
Synthesis and Characterization of Ruthenium-Paraphenylene-Cyclopentadienyl Full-Sandwich Complexes: Cytotoxic Activity against A549 Lung Cancer Cell Line and DNA Binding Properties
 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of the Complexes (1)–(8)
2.1.1. Synthesis
2.1.2. Solution Characterization
2.1.3. Crystal Structure of the Complexes (1), (3) and (4)
2.2. Biological Studies
2.2.1. Fluorescence Quenching Studies of the d(5′-CGCGAATTCGCG-3′)2-Ethidium Bromide Adduct, with the Complexes (1)–(8)
2.2.2. Cytotoxic Activity
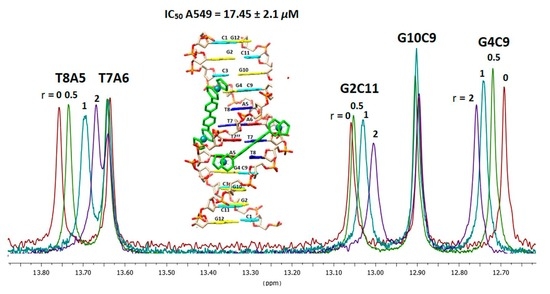
2.2.3. NMR Studies of the Interactions of the Complexes (6)Cl and (7)Cl2 with the d(5′-CGCGAATTCGCG-3′)2
[(η5-C5H5)2Ru2(η6-p-quaterphenyl)]Cl2, (7)Cl2
[(η5-C5H5)Ru2(η6-p-quaterphenyl)]Cl, (6)Cl
3. Experimental
3.1. Materials and Methods
3.2. Fluorescence Measurements
3.3. Cell Culture
3.4. Cell Viability Assay
3.5. Crystal Structure Analysis
3.6. Synthesis of the Complexes
- [(η5-C5H5)Ru(η6-Biphenyl)]PF6, (1): In a 10 mL vial, 10 mg (0.065 mmol) of biphenyl was added to 6 mL CH2Cl2 and the solution was heated for 5 min at 55 °C. After the complete dissolution of biphenyl, 30 mg (0.07 mmol) of [(η5-C5H5)Ru(CH3CN)3]PF6 was added, and the mixture was stirred for 24 h at room temperature. Subsequently, the solvent was removed in vacuo, and the resulting off-white solid was washed with H2O (3 × 2 mL) and 100 μL CH2Cl2. Yield: 60%. Anal. for C17H15PF6Ru: calc.% C, 46.06; H, 4.27; found C, 46.10; H, 4.32. 1H NMR: (500 MHz, dmso-d6, δ in ppm), H1a: 7.48 (t, 1H), H2a6a: 7.49 (t, 2H), H3a5a: 7.73 (d, 2H, 3JH-H = 7.0 Hz), H2b6b: 6.72 (d, 2H, 3JH-H = 6.0 Hz), H3b5b: 6.40 (t, 2H, 3JH-H = 5.9 Hz), H1b: 6.28 (t, 1H, 3JH-H = 5.6 Hz), CpH: 5.42 (s, 5H). Suitable crystals for X-ray analysis were obtained by dissolution of an amount of (1) in 2 mL of CH2Cl2 and allowed to slowly diffuse with diethyl ether vapors. After a few days, grey crystals appeared, which were collected by filtration, washed with diethyl ether (3 × 2 mL), and dried under vacuum.
- [(η5-C5H5)2Ru2(η6-Biphenyl)](PF6)2, (2): Complex (2) was prepared similarly to (1), but approximately 2.5 eq. of [(η5-C5H5)Ru(CH3CN3)]PF6 (65 mg, 0.15 mmol) was added. After solvent removal, the resulting white solid was washed with CH2Cl2 (3 × 2 mL) and H2O (3 × 2 mL). Yield: 56%. Anal. for C22H20P2F12Ru2: calc.% C, 37.33; H, 3.86; found C, 37.30; H, 3.89.1H NMR: (500 MHz, dmso-d6, δ in ppm), H1a/1b: 6.36 (t, 2H, 3JH-H = 5.1 Hz), H2a6a/2b6b: 6.42 (t, 4H, 3JH-H = 6.1 Hz), H3a5a/3b5b: 6.73 (d, 4H, 3JH-H = 6.1 Hz), CpH: 5.55 (s, 10H).
