
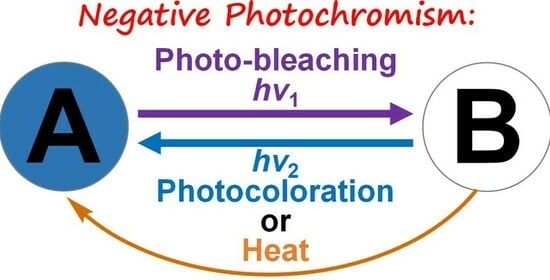
From Visible to Near–Infrared Light–Triggered Photochromism: Negative Photochromism
 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Negatively Photochromic Compounds
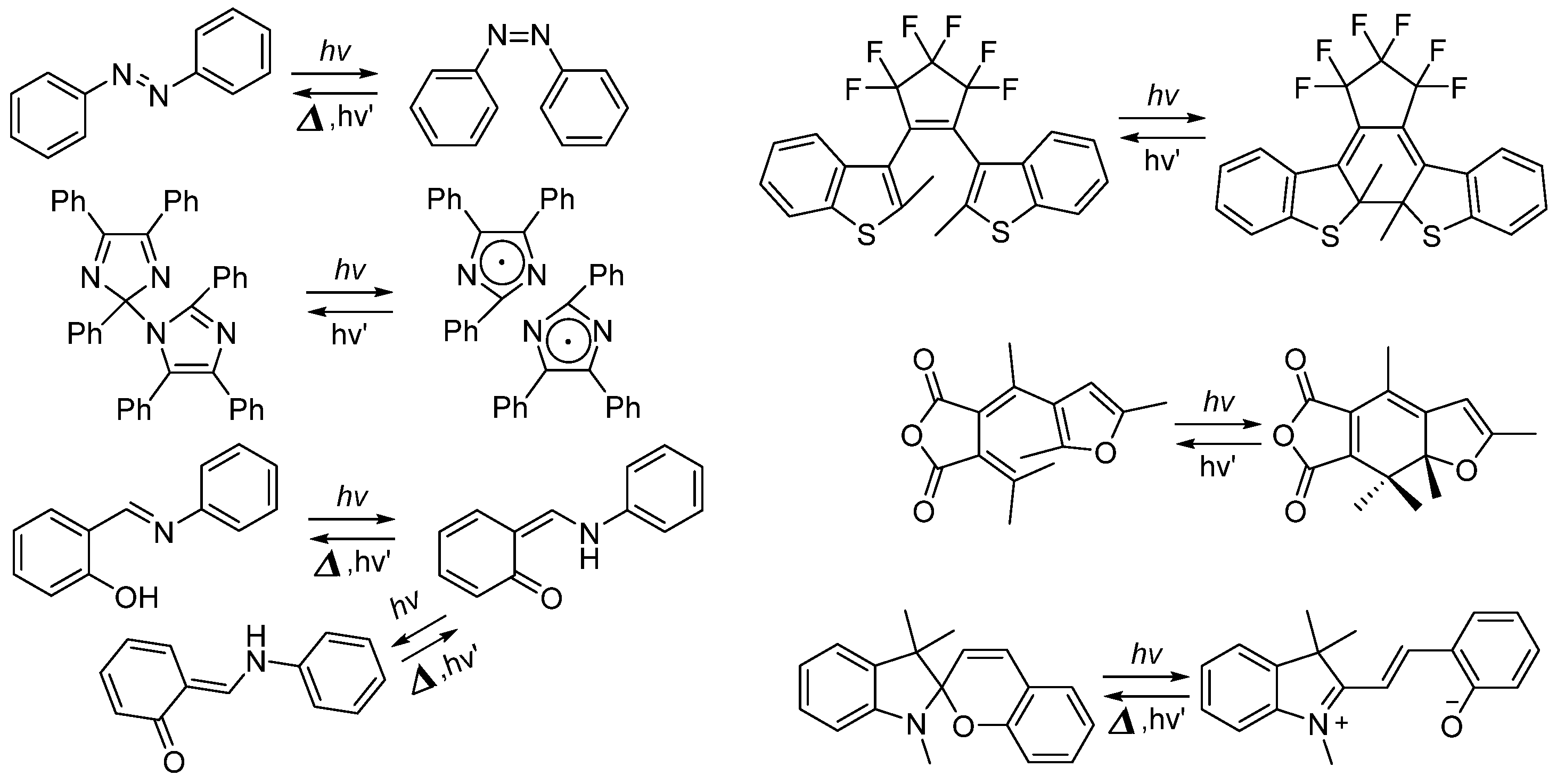
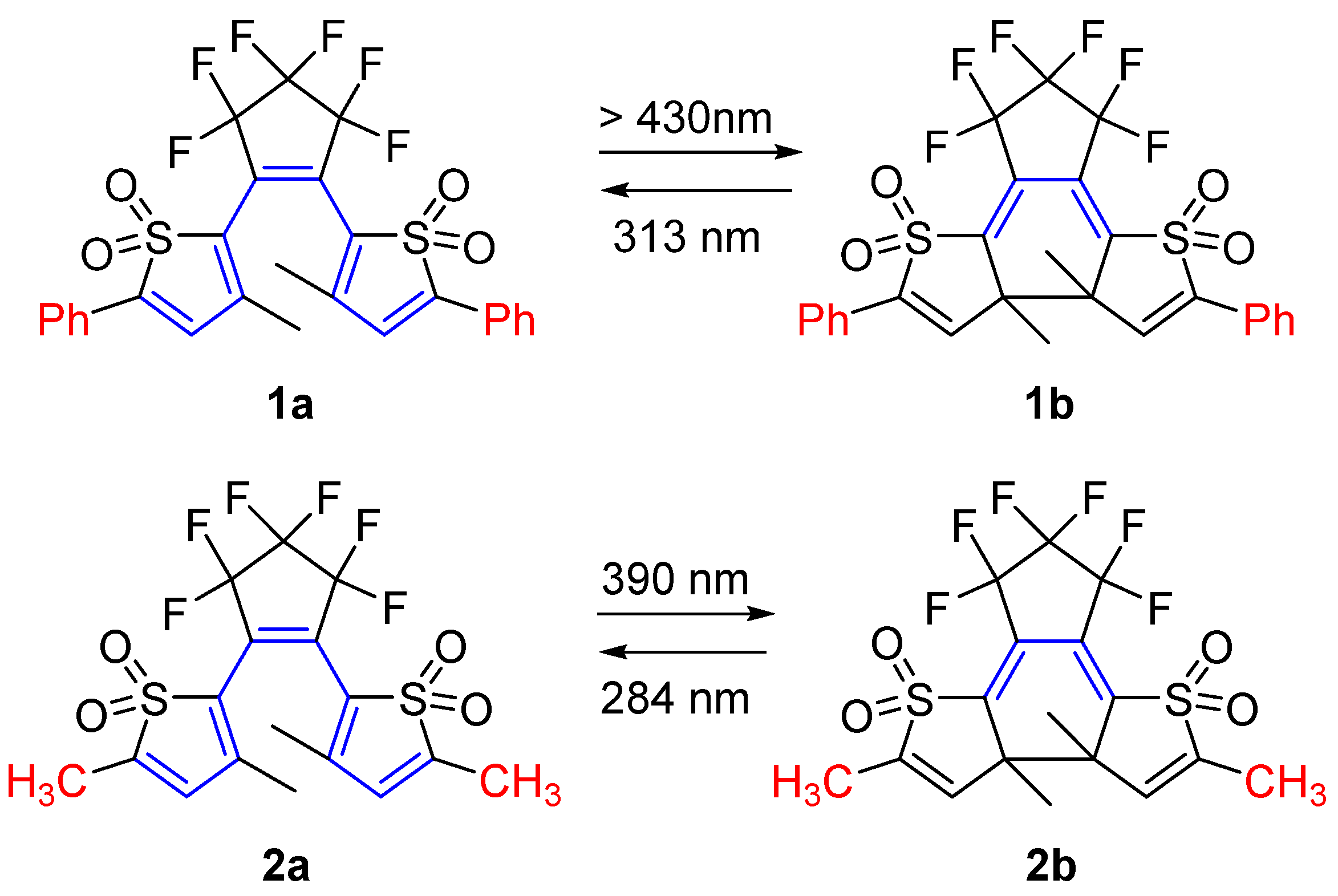
2.1. Diarylethene
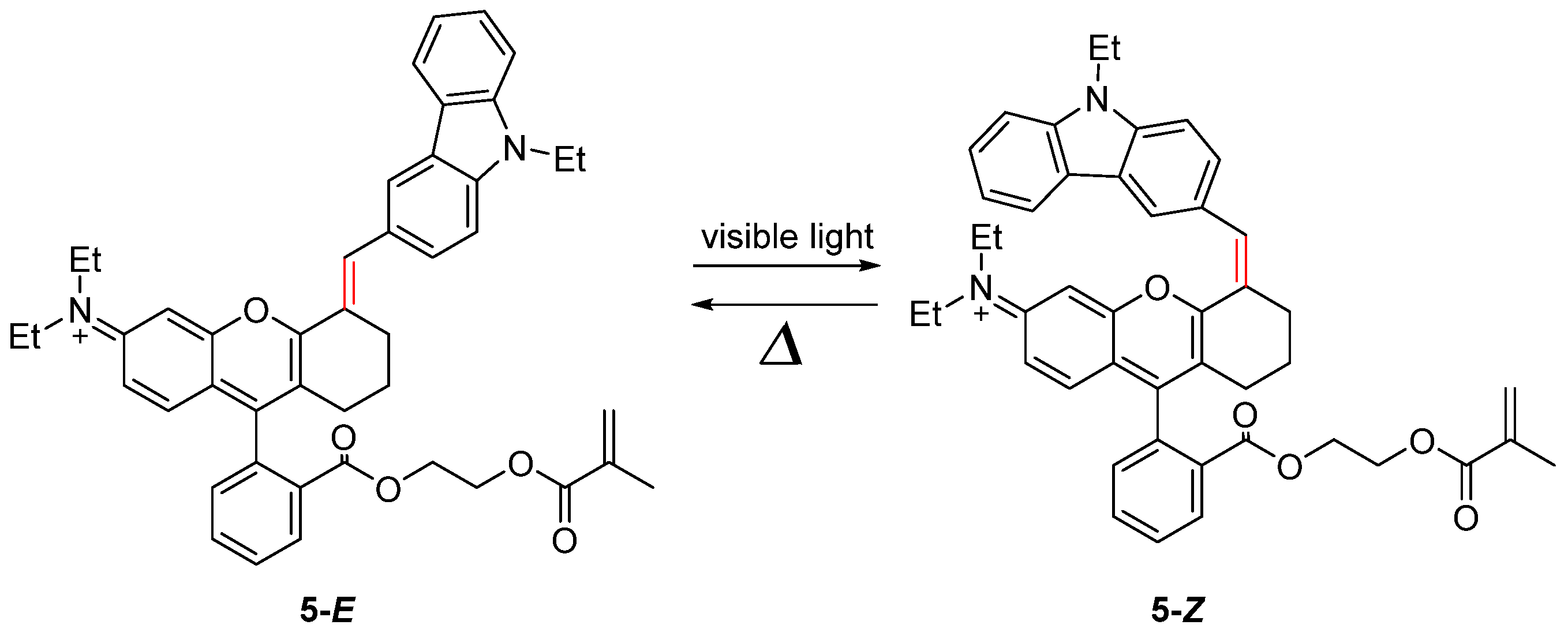
2.2. Azobenzene (or Stilbene) and Derivatives
2.3. Salicylideneaniline (Anils)
2.4. Dihydropyrenes
2.5. Donor–Acceptor Stenhouse Adducts (DASAs)
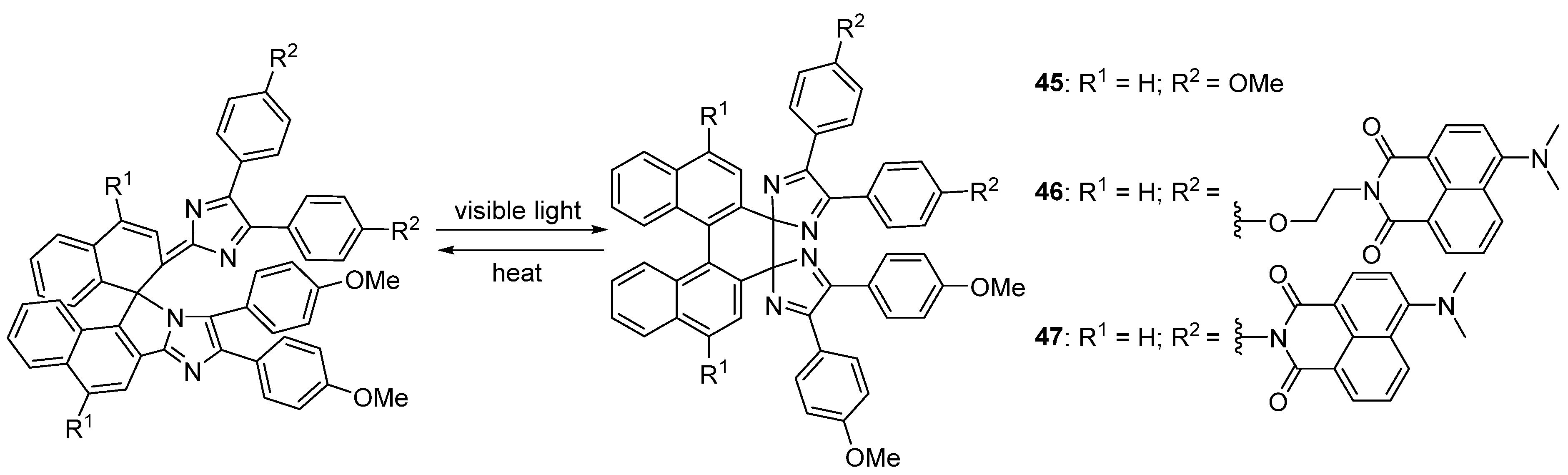
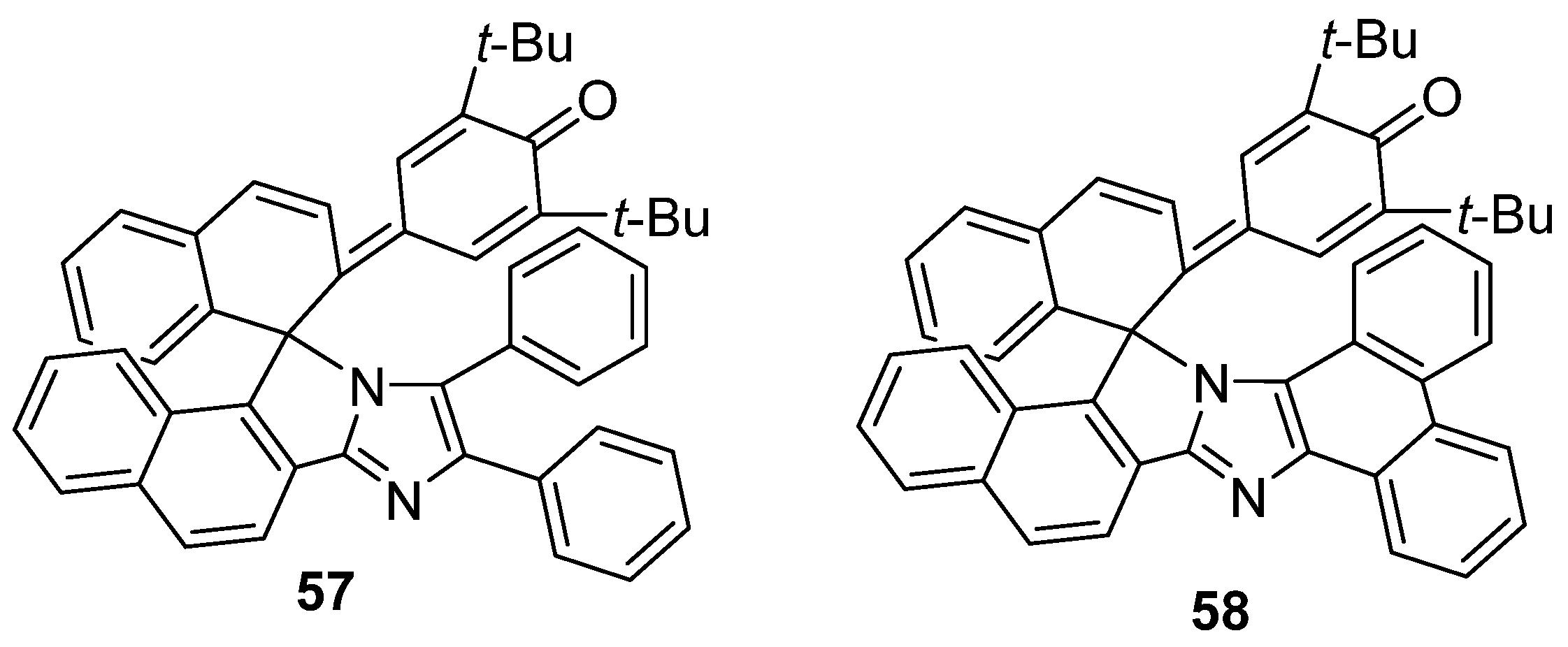
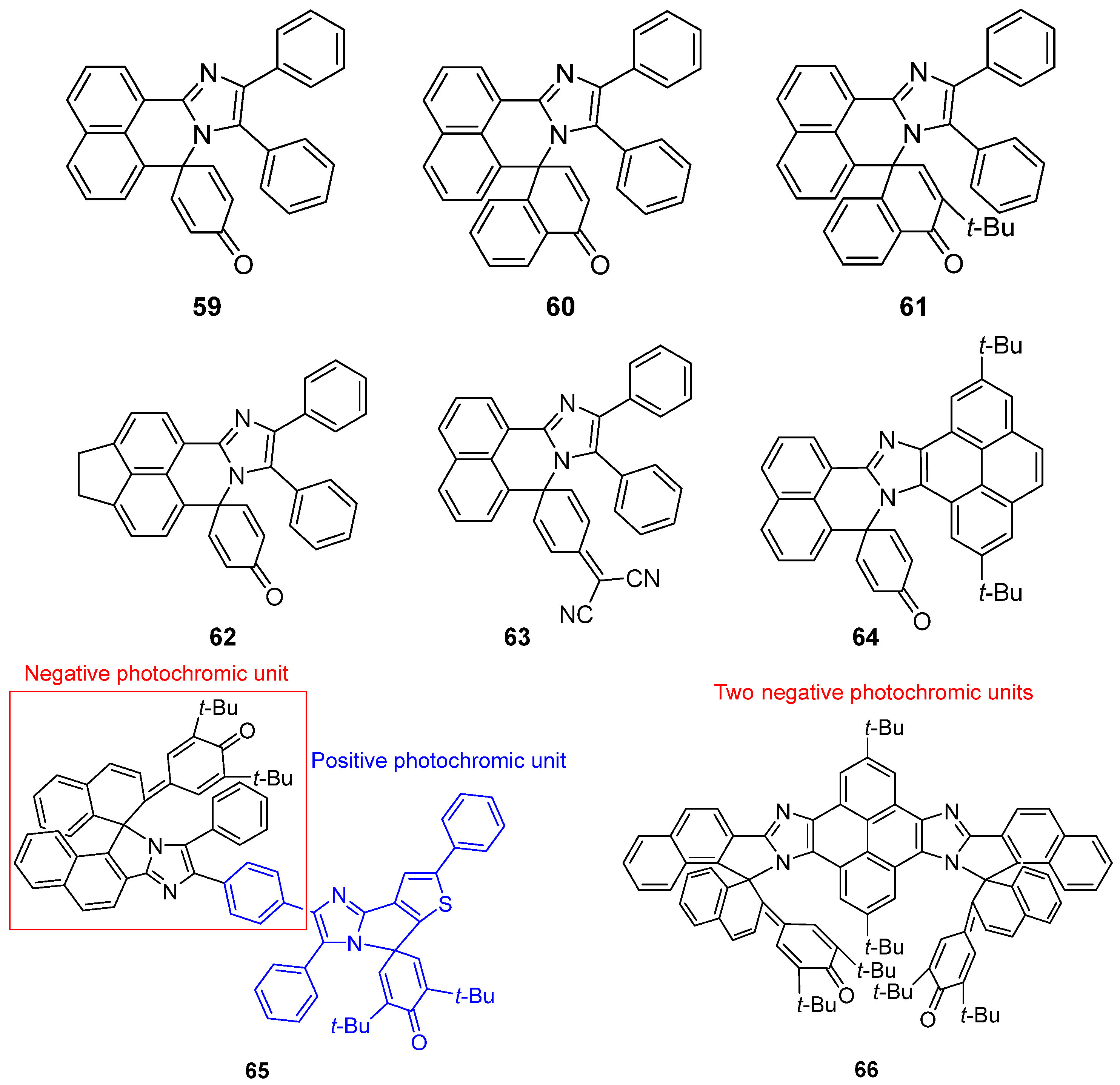
2.6. Imidazole Dimers
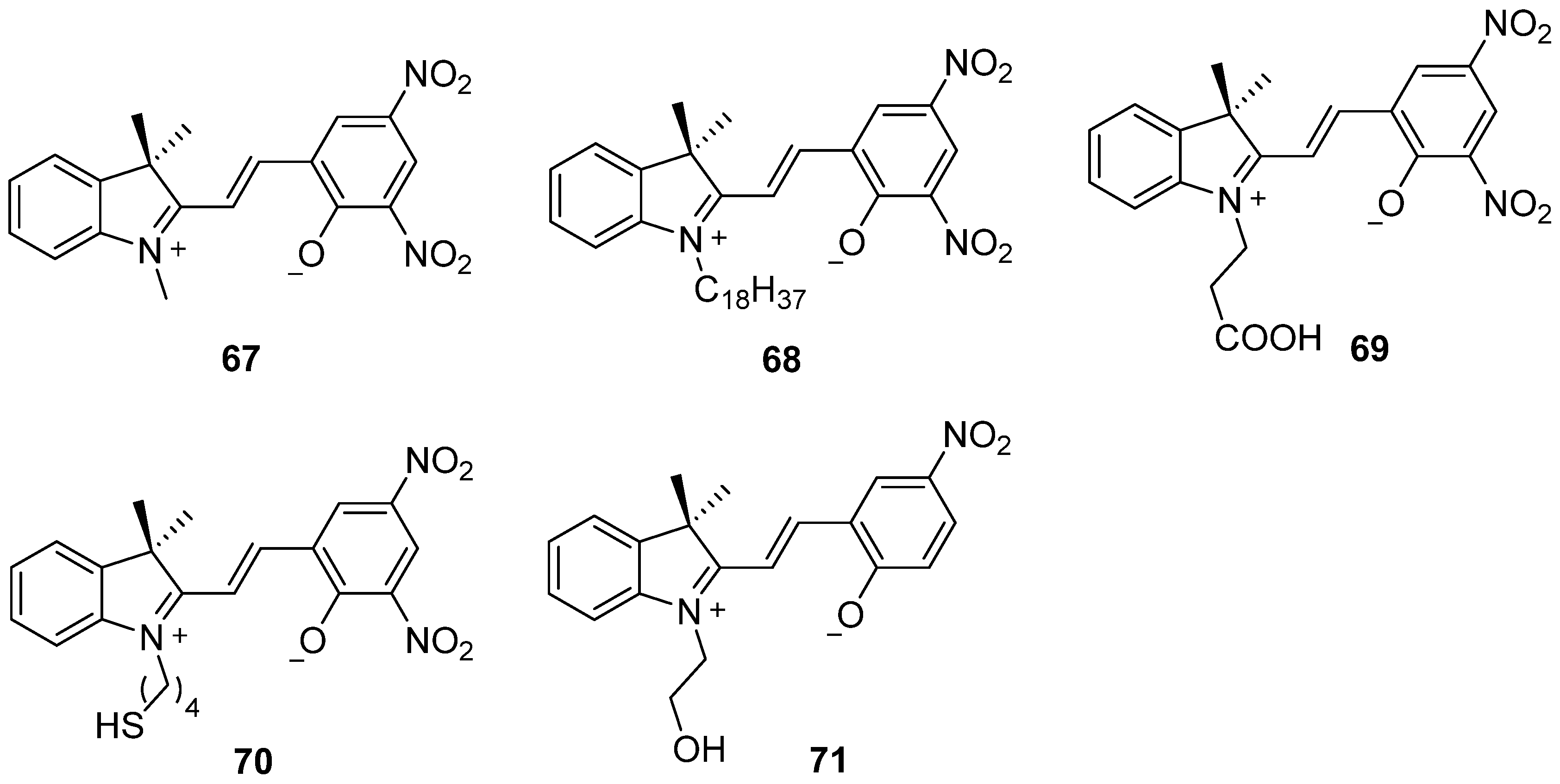
2.7. Spiropyran and Spirooxazine
2.8. Tricyanofuran and Hydroxytricyanopyrrole
2.9. Others
3. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Braslavsky, S.E. Glossary of Terms Used in Photochemistry. Pure Appl. Chem. 2007, 79, 293–465. [Google Scholar] [CrossRef]
- Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Photochromism of Diarylethene Molecules and Crystals: Memories, Switches, and Actuators. Chem. Rev. 2014, 114, 12174–12277. [Google Scholar] [CrossRef] [PubMed]
- Klajn, R. Spiropyran–Based Dynamic Materials. Chem. Soc. Rev. 2014, 43, 148–184. [Google Scholar] [CrossRef]
- Merino, E. Synthesis of Azobenzenes: The Coloured Pieces of Molecular Materials. Chem. Soc. Rev. 2011, 40, 3835–3853. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Maeda, K. Preparation of a New Phototropic Substance. Bull. Chem. Soc. Jpn. 1960, 33, 565–566. [Google Scholar] [CrossRef]
- Yokohama, Y. Fulgides for Memories and Switches. Chem. Rev. 2000, 100, 1717–1740. [Google Scholar] [CrossRef] [PubMed]
- Lerch, M.M.; Szymański, W.; Feringa, B.L. The (Photo)Chemistry of Stenhouse Photoswitches: Guiding Principles and System Design. Chem. Soc. Rev. 2018, 47, 1910–1937. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, J. Photochromic Materials: Preparation, Properties and Applications; Wiley VCH: Weinheim, Germany, 2016. [Google Scholar]
- Barachevsky, V.A. Negative Photochromism in Organic Systems. Rev. J. Chem. 2017, 7, 334–371. [Google Scholar] [CrossRef]
- Aiken, S.; Edgar, R.J.L.; Gabbutt, C.D.; Heron, B.M.; Hobson, P.A. Negatively Photochromic Organic Compounds: Exploring the Dark Side. Dyes Pigm. 2018, 149, 92–121. [Google Scholar] [CrossRef]
- Fukaminato, T.; Tanaka, M.; Kuroki, L.; Irie, M. Invisible Photochromism of Diarylethene Derivatives. Chem. Commun. 2008, 3924–3926. [Google Scholar] [CrossRef]
- Zhang, Z.-X.; Bai, F.-Q.; Li, L.; Zhang, H.-X. Theoretical Investigation on A Series of Novel S,S–dioxide Diarylethenes with Abnormal Photochromic Properties and Design of New Dyads. New J. Chem. 2015, 39, 1634–1642. [Google Scholar] [CrossRef]
