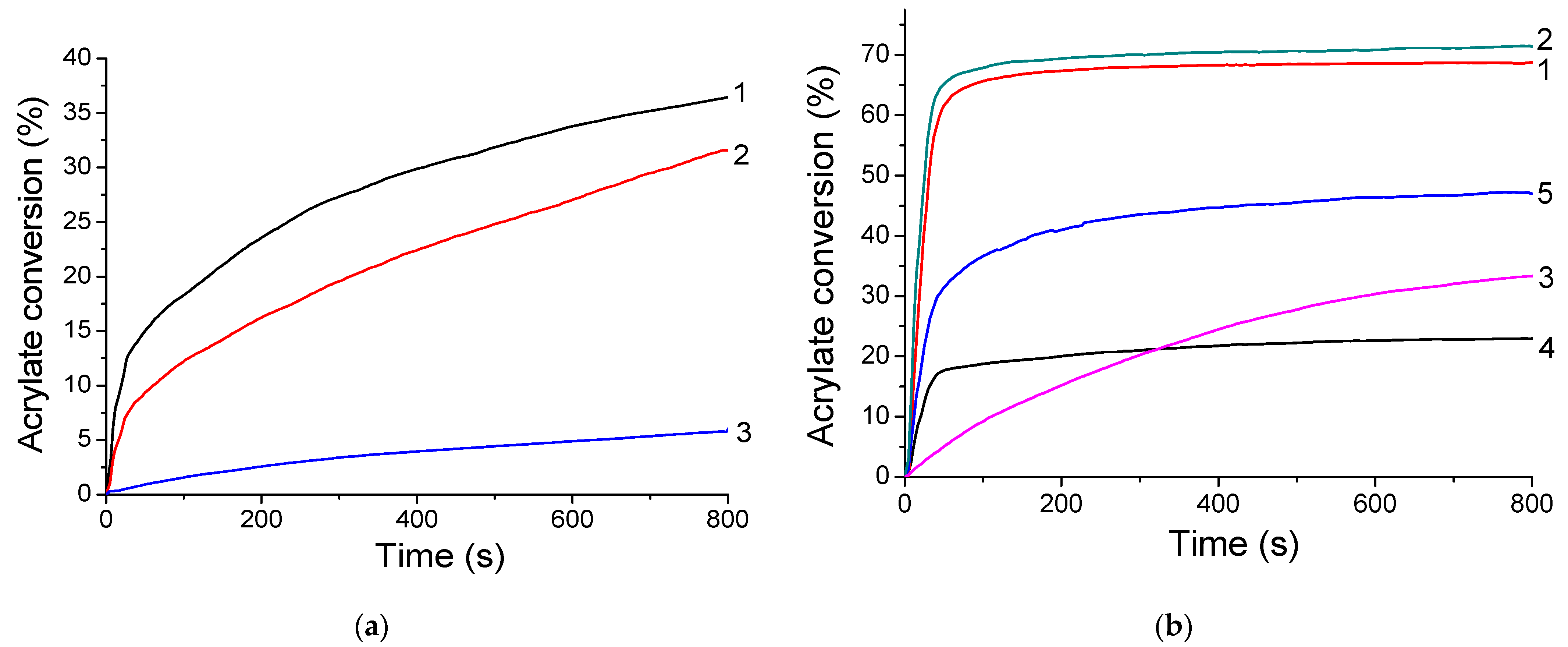
2.4. FRP of MAPTMS with the (TXS/Iod) Photoinitiating System
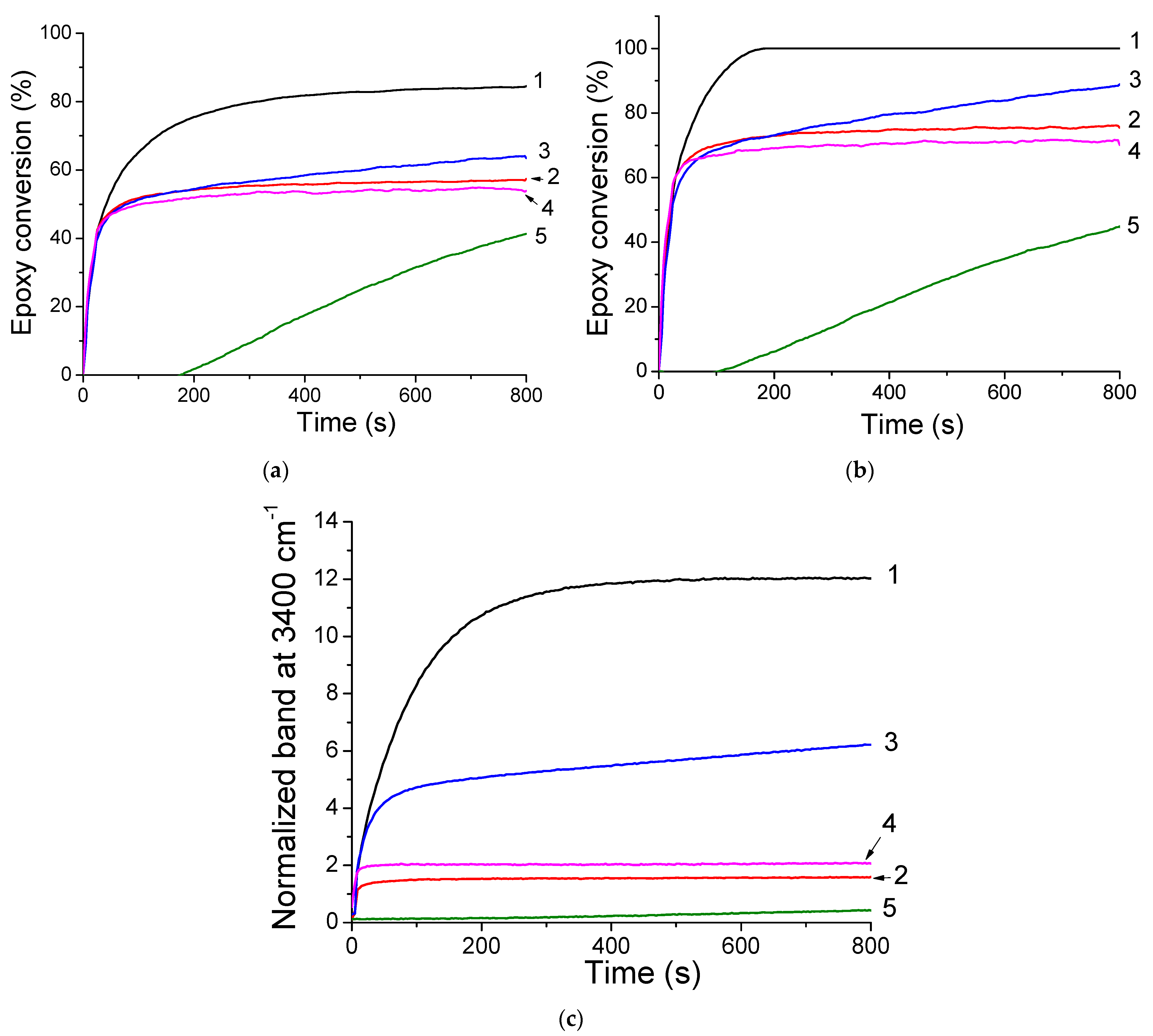
2.5. Cationic Photopolymerization and Hydrolysis/Condensation of GPTMS/Epoxy Monomers with the (TXS/Iod) Photoinitiating System
3. Materials and Methods
3.1. Materials
3.2. Light Sources
3.3. Steady-State Photolysis
3.4. UV-Visible Absorption
3.5. Fluorescence Studies
3.6. Phosphorescence Studies
3.7. Laser Flash Photolysis Experiments
3.8. Triplet Quantum Yield (ΦT)
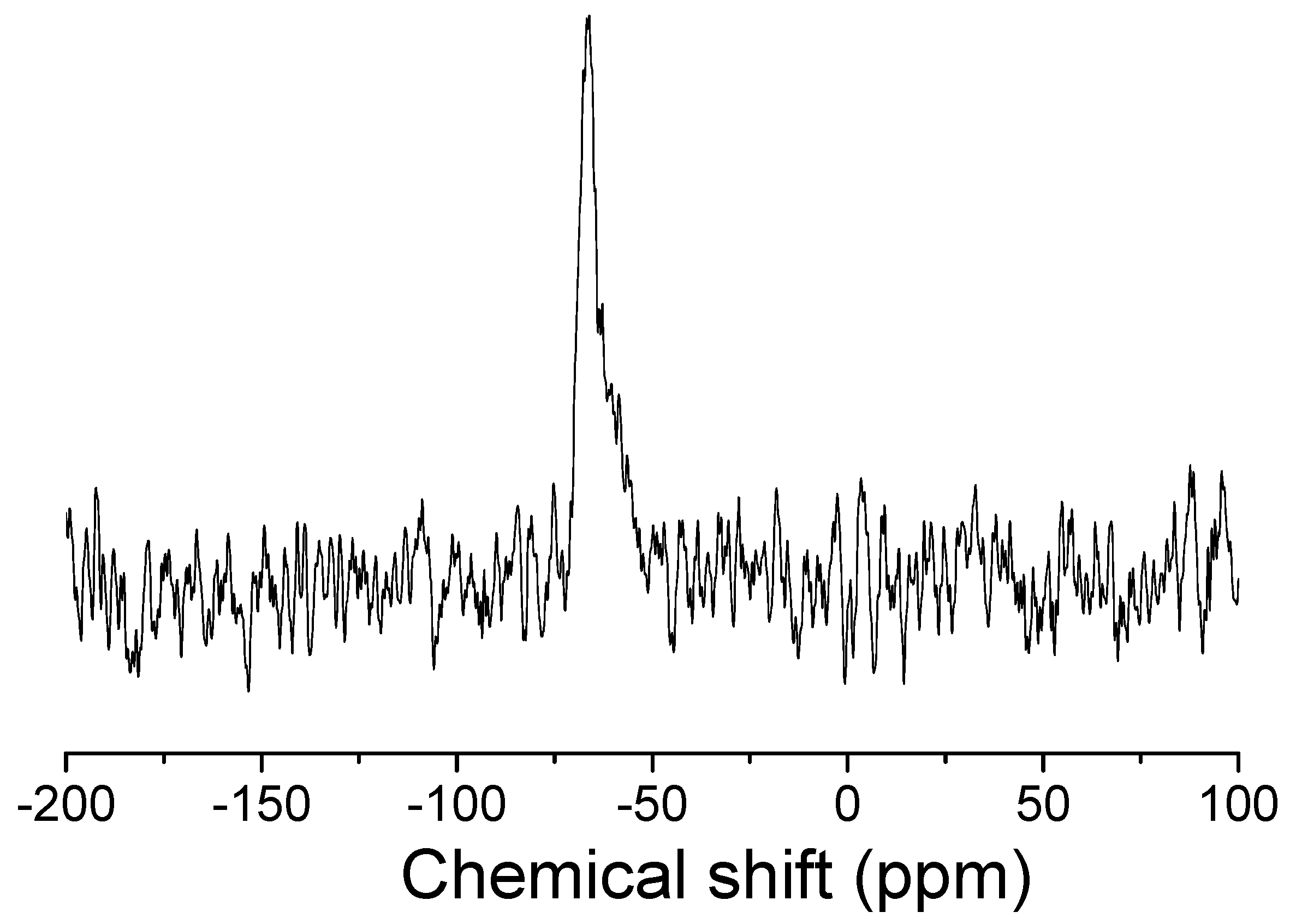
3.9. Electron Paramagnetic Resonance (EPR)
3.10. Cyclic Voltammetry
3.11. Photopolymerization Investigations
3.12. Extraction Study
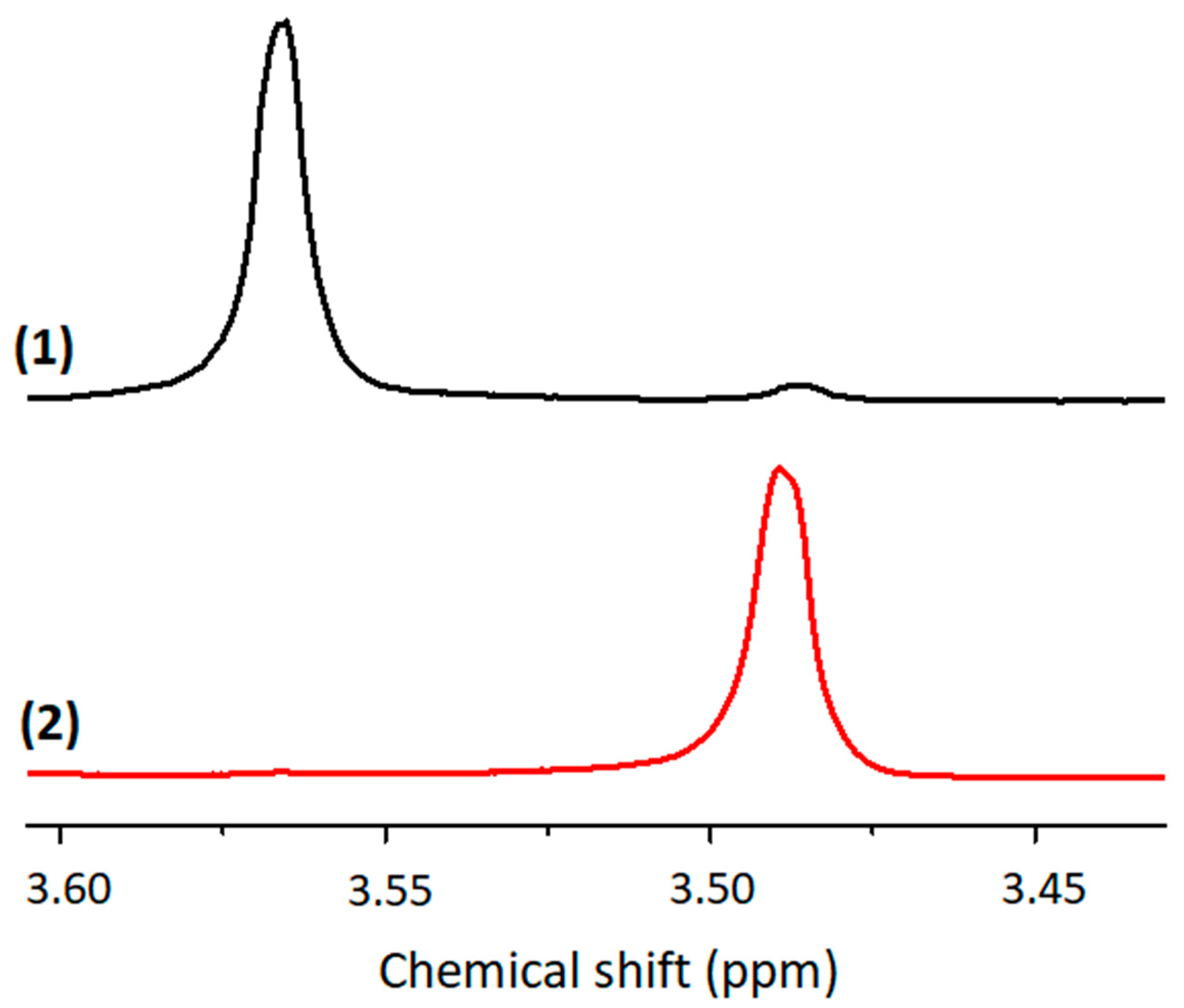
3.13. 29Si Solid-State NMR
3.14. Synthesis of (2-Bromoethyl)thioxanthone (1a + 1b)
3.15. Synthesis of (2-Ethylthioacetate)thioxanthone (2a + 2b)
3.16. Synthesis of (2-Mercaptoethyl)thioxanthone (3a + 3b)
3.17. Synthesis of 2-(2-{[3-(Trimethoxysilyl)propyl]sulfunyl}ethyl-9H-thioxanthen-9-one (TXS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pierau, L.; Elian, C.; Akimoto, J.; Ito, Y.; Caillol, S.; Versace, D.-L. Bio-sourced monomers and cationic photopolymerization–The green combination towards eco-friendly and non-toxic materials. Prog. Polym. Sci. 2022, 127, 101517. [Google Scholar] [CrossRef]
- Yagci, Y.; Jockusch, S.; Turro, N.J. Photoinitiated Polymerization: Advances, Challenges, and Opportunities. Macromolecules 2010, 43, 6245–6260. [Google Scholar] [CrossRef]
- Fouassier, J.P.; Allonas, X.; Burget, D. Photopolymerization reactions under visible lights: Principle, mechanisms and examples of applications. Prog. Org. Coat. 2003, 47, 16–36. [Google Scholar] [CrossRef]
- Xiao, P.; Zhang, J.; Dumur, F.; Tehfe, M.A.; Morlet-Savary, F.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Visible light sensitive photoinitiating systems: Recent progress in cationic and radical photopolymerization reactions under soft conditions. Prog. Polym. Sci. 2015, 41, 32–66. [Google Scholar] [CrossRef]