- [(η5-C5H5)Ru(η6-p-Terphenyl)]PF6, (3): Complex (3) was prepared similarly to (1). Yield: 45%. Anal. for C23H19PF6Ru: calc.% C, 52.54; H, 4.41; found C, 52.52; H, 2.68. 1H NMR: (500 MHz, dmso-d6, δ in ppm), H1a: 6.31 (t, 1H, 3JH-H = 4.5 Hz), H2a6a: 6.44 (t, 2H, 3JH-H = 6.2 Hz), H3a5a, 6.81: (d, 2H, 3JH-H = 6.2 Hz), H2b6b: 7.85 (d, 2H, 3JH-H = 8.4 Hz), H3b5b: 7.81 (d, 2H, 3JH-H = 8.4 Hz), H3c5c: 7.74 (d, 2H, 3JH-H = 7.9 Hz), H2c6c: 7.51 (t, 2H, 3JH-H = 7.5 Hz), H1c: 7.43 (t, 1H, 3JH-H = 7.5 Hz), CpH: 5.44 (s, 5H). Suitable crystals for X-ray analysis were obtained by dissolution of an amount of (3) in 2 mL of CH2Cl2 and allowed to slowly diffuse with diethyl ether vapors. After a few days, grey crystals appeared, which were collected by filtration, washed with diethyl ether (3 × 2 mL), and dried under vacuum.
- [(η5-C5H5)2Ru2(η6-p-Terphenyl)](PF6)2, (4): In a 10 mL vial, 10 mg of p-terphenyl (0.04 mmol) was added to 3 mL of CH2Cl2, and the mixture was heated at 55 °C until it completely dissolved. Then, 1.1 eq. of [(η5-C5H5)Ru(CH3CN3)]PF6 (20 mg, 0.045 mmol) dissolved in 1 mL of CH2Cl2 was added to the reaction mixture, which was stirred for 24 h at room temperature. After the solvent removal, a solution containing 2 eq. (35 mg, 0.08 mmol) of [(η5-C5H5)Ru(CH3CN3)]PF6 in 1 mL of acetone was added. The mixture was heated at 55 °C for an additional 24h, filtered, evaporated to dryness, washed with H2O (2 mL × 2 times) and CH2Cl2 (2 mL × 2 times), and dried under vacuum. Yield: 44%. Anal. for C28H24P2F12Ru2: calc.% C, 42.11; H, 3.98; found C, 42.14; H, 3.95. HR-ESI-MS, positive (m/z): found. 243.3142, calc. 243.3132 for [C28H24Ru2]2+. 1H NMR: (500 MHz, dmso-d6, δ in ppm), H1a/1c: 6.33 (t, 2H, 3JH-H = 5.6 Hz), H2a6a/2c6c: 6.45 (t, 4H, 3JH-H = 5.7 Hz), H3a5a/3c5c: 6.78 (d, 4H, 3JH-H = 6.0 Hz), H2b6b/3b5b: 7.83 (s, 4H), CpH: 5.45 (s, 10H). Suitable crystals for X-ray analysis were obtained by dissolution of an amount of (4) in 2mL of a mixture of methanol:acetone 1:1 and allowed to slow evaporation. After a few days, grey crystals appeared, which were collected by filtration and dried in a vacuum.
- [(η5-C5H5)3Ru3(η6-p-Terphenyl)](PF6)3, (5): In a 10 mL vial, 10 mg of p-terphenyl (0.04 mmol) and 3 mL of CH2Cl2 were added. The solution was heated at 55 °C until complete dissolution and 3 eq. (52 mg, 0.12 mmol) of [(η5-C5H5)Ru(CH3CN3)]PF6 dissolved in 2 mL of CH2Cl2 was added. The mixture was left to react for 24h at room temperature and then evaporated to dryness, resulting in the formation of a gray solid. Following this, 4 eq. (70 mg, 0.16 mmol) of [(η5-C5H5)Ru(CH3CN3)]PF6 dissolved in 3 mL of acetone was added to the crude solid, and the mixture was heated at 55 °C for a further 24 h. Then, the solvent was removed, and the solid was washed with H2O (2 mL × 2 times) and CH2Cl2 (2 mL × 2 times). Yield: 38%. Anal. for C33H29P3F18Ru3: calc.% C, 37.36; H, 3.78; found C, 37.34; H, 3.80. HR-ESI-MS, positive (m/z): found. 243.3142 calc. 243.3132 for [C33H29Ru3]3+. 1H NMR: (500 MHz, acetone-d6, δ in ppm), H1a/1c: 6.56 (t, 2H, 3JH-H = 5.7 Hz), H2a6a/2c6c: 6.63 (t, 4H, 3JH-H = 5.8 Hz), H3a5a/3c5c: 6.97 (d, 4H, 3JH-H = 5.9 Hz), H2b6b/3b5b: 7.15 (s, 4H), Cp1H: 5.63 (s, 10H), Cp2H: 5.73 (s, 5H).