- Hou, I.C.-Y.; Berger, F.; Narita, A.; Müllen, K.; Hecht, S. Proton–Gated Ring–Closure of a Negative Photochromic Azulene Based Diarylethene. Angew. Chem. Int. Ed. 2020, 59, 18532–18536. [Google Scholar] [CrossRef] [PubMed]
- Ravat, P.; Šolomek, T.; Häussinger, D.; Blacque, O.; Juríćek, M. Dimethylcethrene: A Chiroptical Diradicaloid Photoswitch. J. Am. Chem. Soc. 2018, 140, 10839–10847. [Google Scholar] [CrossRef] [PubMed]
- Bandarab, H.M.D.; Burdette, S.C. Photoisomerization in Different Classes of Azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef] [PubMed]
- Feringa, B.L.; van Delden, R.A.; Koumura, N.; Geertsema, E.M. Chiroptical Molecular Switches. Chem. Rev. 2000, 100, 1789–1816. [Google Scholar] [CrossRef] [PubMed]
- Beharry, A.A.; Woolley, G.A. Azobenzene Photoswitches for Biomolecules. Chem. Soc. Rev. 2011, 40, 4422–4437. [Google Scholar] [CrossRef]
- Riefolo, F.; Matera, C.; Garrido–Charles, A.; Gomila, A.M.J.; Sortino, R.; Agnetta, L.; Claro, E.; Masgrau, R.; Holzgrabe, U.; Batlle, M.; et al. Optical Control of Cardiac Function with a Photoswitchable Muscarinic Agonist. J. Am. Chem. Soc. 2019, 141, 7628–7636. [Google Scholar] [CrossRef]
- Kienzler, M.A.; Reiner, A.; Trautman, E.; Yoo, S.; Trauner, D.; Isacoff, E.Y. A Red–Shifted, Fast–Relaxing Azobenzene Photoswitch for Visible Light Control of an Ionotropic Glutamate Receptor. J. Am. Chem. Soc. 2013, 135, 17683–17686. [Google Scholar] [CrossRef]
- Samanta, S.; Beharry, A.A.; Sadovski, O.; McCormick, T.M.; Babalhavaeji, A.; Tropepe, V.; Woolley, G.A. Photoswitching Azo Compounds in Vivo with Red Light. J. Am. Chem. Soc. 2013, 135, 9777–9784. [Google Scholar] [CrossRef]
- Dong, L.; Feng, Y.; Wang, L.; Feng, W. Azobenzene–Based Solar Thermal Fuels: Design, Properties, and Applications. Chem. Soc. Rev. 2018, 47, 7339–7368. [Google Scholar] [CrossRef]
- Velema, W.A.; Szymanski, W.; Feringa, B.L. Photopharmacology: Beyond Proof of Principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. [Google Scholar] [CrossRef] [PubMed]
- Hüll, K.; Morstein, J.; Trauner, D. In Vivo Photopharmacology. Chem. Rev. 2018, 118, 10710–10747. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.S.; Neckers, D.C. Photochemistry of Azobenzene–Containing Polymers. Chem. Rev. 1989, 89, 1915–1925. [Google Scholar] [CrossRef]
- Natansohn, A.; Rochon, P. Photoinduced Motions in Azo–Containing Polymers. Chem. Rev. 2002, 102, 4139–4176. [Google Scholar] [CrossRef] [PubMed]