- Breloy, L.; Losantos, R.; Sampedro, D.; Marazzi, M.; Malval, J.-P.; Heo, Y.; Akimoto, J.; Ito, Y.; Brezová, V.; Versace, D.-L. Allyl amino-thioxanthone derivatives as highly efficient visible light H-donors and co-polymerizable photoinitiators. Polym. Chem. 2020, 11, 4297–4312. [Google Scholar] [CrossRef]
- Sautrot-Ba, P.; Jockusch, S.; Malval, J.-P.; Brezová, V.; Rivard, M.; Abbad-Andaloussi, S.; Blacha-Grzechnik, A.; Versace, D.-L. Quinizarin Derivatives as Photoinitiators for Free-Radical and Cationic Photopolymerizations in the Visible Spectral Range. Macromolecules 2020, 53, 1129–1141. [Google Scholar] [CrossRef]
- Breloy, L.; Yavuz, O.; Yilmaz, I.; Yagci, Y.; Versace, D.-L. Design, synthesis and use of phthalocyanines as a new class of visible-light photoinitiators for free-radical and cationic polymerizations. Polym. Chem. 2021, 12, 4291–4316. [Google Scholar] [CrossRef]
- Breloy, L.; Mhanna, R.; Malval, J.-P.; Brezová, V.; Jacquemin, D.; Pascal, S.; Siri, O.; Versace, D.-L. Azacalixphyrins as an innovative alternative for the free-radical photopolymerization under visible and NIR irradiation without the need of co-initiators. Chem. Commun. 2021, 57, 8973–8976. [Google Scholar] [CrossRef]
- Durmaz, Y.Y.; Moszner, N.; Yagci, Y. Visible Light Initiated Free Radical Promoted Cationic Polymerization Using Acylgermane Based Photoinitiator in the Presence of Onium Salts. Macromolecules 2008, 41, 6714–6718. [Google Scholar] [CrossRef]
- Telitel, S.; Lalevée, J.; Blanchard, N.; Kavalli, T.; Tehfe, M.-A.; Schweizer, S.; Morlet-Savary, F.; Graff, B.; Fouassier, J.-P. Photopolymerization of Cationic Monomers and Acrylate/Divinylether Blends under Visible Light Using Pyrromethene Dyes. Macromolecules 2012, 45, 6864–6868. [Google Scholar] [CrossRef]
- Erdur, S.; Yilmaz, G.; Goen Colak, D.; Cianga, I.; Yagci, Y. Poly(phenylenevinylene)s as Sensitizers for Visible Light Induced Cationic Polymerization. Macromolecules 2014, 47, 7296–7302. [Google Scholar] [CrossRef]