- [(η5-C5H5)Ru(η6-p-Quaterphenyl)]PF6, (6): Complex (6) was prepared similarly to (1), but at 20 mg of p-quaterphenyl (0.06 mmol) in 40 mL of CH2Cl2, 0.2 eq. of [(η5-C5H5)Ru(CH3CN3)]PF6 (10 mg, 0.02 mmol) in 0.5 mL of CH2Cl2 was added. Also, after removing the solvent, the crude product was washed with 2 mL of acetone, and the resulting solution was evaporated to dryness. Yield: 36%. Anal. for C29H23PF6Ru: calc.% C, 57.49; H, 4.51; found C, 57.51; H, 4.49. HR-ESI-MS, positive (m/z): found. 473.0840, calc. 473.0848 for [C29H23Ru2]+. 1H NMR: (500 MHz, dmso-d6, δ in ppm), H1a: 6.32 (t, 1H, 3JH-H = 5.4 Hz), H2a6a: 6.45 (t, 2H, 3JH-H = 5.8 Hz), H3a5a: 6.81 (d, 2H, 3JH-H = 6.4 Hz), H2b6b/3b5b: 7.83 (4H), H2c6c/3c5c: 7.86 (4H), H3d5d: 7.74 (d, 2H, 3JH-H = 7.5 Hz), H2d6d: 7.50 (t, 2H, 3JH-H = 7.5 Hz), H1d: 7.40 (t, 1H, 3JH-H = 7.2 Hz), CpH: 5.46 (s, 5H).
- [(η5-C5H5)2Ru2(η6-p-Quaterphenyl)](PF6)2, (7): In a 50 mL round-bottom flask, 35 mg of p-quaterphenyl (0.11 mmol) and 40 mL of CH2Cl2 were added. The mixture was heated at 55 °C until complete dissolution. Then, 1 mL of a CH2Cl2 solution containing 0.3 eq. of [(η5-C5H5)Ru(CH3CN)3]PF6 (15 mg, 0.35 mmol) was added, and the mixture was allowed to react for 1 h at room temperature. Next, 1.5 eq. [(η5-C5H5)Ru(CH3CN3)]PF6 in 0.5 mL of acetone was added, and the mixture was stirred for 24 h at room temperature. Subsequently, the solvent evaporated to dryness, and the grey solid was washed with H2O (2 mL × 2 times) and CH2Cl2 (2 mL × 2 times). Yield: 30%. Anal. for C34H28P2F12Ru2: calc.% C, 46.66; H, 4.32; found C, 46.68; H, 4.30. HR-ESI-MS, positive (m/z): found. 320.0133, calc. 320.0100 for [C34H27Ru2]2+. 1H NMR: (500 MHz, dmso-d6, δ in ppm), Ha1/d1: 6.32 (t, 2H, 3JH-H = 5.7 Hz), Ha2a6/d2d6: 6.44 (t, 4H, 3JH-H = 6.1 Hz), Ha3a5/d3d5: 6.79 (d, 4H, 3JH-H = 6.2 Hz), H2b6b/3b5b and H2c6c/3c5c: 7.86 (s, 8H), CpH: 5.45 (s, 10H).