- Borré, E.; Stumbé, J.-F.; Bellemin–Laponnaz, S.; Mauro, M. Light–Powered Self–Healable Metallosupramolecular Soft Actuators. Angew. Chem. Int. Ed. 2016, 55, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Uchida, E.; Sakaki, K.; Nakamura, Y.; Azumi, R.; Hirai, Y.; Akiyama, H.; Yoshida, M.; Norikane, Y. Control of the Orientation and Photoinduced Phase Transitions of Macrocyclic Azobenzene. Chem. Eur. J. 2013, 19, 17391–17397. [Google Scholar] [CrossRef]
- Siewertsen, R.; Neumann, H.; Buchheim–Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. Highly Efficient Reversible Z–E Photoisomerization of a Bridged Azobenzene with Visible Light through Resolved S1(nπ*) Absorption Bands. J. Am. Chem. Soc. 2009, 131, 15594–15595. [Google Scholar] [CrossRef]
- Lentes, P.; Stadler, E.; Röhricht, F.; Brahms, A.; Gröbner, J.; Sönnichsen, F.D.; Gescheidt, G.; Herges, R. Nitrogen Bridged Diazocines: Photochromes Switching within the Near–Infrared Region with High Quantum Yields in Organic Solvents and in Water. J. Am. Chem. Soc. 2019, 141, 13592–13600. [Google Scholar] [CrossRef]
- Coelho, P.J.; Carvalho, L.M.; Fonseca, A.M.C.; Raposo, M.M.M. Photochromic Properties of Thienylpyrrole Azo Dyes in Solution. Tetrahedron Lett. 2006, 47, 3711–3714. [Google Scholar] [CrossRef]
- Coelho, P.J.; Carvalho, L.M.; Moura, J.C.V.P.; Raposo, M.M.M. Novel Photochromic 2,2’–Bithiophene Azo Dyes. Dyes Pigm. 2009, 82, 130–133. [Google Scholar] [CrossRef]
- Nishihara, H. Multi–Mode Molecular Switching Properties and Functions of Azo–Conjugated Metal Complexes. Bull. Chem. Soc. Jpn. 2004, 77, 407–428. [Google Scholar] [CrossRef]
- Bleger, D.; Schwarz, J.; Brouwer, A.M.; Hecht, S. o-Fluoroazobenzenes as Readily Synthesized Photoswitches Offering Nearly Quantitative Two–Way Isomerization with Visible Light. J. Am. Chem. Soc. 2012, 134, 20597–20600. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Babalhavaeji, A.; Dong, M.-X.; Woolley, G.A. Photoswitching of ortho–Substituted Azonium Ions by Red Light in Whole Blood. Angew. Chem. Int. Ed. 2013, 52, 14127–14130. [Google Scholar] [CrossRef] [PubMed]
- Knie, C.; Utecht, M.; Zhao, F.; Kulla, H.; Kovalenko, S.; Brouwer, A.M.; Saalfrank, P.; Hecht, S.; Bléger, D. ortho–Fluoroazobenzenes: Visible Light Switches with Very Long–Lived Z Isomers. Chem. Eur. J. 2014, 20, 16492–16501. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.; Gerecke, M.; Grubert, L.; Kovalenko, S.A.; Hecht, S. Sensitized Two–NIR–Photon Z→E Isomerization of a Visible–Light Addressable Bistable Azobenzene Derivative. Angew. Chem. Int. Ed. 2015, 55, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, K.; Enoki, M.; Katoh, R.; Suzuki, K.; Murase, T.; Imazeki, S. Negative Photochromism of a Blue Cyanine Dye. Chem. Commun. 2020, 56, 15205–15207. [Google Scholar] [CrossRef] [PubMed]