- Dumur, F. Recent advances on anthracene-based photoinitiators of polymerization. Eur. Polym. J. 2022, 169, 111139. [Google Scholar] [CrossRef]
- Noirbent, G.; Dumur, F. Photoinitiators of polymerization with reduced environmental impact: Nature as an unlimited and renewable source of dyes. Eur. Polym. J. 2021, 142, 110109. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, P.; Dumur, F.; Guo, C.; Hong, W.; Li, Y.; Gigmes, D.; Graff, B.; Fouassier, J.-P.; Lalevée, J. Polymeric Photoinitiators: A New Search toward High Performance Visible Light Photoinitiating Systems. Macromol. Chem. Phys. 2016, 217, 2145–2153. [Google Scholar] [CrossRef]
- Balta, D.K.; Temel, G.; Goksu, G.; Ocal, N.; Arsu, N. Thioxanthone–Diphenyl Anthracene: Visible Light Photoinitiator. Macromolecules 2012, 45, 119–125. [Google Scholar] [CrossRef]
- Cokbaglan, L.; Arsu, N.; Yagci, Y.; Jockusch, S.; Turro, N.J. 2-Mercaptothioxanthone as a Novel Photoinitiator for Free Radical Polymerization. Macromolecules 2003, 36, 2649–2653. [Google Scholar] [CrossRef]
- Cook, W.D.; Chen, F. Enhanced visible radiation photopolymerization of dimethacrylates with the three component thioxanthone (CPTXO)–amine–iodonium salt system. Polym. Chem. 2015, 6, 1325–1338. [Google Scholar] [CrossRef]
- Hola, E.; Pilch, M.; Ortyl, J. Thioxanthone Derivatives as a New Class of Organic Photocatalysts for Photopolymerisation Processes and the 3D Printing of Photocurable Resins under Visible Light. Catalysts 2020, 10, 903. [Google Scholar] [CrossRef]
- Yang, J.; Zeng, Z.; Chen, Y. Amine-linked thioxanthones as water-compatible photoinitiators. J. Polym. Sci. Part A Polym. Chem. 1998, 36, 2563–2570. [Google Scholar] [CrossRef]
- Liska, R. Photoinitiators with functional groups. V. New water-soluble photoinitiators containing carbohydrate residues and copolymerizable derivatives thereof. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 1504–1518. [Google Scholar] [CrossRef]