- [(η5-C5H5)3Ru(η6-p-Quaterphenyl)](PF6)3, (8): In a 50 mL round bottom flask, 20 mg of p-quaterphenyl (0.06 mmol) and 40 mL of CH2Cl2 were added. The mixture was heated at 55 °C until complete dissolution. Next, 1 mL of a CH2Cl2 solution containing 2 eq. of [(η5-C5H5)Ru(CH3CN)3]PF6 (52 mg, 0.12 mmol) was added and the mixture was allowed to react for 1 h at room temperature. Subsequently, 3 eq. [(η5-C5H5)Ru(CH3CN3)]PF6 (80 mg, 0.18 mmol) dissolved in 1 mL of acetone was added, and the mixture was stirred for 24 h at room temperature. Then, the solvent was evaporated to dryness, and the resulting grey solid was washed with H2O (2 mL × 2 times) and CH2Cl2 (2 mL × 2 times). Yield: 50%. Anal. for C39H33P3F18Ru3: calc.% C, 41.08; H, 4.05; found C, 41.06; H, 4.07. HR-ESI-MS, positive (m/z): found. 268.6579 calc. 268.6570 for [C39H33Ru3]3+. 1H NMR: (500 MHz, acetone-d6, δ in ppm), H1a: 6.61 (t, 1H), H2a6a: 7.03 (t, 2H, 3JH-H = 6.0 Hz), H3a5a: 6.63 (d, 2H), H2b6b: 7.17 (d, 2H, 3JH-H = 6.3 Hz), H3b5b: 7.09 (d, 2H, 3JH-H = 6.4 Hz), H2c6c/3c5c: 7.99 (s, 4H), H2d6d: 6.89, (d, 2H, 3JH-H = 6.1 Hz), H3d5d: 6.58 (t, 2H), H1d: 6.48 (t, 1H, 3JH-H = 5.9 Hz), HCp1: 5.67 (s, 5H), HCp2: 5.62 (s, 5H), HCp3: 5.54 (s, 5H).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Division, C.; Products, E.; Electrode, P. Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 20, 698–699. [Google Scholar]
- Scott, R.B. Cancer Chemotherapy—The First Twenty-five Years. Br. Med. J. 1970, 4, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Shang, Q.; Li, W.; Guo, W.; Stojadinovic, A.; Mannion, C.; Man, Y.G.; Chen, T. Antibiotics for cancer treatment: A double-edged sword. J. Cancer 2020, 11, 5135–5149. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Emadi, A. Ruthenium-based chemotherapeutics: Are they ready for prime time? Cancer Chemother. Pharmacol. 2010, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Elgar, C.E.; Yusoh, N.A.; Tiley, P.R.; Kolozsvári, N.; Bennett, L.G.; Gamble, A.; Péan, E.V.; Davies, M.L.; Staples, C.J.; Ahmad, H.; et al. Ruthenium(II) Polypyridyl Complexes as FRET Donors: Structure- and Sequence-Selective DNA-Binding and Anticancer Properties. J. Am. Chem. Soc. 2023, 145, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Berners-Price, S.J.; Filipovska, A. The design of gold-based, mitochondria-targeted chemotherapeutics. Aust. J. Chem. 2008, 61, 661–668. [Google Scholar] [CrossRef]
- Dubarle-Offner, J.; Clavel, C.M.; Gontard, G.; Dyson, P.J.; Amouri, H. Selenoquinones stabilized by ruthenium(II) arene complexes: Synthesis, structure, and cytotoxicity. Chem.-A Eur. J. 2014, 20, 5795–5801. [Google Scholar] [CrossRef]
- Loughrey, B.T.; Williams, M.L.; Parsons, P.G.; Healy, P.C. Nucleophilic substitution reactions of [(η5-Cp∗)Ru(η6-C6H5CO2H)]+: Synthesis, characterization and cytotoxicity of organoruthenium ester and amide complexes. J. Organomet. Chem. 2016, 819, 1–10. [Google Scholar] [CrossRef]
- Loughrey, B.T.; Healy, P.C.; Parsons, P.G.; Williams, M.L. Selective cytotoxic Ru(II) arene Cp* complex salts [R-PhRuCp*]+X- for X = BF4-, PF6-, and BPh4-. Inorg. Chem. 2008, 47, 8589–8591. [Google Scholar] [CrossRef]