- Senier, A.; Shepheard, F.G. CCVII.—Studies in Phototropy and Thermotropy. Part I. Arylidene– and Naphtylidene–Amines. J. Chem. Soc., Trans. 1909, 95, 1943–1955. [Google Scholar] [CrossRef]
- Senier, A.; Shepheard, F.G.; Clarke, R. CCVII.—Studies in Phototropy and Thermotropy. Part III. Arylideneamines. J. Chem. Soc. Trans. 1912, 101, 1950–1958. [Google Scholar] [CrossRef]
- Cohen, M.D.; Schmidt, G.M.J.; Flavian, S. 338. Topochemistry. Part VI. Experiments on Photochromy and Thermochromy of Crystalline Anils of Salicylaldehydes. J. Chem. Soc. 1964, 2041–2051. [Google Scholar] [CrossRef]
- Ritter, E.; Przybylski, P.; Brzezinski, B.; Bartl, F. Schiff Bases in Biological Systems. Curr. Org. Chem. 2009, 13, 241–249. [Google Scholar] [CrossRef]
- Sliwa, M.; Mouton, N.; Ruckebusch, C.; Aloïse, S.P.; Poizat, O.; Buntinx, G.; Métivier, R.M.; Nakatani, K.; Masuhara, H.; Asahi, T. Comparative Investigation of Ultrafast Photoinduced Processes in Salicylidene–Aminopyridine in Solution and Solid State. J. Phys. Chem. C 2009, 113, 11959–11968. [Google Scholar] [CrossRef]
- Mitra, S.; Tamai, N. Dynamics of Photochromism in Salicylideneaniline: A Femtosecond Spectroscopic Study. Phys. Chem. Chem. Phys. 2003, 5, 4647–4652. [Google Scholar] [CrossRef]
- Hadjoudis, E.K.; Hayon, E. Flash Photolysis of Some Photochromic N–Benzylideneanilines. J. Phys. Chem. 1970, 74, 3184–3188. [Google Scholar] [CrossRef]
- Hadjoudis, E.; Mavridis, I.M. Photochromism and Thermochromism of Schiff Bases in the Solid State: Structural Aspects. Chem. Soc. Rev. 2004, 33, 579–588. [Google Scholar] [CrossRef]
- Safin, D.A.; Bolte, M.; Garcia, Y. Photoreversible Solid State Negative Photochromism of N–(3,5–dichlorosalicylidene)–1–Aminopyrene. CrystEngComm 2014, 16, 5524–5526. [Google Scholar] [CrossRef]
- Jana, P.; Koppayithodi, S.; Mahato, S.; Molla, S.; Bandyopadhyay, S. Stable Diradical on the Dimethyldihydropyrene Scaffold. J. Phys. Chem. Lett. 2023, 14, 7433–7439. [Google Scholar] [CrossRef]
- Sheepwash, M.A.L.; Mitchell, R.H.; Bohne, C. Mechanistic Insights into the Photochromism of trans–10b,10c–Dimethyl–10b,10c–dihydropyrene Derivatives. J. Am. Chem. Soc. 2002, 124, 4693–4700. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.H.; Iyer, V.S.; Mahadevan, R.; Venugopalan, S.; Zhou, P. Annelated Dimethyldihydropyrene. Electrophilic Substitution and Valence Isomerization to Metacyclophanedienes. J. Org. Chem. 1996, 61, 5116–5120. [Google Scholar] [CrossRef]
- Ziani, Z.; Cobo, S.; Loiseau, F.; Jouvenot, D.; Lognon, E.; Boggio–Pasqua, M.; Royal, G. All Visible Light Photoswitch Based on the dimethyldihydropyrene Unit Operate in Aqueous Solutions with High Quantum Yields. JACS Au 2023, 3, 131–142. [Google Scholar] [CrossRef]