- Wu, Q.; Liao, W.; Xiong, Y.; Yang, J.; Li, Z.; Tang, H. Silicone-Thioxanthone: A Multifunctionalized Visible Light Photoinitiator with an Ability to Modify the Cured Polymers. Polymers 2019, 11, 695. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wang, X.; Xiong, Y.; Yang, J.; Tang, H. Thioxanthone based one-component polymerizable visible light photoinitiator for free radical polymerization. RSC Adv. 2016, 6, 66098–66107. [Google Scholar] [CrossRef]
- Yilmaz, G.; Aydogan, B.; Temel, G.; Arsu, N.; Moszner, N.; Yagci, Y. Thioxanthone–Fluorenes as Visible Light Photoinitiators for Free Radical Polymerization. Macromolecules 2010, 43, 4520–4526. [Google Scholar] [CrossRef]
- Hola, E.; Fiedor, P.; Dzienia, A.; Ortyl, J. Visible-Light Amine Thioxanthone Derivatives as Photoredox Catalysts for Photopolymerization Processes. ACS Appl. Polym. Mater. 2021, 3, 5547–5558. [Google Scholar] [CrossRef]
- Corrales, T.; Catalina, F.; Peinado, C.; Allen, N.S.; Rufs, A.M.; Bueno, C.; Encinas, M.V. Photochemical study and photoinitiation activity of macroinitiators based on thioxanthone. Polymer 2002, 43, 4591–4597. [Google Scholar] [CrossRef]
- Corrales, T.; Catalina, F.; Allen, N.S.; Peinado, C. Novel water soluble copolymers based on thioxanthone: Photochemistry and photoinitiation activity. J. Photochem. Photobiol. A Chem. 2004, 169, 95–100. [Google Scholar] [CrossRef]
- Shimizu, I.; Yoshino, A.; Okabayashi, H.; Nishio, E.; O’Connor, C.J. Kinetics of interaction of 3-aminopropyltriethoxysilane on a silica gel surface using elemental analysis and diffuse reflectance infrared Fourier transform spectra. J. Chem. Soc. Faraday Trans. 1997, 93, 1971–1979. [Google Scholar] [CrossRef]
- Rehm, D.; Weller, A. Kinetics of Fluorescence Quenching by Electron and H-Atom Transfer. Isr. J. Chem. 1970, 8, 259–271. [Google Scholar] [CrossRef]