- Loughrey, B.T.; Williams, M.L.; Carruthers, T.J.; Parsons, P.G.; Healy, P.C. Synthesis, structure, and selective cytotoxicity of organometallic Cp *RuII O-alkyl-N-phenylcarbamate sandwich complexes. Aust. J. Chem. 2010, 63, 245–251. [Google Scholar] [CrossRef]
- Loughrey, B.T.; Williams, M.L.; Healy, P.C.; Innocenti, A.; Vullo, D.; Supuran, C.T.; Parsons, P.G.; Poulsen, S.A. Novel organometallic cationic ruthenium(II) pentamethylcyclopentadienyl benzenesulfonamide complexes targeted to inhibit carbonic anhydrase. J. Biol. Inorg. Chem. 2009, 14, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Lamač, M.; Horáček, M.; Šťastná, L.Č.; Karban, J.; Sommerová, L.; Skoupilová, H.; Hrstka, R.; Pinkas, J. Harmless glucose-modified ruthenium complexes suppressing cell migration of highly invasive cancer cell lines. Appl. Organomet. Chem. 2020, 34, 1–5. [Google Scholar] [CrossRef]
- Garcia, M.H.; Valente, A.; Florindo, P.; Morais, T.S.; Piedade, M.F.M.; Duarte, M.T.; Moreno, V.; Avilés, F.X.; Loreno, J. New ruthenium(II) mixed metallocene derived complexes: Synthesis, characterization by X-ray diffraction and evaluation on DNA interaction by atomic force microscopy. Inorganica Chim. Acta 2010, 363, 3765–3775. [Google Scholar] [CrossRef]
- Gross, A.; Schur, J.; Ott, I.; Metzler-nolte, N. A Ruthenocene–PNA Bioconjugate Synthesis, Characterization, Cytotoxicity, and AAS-Detected Cellular Uptake. Bioconjug. Chem. 2012, 23, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Loughrey, B.T.; Cunning, B.V.; Healy, P.C.; Brown, C.L.; Parsons, P.G.; Williams, M.L. Selective, cytotoxic organoruthenium(II) full-sandwich complexes: A structural, computational and in vitro biological study. Chem.—An Asian J. 2012, 7, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Vanecek, A.S.; Tepe, J.J.; Odom, A.L. Synthesis, structure, properties, and cytotoxicity of a (quinoline)RuCp+ complex. Dalt. Trans. 2022, 52, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Micallef, L.S.; Loughrey, B.T.; Healy, P.C.; Parsons, P.G.; Williams, M.L. Synthesis, spectroscopic characterization, and cytotoxic evaluation of pentasubstituted ruthenocenyl esters. Organometallics 2010, 29, 6237–6244. [Google Scholar] [CrossRef]
- Micallef, L.S.; Loughrey, B.T.; Healy, P.C.; Parsons, P.G.; Williams, M.L. Mono- and 1,1′-disubstituted organoruthenium cyclopentadiene complexes: Synthesis, structural characterization, and antitumoral evaluation. Organometallics 2011, 30, 1395–1403. [Google Scholar] [CrossRef]
- Gozzi, M.; Schwarze, B.; Sárosi, M.B.; Lönnecke, P.; Drača, D.; Maksimović-Ivanić, D.; Mijatović, S.; Hey-Hawkins, E. Antiproliferative activity of (η6-arene)ruthenacarborane sandwich complexes against HCT116 and MCF7 cell lines. Dalt. Trans. 2017, 46, 12067–12080. [Google Scholar] [CrossRef]
- Fagan, P.A.; Spielmann, H.P.; Sigurdsson, S.T.; Rink, S.M.; Hopkins, P.B.; Wemmer, D.E. An NMR study of [d(CGCGAATTCGCG)]2 containing an interstrand cross-link derived from a distamycin-pyrrole conjugate. Nucleic Acids Res. 1996, 24, 1566–1573. [Google Scholar] [CrossRef]