- Mitchell, R.H.; Iyer, V.S.; Khalifa, N.; Mahadevan, R.; Venugopalan, S.; Weerawarna, S.A.; Zhou, P. An Experimental Estimation of Aromaticity Relative to That of Benzene. The Synthesis and NMR Properties of a Series of Highly Annelated Dimethyldihydropyrenes: Bridged Benzannulenes. J. Am. Chem. Soc. 1995, 117, 1514–1532. [Google Scholar] [CrossRef]
- Williams, R.V.; Edwards, W.D.; Vij, A.; Tolbert, R.W.; Mitchell, R.H. Theoretical Study and X–ray Structure Determination of Dimethyldihydropyrene. J. Org. Chem. 1998, 63, 3125–3127. [Google Scholar] [CrossRef]
- Mitchell, R.H.; Brkic, Z.; Sauro, V.A.; Berg, D.J. A Photochromic, Electrochromic, Thermochromic Ru Complexed Benzannulene: An Organometallic Example of the Dimethyldihydropyrene–Metacyclophanediene Valence Isomerization. J. Am. Chem. Soc. 2003, 125, 7581–7585. [Google Scholar] [CrossRef] [PubMed]
- Bakkar, A.; Cobo, S.; Lafolet, F.; Roldan, D.; Jacquet, M.; Bucher, C.; Royal, G.; Saint–Aman, E. Dimethyldihydropyrene–Cyclophanediene Photochromic Couple Functionalized with Terpyridyl Metal Complexes as Multi–Addressable Redox– and Photo–Switches. Dalton Trans. 2016, 45, 13700–13708. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, M.; Uriarte, L.M.; Lafolet, F.; Boggio–Pasqua, M.; Sliwa, M.; Loiseau, F.; Saint–Aman, E.; Cobo, S.; Royal, G. All Visible Light Switch Based on the Dimethyldihydropyrene Photochromic Core. J. Phys. Chem. Lett. 2020, 11, 2682–2688. [Google Scholar] [CrossRef] [PubMed]
- Courtois, J.; Wang, C.; Tian, Q.; Wang, B.; Feng, W. Nanostructured photoswitchable colloidal particles made of coordination polymer containing dimethyldihydropyrene units. Colloids Surf. A Physicochem. Eng. Asp. 2023, 662, 131032. [Google Scholar] [CrossRef]
- Klaue, K.; Garmshausen, Y.; Hecht, S. Taking Photochromism Beyond Visible: Direct One–Photon NIR Photoswitches Operating in the Biological Window. Angew. Chem. Int. Ed. 2018, 57, 1414–1417. [Google Scholar] [CrossRef]
- Klaue, K.; Han, W.; Liesfeld, P.; Berger, F.; Garmshausen, Y.; Hecht, S. Donor–Acceptor Dihydropyrenes Switchable with Near–Infrared Light. J. Am. Chem. Soc. 2020, 142, 11857–11864. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Kasuya, K.; Oketani, R.; Hisaki, I. Construction of Hydrogen–bonded Crystalline Frameworks Using Tetrakis(carboxyphenyl)dimethyldihydropyrene Derivative. Chem. Lett. 2023, 52, 542–545. [Google Scholar] [CrossRef]
- Helmy, S.; Leibfarth, F.A.; Oh, S.; Poelma, J.E.; Hawker, C.J.; Read de Alaniz, J. Photoswitching Using Visible Light: A New Class of Organic Photochromic Molecules. J. Am. Chem. Soc. 2014, 136, 8169–8172. [Google Scholar] [CrossRef]