- Romańczyk, P.P.; Kurek, S.S. Reliable reduction potentials of diaryliodonium cations and aryl radicals in acetonitrile from high-level ab initio computations. Electrochim. Acta 2020, 351, 136404. [Google Scholar] [CrossRef]
- Chen, S.; Pan, H.; Wan, D.; Jin, M. High-performance LED induces cationic photopolymerization using novel 1,3,5-triaryl-2-pyrazoline as photosensitizer. Prog. Org. Coat. 2021, 161, 106460. [Google Scholar] [CrossRef]
- Peyrot, F.; Lajnef, S.; Versace, D.-L. Electron Paramagnetic Resonance Spin Trapping (EPR–ST) Technique in Photopolymerization Processes. Catalysts 2022, 12, 772. [Google Scholar] [CrossRef]
- Buettner, G.R. Spin trapping: ESR parameters of spin adducts. Free Rad. Biol. Med. 1987, 3, 259–303. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.K.; Bowman, C.N. Impact of Oxygen on Photopolymerization Kinetics and Polymer Structure. Macromolecules 2006, 39, 2501–2506. [Google Scholar] [CrossRef]
- Crivello, J.V.; Bi, D.; Lu, Y. Cationic photopolymerization of ambifunctional monomers. Macromol. Symp. 1995, 95, 79–89. [Google Scholar] [CrossRef]
- Crivello, J.V.; Mao, Z. Synthesis of Novel Multifunctional Siloxane Oligomers Using Sol–Gel Techniques and Their Photoinitiated Cationic Polymerization. Chem. Mater. 1997, 9, 1554–1561. [Google Scholar] [CrossRef]
- Crivello, J.V. The discovery and development of onium salt cationic photoinitiators. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 4241–4254. [Google Scholar] [CrossRef]
- Soucek, M.D.; Johnson, A.H.; Meemken, L.E.; Wegner, J.M. Preparation of nano-sized UV-absorbing titanium-oxo-clusters via a photo-curing ceramer process. Polym. Adv. Technol. 2005, 16, 257–261. [Google Scholar] [CrossRef]
- Zou, K.; Soucek, M.D. UV-Curable Organic-Inorganic Hybrid Film Coatings Based on Epoxidized Cyclohexene Derivatized Linseed Oil. Macromol. Chem. Phys. 2004, 205, 2032–2039. [Google Scholar] [CrossRef]
- Zvonkina, I.J.; Soucek, M.D. Inorganic–organic hybrid coatings: Common and new approaches. Curr. Opin. Chem. Eng. 2016, 11, 123–127. [Google Scholar] [CrossRef]