- Georgakopoulou, C.; Thomos, D.; Tsolis, T.; Ypsilantis, K.; Plakatouras, J.C.; Kordias, D.; Magklara, A.; Kouderis, C.; Kalampounias, A.G.; Garoufis, A. Synthesis, characterization, interactions with the DNA duplex dodecamer d(5′-CGCGAATTCGCG-3′)2 and cytotoxicity of binuclear η6-arene-Ru(ii) complexes. Dalt. Trans. 2022, 51, 13808–13825. [Google Scholar] [CrossRef] [PubMed]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Parks, W.P. In vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Bade, B.C.; Cruz, C.S.D. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2020, 41, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.D.; Fagan, P.J.; Calabrese, J.C.; Johnson, D.C. Electrostatic Structural Enforcement in Low-Dimensional Solids: Synthesis, Structure, and Electronic Properties of Polycationic Ruthenium Complexes with Polycyanoanions. J. Am. Chem. Soc. 1989, 111, 1719–1732. [Google Scholar] [CrossRef]
- Harre, K.; Enkelmann, V.; Schulze, M.; Bunz, U.H.F. Suzaki-coupling of Cp*Ru(para-C6M4Br2) with phenyl boronic acid: A model recation for the synthesis of organometallic polymers. Chem. Ber. 1996, 129, 1323–1325. [Google Scholar] [CrossRef]
- Ziemann, E.A.; Baljak, S.; Steffens, S.; Stein, T.; Van Steerteghem, N.; Asselberghs, I.; Clays, K.; Heck, J. Stille cross-coupling reaction with cationic [(η5-Cp)( eta;6-C6H6- xIx)Ru]+ complexes as key for ethynyl-bridged homo- and heteronuclear sandwich compounds. Organometallics 2015, 34, 1692–1700. [Google Scholar] [CrossRef]
- Perekalin, D.S.; Karslyan, E.E.; Petrovskii, P.V.; Borissova, A.O.; Lyssenko, K.A.; Kudinov, A.R. Arene exchange in the ruthenium-naphthalene complex [CpRu(C 10H8)]+. Eur. J. Inorg. Chem. 2012, 2012, 1485–1492. [Google Scholar] [CrossRef]
- Tsolis, T.; Nikolaou, N.; Ypsilantis, K.; Kougioumtzi, A.; Kordias, D.; Magklara, A.; Garoufis, A. Synthesis, characterization, interactions with 9-MeG and cytotoxic activity of heterobimetallic RuII-PtII complexes bridged with 2,2′-bipyrimidine. J. Inorg. Biochem. 2021, 219, 111435. [Google Scholar] [CrossRef]
- Ramana, M.M.V.; Betkar, R.; Nimkar, A.; Ranade, P.; Mundhe, B.; Pardeshi, S. In vitro DNA binding studies of antiretroviral drug nelfinavir using ethidium bromide as fluorescence probe. J. Photochem. Photobiol. B Biol. 2015, 151, 194–200. [Google Scholar] [CrossRef]
- Bittman, R. Studies of the binding of ethidium bromide to transfer ribonucleic acid: Absorption, fluorescence, ultracentrifugation and kinetic investigations. J. Mol. Biol. 1969, 46, 251–268. [Google Scholar] [CrossRef]
- Waring, M.J. Complex formation between ethidium bromide and nucleic acids. J. Mol. Biol. 1965, 13, 269–282. [Google Scholar] [CrossRef]
- Lepecq, J.B.; Paoletti, C. A fluorescent complex between ethidium bromide and nucleic acids. Physical-Chemical characterization. J. Mol. Biol. 1967, 27, 87–106. [Google Scholar] [CrossRef]
- Aramesh-Boroujeni, Z.; Jahani, S.; Khorasani-Motlagh, M.; Kerman, K.; Aramesh, N.; Asadpour, S.; Noroozifar, M. Experimental and theoretical investigations of Dy(III) complex with 2,2′-bipyridine ligand: DNA and BSA interactions and antimicrobial activity study. J. Biomol. Struct. Dyn. 2020, 38, 4746–4763. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Cheatham, T.E. Ethidium bromide interactions with DNA: An exploration of a classic DNA-ligand complex with unbiased molecular dynamics simulations. Nucleic Acids Res. 2021, 49, 3735–3747. [Google Scholar] [CrossRef]
- Anbu, S.; Kandaswamy, M.; Kamalraj, S.; Muthumarry, J.; Varghese, B. Phosphatase-like activity, DNA binding, DNA hydrolysis, anticancer and lactate dehydrogenase inhibition activity promoting by a new bis-phenanthroline dicopper(II) complex. Dalt. Trans. 2011, 40, 7310–7318. [Google Scholar] [CrossRef]