- Helmy, S.; Oh, S.; Leibfarth, F.A.; Hawker, C.J.; Read de Alaniz, J. Design and Synthesis of Donor–Acceptor Stenhouse Adducts: A Visible Light Photoswitch Derived from Furfural. J. Org. Chem. 2014, 79, 11316–11329. [Google Scholar] [CrossRef]
- Lerch, M.M.; Donato, M.D.; Laurent, A.D.; Medved, M.; Iagatti, A.; Bussotti, L.; Lapini, A.; Buma, W.J.; Foggi, P.; Szymański, W.; et al. Solvent Effects on the Actinic Step of Donor–Acceptor Stenhouse Adduct Photoswitching. Angew. Chem. Int. Ed. 2018, 57, 8063–8068. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.; Wiedbrauk, S.; Gräfe, D.; Walden, S.L.; Blinco, J.P.; Barner–Kowollik, C. It’s a Trap: Thiol–Michael Chemistry on a DASA Photoswitch. Chem. Eur. J. 2020, 26, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Mallo, N.; Brown, P.T.; Iranmanesh, H.; MacDonald, T.S.C.; Teusner, M.J.; Harper, J.B.; Ball, G.E.; Beves, J.E. Photochromic Switching Behaviour of Donor–Acceptor Stenhouse Adducts in Organic Solvents. Chem. Commun. 2016, 52, 13576–13579. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, J.R.; Page, Z.A.; Clark, D.K.; Stricker, F.; Dolinski, D.N.; Hawker, C.J.; Read de Alaniz, J. Controlling Dark Equilibria and Enhancing DASA Photoswitching Properties Through Carbon Acid Design. J. Am. Chem. Soc. 2018, 140, 10425–10429. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, J.R.; Poelma, S.O.; Treat, N.; Page, Z.A.; Dolinski, N.; Diaz, Y.J.; Tomlinson, W.; Clark, D.K.; Hooper, J.P.; Hawker, C.J.; et al. Tunable Visible and Near Infrared Photoswitches. J. Am. Chem. Soc. 2016, 138, 13960–13966. [Google Scholar] [CrossRef]
- Castagna, R.; Maleeva, G.; Pirovano, D.; Matera, C.; Gorostiza, P. Donor–Acceptor Stenhouse Adduct Displaying Reversible Photoswitching in Water and Neuronal Activity. J. Am. Chem. Soc. 2022, 144, 15595–15602. [Google Scholar] [CrossRef]
- Mao, L.; Wang, Z.; Duan, Y.; He, C.; Deng, X.; Zheng, Y.; Wang, D. Designing of Rewritable paper by Hydrochromic Donor–Acceptor Stenhouse Adducts. ACS Nano 2021, 15, 10384–10392. [Google Scholar] [CrossRef]
- Dubuis, S.; Dellai, A.; Courdurié, C.; Owona, J.; Kalafatis, A.; Vellutini, L.; Genin, E.; Rodriguez, V.; Castet, F. Nonlinear Optical Responses of Photoswitchable Donor–Acceptor Stenhouse Adducts. J. Am. Chem. 2023, 145, 10861–10871. [Google Scholar] [CrossRef]
- Lui, B.F.; Tierce, N.T.; Tong, F.; Sroda, M.M.; Lu, H.; Read de Alaniz, J.; Bardeen, C.J. Unusual Concentration Dependence of the Photoisomerization Reaction in Donor–Acceptor Stenhouse Adducts. Photochem. Photobiol. Sci. 2019, 18, 1587–1595. [Google Scholar] [CrossRef]
- McDonough, R.; Rudgley, N.; Majewski, O.; Perkins, M.V.; Evans, R.A.; Lewis, D.A. Fatigue of Donor–Acceptor Stenhouse Adducts in Polymer Matrices and Solution. ChemPhotoChem 2023, 7, e202200247. [Google Scholar] [CrossRef]