- Teng, G.; Wegner, J.R.; Hurtt, G.J.; Soucek, M.D. Novel inorganic/organic hybrid materials based on blown soybean oil with sol–gel precursors. Prog. Org. Coat. 2001, 42, 29–37. [Google Scholar] [CrossRef]
- Amerio, E.; Sangermano, M.; Malucelli, G.; Priola, A.; Voit, B. Preparation and characterization of hybrid nanocomposite coatings by photopolymerization and sol–gel process. Polymer 2005, 46, 11241–11246. [Google Scholar] [CrossRef]
- Sangermano, M.; Gaspari, E.; Vescovo, L.; Messori, M. Enhancement of scratch-resistance properties of methacrylated UV-cured coatings. Prog. Org. Coat. 2011, 72, 287–291. [Google Scholar] [CrossRef]
- Sangermano, M.; Amerio, E.; Epicoco, P.; Priola, A.; Rizza, G.; Malucelli, G. Preparation and Characterization of Hybrid Nanocomposite Coatings by Cationic UV-Curing and the Sol–gel Process of a Vinyl Ether Based System. Macromol. Mater. Eng. 2007, 292, 634–640. [Google Scholar] [CrossRef]
- Morselli, D.; Bondioli, F.; Sangermano, M.; Messori, M. Photo-cured epoxy networks reinforced with TiO2 in-situ generated by means of non-hydrolytic sol–gel process. Polymer 2012, 53, 283–290. [Google Scholar] [CrossRef]
- Amerio, E.; Malucelli, G.; Sangermano, M.; Priola, A. Nanostructured hybrid materials obtained by UV curing and sol–gel processes involving alkoxysilane groups. e-Polymers 2009, 9, 059. [Google Scholar] [CrossRef]
- Chemtob, A.; Belon, C.; Croutxé-Barghorn, C.; Brendlé, J.; Soulard, M.; Rigolet, S.; Le Houérou, V.; Gauthier, C. Bridged polysilsesquioxane films via photoinduced sol–gel chemistry. New J. Chem. 2010, 34, 1068–1072. [Google Scholar] [CrossRef]
- Chemtob, A.; Peter, M.; Belon, C.; Dietlin, C.; Croutxé-Barghorn, C.; Vidal, L.; Rigolet, S. Macroporous organosilica films via a template-free photoinduced sol–gel process. J. Mater. Chem. 2010, 20, 9104–9112. [Google Scholar] [CrossRef]