- Gürses, C.; Aktaş, A.; Balcıoğlu, S.; Fadhilah, A.; Gök, Y.; Ateş, B. Synthesis, characterization, DNA binding and anticancer activities of the imidazolidine-functionalized (NHC)Ru(II) complexes. J. Mol. Struct. 2022, 1247, 131350. [Google Scholar] [CrossRef]
- Afrati, T.; Pantazaki, A.A.; Dendrinou-Samara, C.; Raptopoulou, C.; Terzis, A.; Kessissoglou, D.P. Copper inverse-9-metallacrown-3 compounds interacting with DNA. Dalt. Trans. 2010, 39, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Khanvilkar, P.; Dash, S.R.; Vohra, A.; Devkar, R.; Chakraborty, D. Evaluation of biomolecular interactions and cytotoxic activity of organometallic binuclear Ru(II) complexes of ferrocenyl thiosemicarbazones. J. Biomol. Struct. Dyn. 2021, 39, 6044–6055. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.J.; Kozlowski, S.A.; Rice, J.A.; Marky, L.A.; Breslauer, K.J.; Broka, C.; Itakura, K. Premelting and Melting Transitions in the d(CGCGAATTCGCG) Self-Complementary Duplex in Solution. Biochemistry 1982, 21, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.R.; Wemmer, D.E.; Chou, S.H.; Drobny, G.; Reid, B.R. Assignment of the non-exchangeable proton resonances of d(C-G-C-G-A-A-T-T-C-G-C-G) using two-dimensional nuclear magnetic resonance methods. J. Mol. Biol. 1983, 171, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.; Lygo, B.; Procter, G. Advanced Practical Organic Chemistry; Taylor & Francis: Oxfordshire, UK, 2013. [Google Scholar]
- Trost, B.M.; Older, C.M. A convenient synthetic route to [CpRu(CH3CN3)]PF6. Organometallics 2002, 21, 2544–2546. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Rehman, S.U.; Yaseen, Z.; Husain, M.A.; Sarwar, T.; Ishqi, H.M.; Tabish, M. Interaction of 6 mercaptopurine with calf thymus DNA—Deciphering the binding mode and photoinduced DNA damage. PLoS ONE 2014, 9, e93913. [Google Scholar] [CrossRef] [PubMed]
- APEX3; SAINT, SHELXT; Bruker AXS Inc.: Madison, WI, USA. 2016. Available online: https://xray.uky.edu/Resources/manuals/%0AApex3-manual.pdf (accessed on 6 November 2023).
- Sheldrick, G.M. SADABS; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Barbour, L.J. X-Seed—A Software Tool for Supramolecular Crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (1) ǂ | (3) | (4) | |
|---|---|---|---|
| Mean Ru–C (C5) | 2.182, 2.148 | 2.193 | 2.179 |
| Mean Ru–C (C6) | 2.198, 2.202 | 2.208 | 2.208 |
| Ru–Centroid C5 | 1.784, 1.766 | 1.829 | 1.814 |
| Ru–LS plane C5 | 1.782, 1.765 | 1.829 | 1.814 |
| Ru–Centroid C6 | 1.693, 1.696 | 1.700 | 1.701 |
| Ru–LS plane C5 | 1.693, 1.696 | 1.700 | 1.701 |
| Centroid C5–Ru–Centroid C6 | 176.77, 175.65 | 179.43 | 178.52 |
| Dihedral angle LS planes C5 and C6A | 3.278, 5.712 | 1.325 | 1.085 |
| Dihedral angle LS planes C6A and C6B | 39.02, 32.77 | 39.24 | 41.74 |
| Dihedral angle LS planes C6B and C6C | 50.93 | ||
| Dihedral angle LS planes C6A and C6C | 89.87 |
| Complex | Ksv (103 M−1) | Kb (103 M−1) | n | Quenching (%) |
|---|---|---|---|---|
| (1)Cl | 1.81 ± 0.053 | 2.330 ± 0.001 | 1.02 | 24.98 |
| (3)Cl | 1.04 ± 0.029 | 3.532 ± 0.001 | 1.15 | 26.97 |
| (4)Cl2 | 0.83 ± 0.032 | 0.815 ± 0.001 | 1.09 | 11.31 |