- Chen, Q.; Diaz, Y.J.; Hawker, M.C.; Martinez, M.R.; Page, Z.A.; Zhang, S.X.-A.; Hawker, C.J.; Read de Alaniz, J. Stable Activated Furan and Donor–Acceptor Stenhouse Adduct Polymer Conjugates as Chemical and Thermal Sensors. Macromolecules 2019, 52, 4370–4375. [Google Scholar] [CrossRef]
- Yap, J.E.; Zhang, L.; Lovegrove, J.T.; Beves, J.E.; Stenzel, M.H. Visible Light–Responsive Drug Delivery Nanoparticle via Donor–Acceptor Stenhouse Adducts. Macromol. Rapid Commun. 2020, 41, 2000236. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Li, W.; Fan, H.; Xiang, J. A Red–Light–Responsive DASA–Polymer with High Water Stability for Controlled Release. Polymers 2023, 15, 2489. [Google Scholar] [CrossRef]
- Clerc, M.; Sandlass, S.; Rifaie–Graham, O.; Peterson, J.A.; Bruns, N.; Read de Alaniz, J.; Boesel, L.F. Visible light–responsive materials: The (photo)chemistry and applications of donor–acceptor Stenhouse adducts in polymer science. Chem. Soc. Rev. 2023, 52, 8245–8294. [Google Scholar] [CrossRef] [PubMed]
- Hatano, S.; Horino, T.; Tokita, A.; Oshima, T.; Abe, J. Unusual Negative Photochromism via a Short–Lived Imidazolyl Radical of 1,1′–Binaphthyl–Bridged Imidazole Dimer. J. Am. Chem. Soc. 2013, 135, 3164–3172. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, K.; Miyashita, N.; Arai, K.; Abe, J. Turn–On Mode Fluorescence Switch by Using Negative Photochromic Imidazole Dimer. J. Am. Chem. Soc. 2019, 141, 5650–5654. [Google Scholar] [CrossRef] [PubMed]
- Kometani, A.; Inagaki, Y.; Mutoh, K.; Abe, J. Red or NIR Light Operating Negative Photochromism of Binaphthyl–Bridged Imidazole Dimer. J. Am. Chem. Soc. 2020, 142, 7995–8005. [Google Scholar] [CrossRef]
- Ito, H.; Mutoh, K.; Abe, J. Bridged–Imidazole Dimer Exhibiting Three–State Negative Photochromism with a Single Photochromic Unit. J. Am. Chem. Soc. 2023, 145, 6498–6506. [Google Scholar] [CrossRef]
- Moriyama, N.; Abe, J. Negative Photochromic 3-Phenylperylenyl–Bridged Imidazole Dimer Offering Quantitative and Selective Bidirectional Photoisomerization with Visible and Near–Infrared Light. J. Am. Chem. Soc. 2023, 145, 3318–3322. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kobayashi, Y.; Abe, J. Fast Negative Photochromism of 1,1′–Binaphthyl–Bridged Phenoxyl–Imidazolyl Radical Complex. J. Am. Chem. Soc. 2016, 138, 906–913. [Google Scholar] [CrossRef]