- De Paz, H.; Chemtob, A.; Croutxé-Barghorn, C.; Le Nouen, D.; Rigolet, S. Insights into Photoinduced Sol–Gel Polymerization: An in Situ Infrared Spectroscopy Study. J. Phys. Chem. B 2012, 116, 5260–5268. [Google Scholar] [CrossRef]
- Brzezińska, K.; Szymański, R.; Kubisa, P.; Penczek, S. Activated monomer mechanism in cationic polymerization, 1. Ethylene oxide, formulation of mechanism. Makromol. Chem. Rapid Commun. 1986, 7, 1–4. [Google Scholar] [CrossRef]
- Bednarek, M.; Kubisa, P.; Penczek, S. Multihydroxyl Branched Polyethers. 2. Mechanistic Aspects of Cationic Polymerization of 3-Ethyl-3-(hydroxymethyl)oxetane. Macromolecules 2001, 34, 5112–5119. [Google Scholar] [CrossRef]
- Breloy, L.; Negrell, C.; Mora, A.S.; Li, W.S.J.; Brezová, V.; Caillol, S.; Versace, D.L. Vanillin derivative as performing type I photoinitiator. Eur. Polym. J. 2020, 132, 109727. [Google Scholar] [CrossRef]
- Sautrot-Ba, P.; Brezová, V.; Malval, J.-P.; Chiappone, A.; Breloy, L.; Abbad-Andaloussi, S.; Versace, D.-L. Purpurin derivatives as visible-light photosensitizers for 3D printing and valuable biological applications. Polym. Chem. 2021, 12, 2627–2642. [Google Scholar] [CrossRef]
- Sato, T.; Hamada, Y.; Sumikawa, M.; Araki, S.; Yamamoto, H. Solubility of Oxygen in Organic Solvents and Calculation of the Hansen Solubility Parameters of Oxygen. Ind. Eng. Chem. Res. 2014, 53, 19331–19337. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Niu, S.; Schneider, R.; Vidal, L.; Balan, L. Thioxanthone functionalized silver nanorods as smart photoinitiating assemblies to generate photopolymer/metal nano-objects. Nanoscale 2013, 5, 6538–6544. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
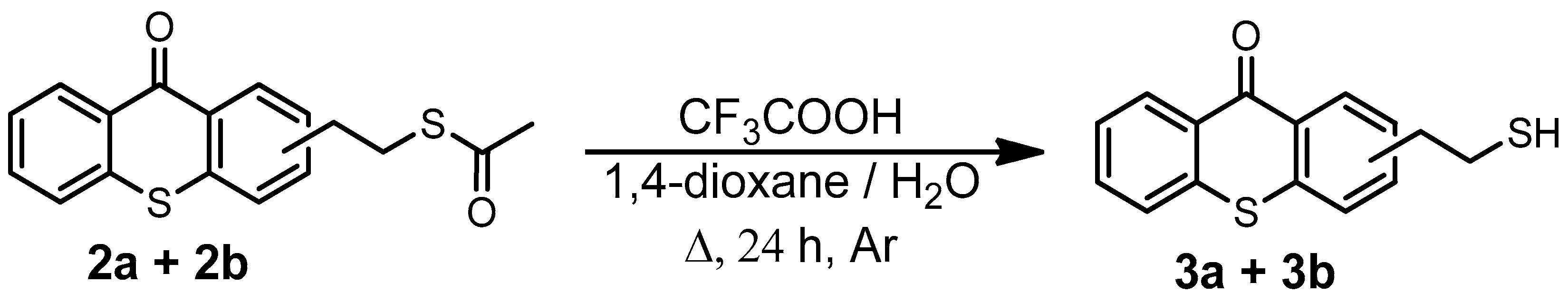{kind=link}
{kind=link}
| Functions | LED@385 nm | LED@405 nm | LED@455 nm | ||||
|---|---|---|---|---|---|---|---|
| Air | Laminate | Air | Laminate | Air | Laminate | ||
| (TXS/Iod)/ GPTMS 100% | Epoxy GPTMS | 91% | 100% | 98% | 100% | 80% | 95% |