| (6)Cl | 4.30 ± 0.075 | 8.111 ± 0.001 | 1.07 | 44.38 |
| (7)Cl2 | 5.91 ± 0.024 | 5.933 ± 0.001 | 1.07 | 56.17 |
| (8)Cl3 | 2.66 ± 0.014 | 2.803 ± 0.121 | 1.00 | 36.72 |
| Complexes | A549 | HFL-1 |
|---|---|---|
| Cisplatin | 5.49 ± 2.1 (0.8) | 4.40 ± 2.1 |
| (6)Cl | 17.45 ± 2.1 (1.1) | 19.35 ± 2.1 |
| (7)Cl2 | 65.83 ± 1.8 (4.8) | 318.7 ± 1.8 |
| Compound | 1 | 3 | 4 |
|---|---|---|---|
| Empirical formula | C34H30F12P2Ru2 | C23H19F6PRu | C28H24F12P2Ru2 |
| Formula weight | 930.66 | 541.42 | 852.55 |
| Temperature (K) | 296(2) | ||
| Wavelength (Å) | 0.71073 | ||
| Crystal system | Triclinic | Monoclinic | Monoclinic |
| Space group | P | C2/c | P21/n |
| Unit cell dimensions a, b, c (Å), α, β, γ (°) | 10.5346(15), 12.1661(16), 14.845(2), 104.711(8), 104.567(7) 101.525(8) | 14.4714(5), 15.8121(6), 18.8661(6), 90, 95.535(1), 90 | 10.5652(6), 13.4766(8), 10.7065(6), 90, 101.663(3), 90 |
| Volume (Å3) | 1708.9(4) | 4296.9(3) | 1492.95(15) |
| Z | 2 | 8 | 2 |
| Density (calcd.) (g/cm3) | 1.809 | 1.674 | 1.897 |
| Absorption coefficient (mm−1) | 1.068 | 0.863 | 1.213 |
| F(000) | 920 | 2160 | 836 |
| Crystal size (mm3) | 0.30 × 0.10 × 0.06 | 0.60 × 0.50 × 0.50 | 0.20 × 0.15 × 0.02 |
| θ range for data collection (°) | 2.339 to 24.998 | 2.788 to 24.998 | 2.896 to 24.998 |
| Index ranges | −12 ≤ h ≤ 12, −14 ≤ k ≤ 14, −17 ≤ l ≤ 17 | −17 ≤ h ≤ 17, −18 ≤ k ≤ 18, −20 ≤ l ≤ 22 | −12 ≤ h ≤ 12, −16 ≤ k ≤ 16, −12 ≤ l ≤ 12 |
| Reflections collected | 90,759 | 62,541 | 43,243 |
| Independent reflections | 6029 [Rint = 0.1691] | 3774 [Rint = 0.0334] | 2629 [Rint = 0.1992] |
| Completeness to θ (%) | 99.9 | 99.4 | 99.9 |
| Refinement method | Full-matrix least-squares on F2 | ||
| Data/restraints/parameters | 6029/312/525 | 3774/72/310 | 2629/258/291 |
| Goodness-of-fit | 1.004 | 1.091 | 1.069 |
| Final R indices [I > 2σ(I)] | Robs = 0.0403, wRobs = 0.0910 | Robs = 0.0417, wRobs = 0.1010 | Robs = 0.0603, wRobs = 0.1053 |
| R indices [all data] | Rall = 0.0791, wRall = 0.1031 | Rall = 0.0440, wRall = 0.1021 | Rall = 0.0955, wRall = 0.1151 |
| Largest diff. peak and hole (e·Å−3) | 0.508 and −0.622 | 1.256 and −0.719 | 0.529 and −0.781 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sifnaiou, E.; Tsolis, T.; Ypsilantis, K.; Roupakia, E.; Kolettas, E.; Plakatouras, J.C.; Garoufis, A. Synthesis and Characterization of Ruthenium-Paraphenylene-Cyclopentadienyl Full-Sandwich Complexes: Cytotoxic Activity against A549 Lung Cancer Cell Line and DNA Binding Properties. Molecules 2024, 29, 17. https://doi.org/10.3390/molecules29010017
Sifnaiou E, Tsolis T, Ypsilantis K, Roupakia E, Kolettas E, Plakatouras JC, Garoufis A. Synthesis and Characterization of Ruthenium-Paraphenylene-Cyclopentadienyl Full-Sandwich Complexes: Cytotoxic Activity against A549 Lung Cancer Cell Line and DNA Binding Properties. Molecules. 2024; 29(1):17. https://doi.org/10.3390/molecules29010017
Chicago/Turabian StyleSifnaiou, Evangelia, Theodoros Tsolis, Konstantinos Ypsilantis, Eugenia Roupakia, Evangelos Kolettas, John C. Plakatouras, and Achilleas Garoufis. 2024. "Synthesis and Characterization of Ruthenium-Paraphenylene-Cyclopentadienyl Full-Sandwich Complexes: Cytotoxic Activity against A549 Lung Cancer Cell Line and DNA Binding Properties" Molecules 29, no. 1: 17. https://doi.org/10.3390/molecules29010017