| (TX/Iod)/ GPTMS 100% | Epoxy GPTMS | 100% | 99% | 100% | 82% | 70% | 30% |
| (TXS/Iod)/ GPTMS 25%/ DPDO 75% | Epoxy GPTMS Epoxy DPDO | 100% 84% | 75% 57% | 90% 65% | 70% 55% | 45% 41% | np np |
| (TX/Iod)/ GPTMS 25%/ DPDO 75% | Epoxy GPTMS Epoxy DPDO | 100% 86% | 95% 65% | 100% 92% | 100% 59% | np np | np np |
| (TXS/Iod)/ GPTMS 25%/ EPOX 75% | Epoxy GPTMS Epoxy EPOX | - 91% | - 74% | - 88% | - 60% | np np | np np |
| (TX/Iod)/ GPTMS 25%/ EPOX 75% | Epoxy GPTMS Epoxy EPOX | - 97% | - 65% | - 93% | - 67% | np np | np np |
| Compound | Structure |
|---|---|
| Dipentene diepoxide (DPDO) |  |
| 3,4-Epoxycyclohexylmethyl 3,4-epoxycyclohexanecarboxylate (EPOX) |  |
| 3-(Trimethoxysilyl)propyl methacrylate (MAPTMS) |  |
| (3-Glycidyloxypropyl)trimethoxysilane (GPTMS) |  |
| Soybean oil acrylate (SOA) |  |
| Bis(4-methylphenyl)iodonium hexafluorophosphate (Iod) |  |
| Thioxanthone (TX) |  |
| 2,2-Dimethyl-4-phenyl-2H-imidazole 1-oxide (DMPIO) |  |
| N-tert-Butyl-α-phenylnitrone (PBN) |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.-T.-T.; Breloy, L.; Rios De Anda, A.; Hayek, H.; Chiappone, A.; Malval, J.-P.; Grande, D.; Versace, D.-L. Thioxanthone-Based Siloxane Photosensitizer for Cationic/Radical Photopolymerization and Photoinduced Sol–Gel Reactions. Molecules 2024, 29, 255. https://doi.org/10.3390/molecules29010255
Nguyen T-T-T, Breloy L, Rios De Anda A, Hayek H, Chiappone A, Malval J-P, Grande D, Versace D-L. Thioxanthone-Based Siloxane Photosensitizer for Cationic/Radical Photopolymerization and Photoinduced Sol–Gel Reactions. Molecules. 2024; 29(1):255. https://doi.org/10.3390/molecules29010255
Chicago/Turabian StyleNguyen, Thi-Thanh-Tam, Louise Breloy, Agustin Rios De Anda, Hassan Hayek, Annalisa Chiappone, Jean-Pierre Malval, Daniel Grande, and Davy-Louis Versace. 2024. "Thioxanthone-Based Siloxane Photosensitizer for Cationic/Radical Photopolymerization and Photoinduced Sol–Gel Reactions" Molecules 29, no. 1: 255. https://doi.org/10.3390/molecules29010255