- Mutoh, K.; Kobayashi, Y.; Hirao, Y.; Kubo, T.; Abe, J. Stealth Fast Photoswitching of Negative Photochromic Naphthalene–Bridged Phenoxyl–Imidazolyl Radical Complexes. Chem. Commun. 2016, 52, 6797–6800. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Mutoh, K.; Abe, J. Enhancement of Negative Photochromic Properties of Naphthalene–Bridged Phenoxyl–Imidazolyl Radical Complex. ChemPhysChem 2020, 21, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Yonekawa, I.; Mutoh, K.; Kobayashi, Y.; Abe, J. Intensity–Dependent Photoresponse of Biphotochromic Molecule Composed of a Negative and a Positive Photochromic Unit. J. Am. Chem. Soc. 2018, 140, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Yonekawa, I.; Mutoh, K.; Abe, J. Visible Light Intensity Dependent Negative Photochromism of a Binaphthyl–Bridged Phenoxyl–Imidazolyl Radical Complex. Chem. Commun. 2019, 55, 1221–1224. [Google Scholar] [CrossRef] [PubMed]
- Koelsch, C.F.; Workman, W.R. Some Thermochromic Spirans. J. Am. Chem. Soc. 1952, 74, 6288–6289. [Google Scholar] [CrossRef]
- Silvia, T.R.; Ana, V.S.L.; González, E.A.S. Novel Syntheses of Spiropyran Photochromatic Compounds Using Ultrasound. Synth. Commun. 1995, 25, 105–110. [Google Scholar] [CrossRef]
- Buback, J.; Kullmann, M.; Langhojer, F.; Nuernberger, P.; Schmidt, R.; Würthner, F.; Brixner, T. Ultrafast Bidirectional Photoswitching of a Spiropyran. J. Am. Chem. Soc. 2010, 132, 16510–16519. [Google Scholar] [CrossRef]
- Hobley, J.; Malatesta, V.; Millini, R.; Montanari, L.; Parker, W.O.N. Proton Exchange and Isomerisation Reactions of Photochromic and Reverse Photochromic Spiro–Pyrans and Their Merocyanine Forms. Phys. Chem. Chem. Phys. 1999, 1, 3259–3267. [Google Scholar] [CrossRef]
- Takeda, J.; Ikeda, Y.; Mihara, D.; Kurita, S.; Sawada, A.; Yokoyama, Y. Transient Absorption Spectroscopy for Photochemical Reactions of a Negative Photochromic Spiropyran. Mol. Cryst. Liq. Cryst. 2000, 345, 191–196. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Hara, W.; Inoue, T.; Ubukata, T.; Sakomura, M.; Tukada, H. Negative Photochromism of a Spiropyran in a Langmuir–Blodgett Film. Chem. Lett. 2005, 34, 1622–1623. [Google Scholar] [CrossRef]
- Hobley, J.; Pfeifer–Fukumura, U.; Bletz, M.; Asahi, T.; Masuhara, H.; Fukumura, H. Ultrafast Photo–Dynamics of a Reversible Photochromic Spiropyran. J. Phys. Chem. A 2002, 106, 2265–2270. [Google Scholar] [CrossRef]
- Blonder, R.; Levi, S.; Tao, G.; Ben–Dov, I.; Willner, I. Development of Amperometric and Microgravimetric Immunosensors and Reversible Antigen Monolayer Electrodes. J. Am. Chem. Soc. 1997, 119, 10467–10478. [Google Scholar] [CrossRef]
- Zhang, H.; Fan, J.; Wang, W.; Yu, D. Positive and negative photochromic and fluorescent color–changing cotton fabrics for information switching and encryption. Ind. Crops Prod. 2023, 205, 117524. [Google Scholar] [CrossRef]
- Tautges, B.; Or, V.; Garcia, J.; Shaw, J.T.; Louie, A.Y. Preparation of a Conjugation–Ready Thiol Responsive Molecular Switch. Tetrahedron Lett. 2015, 56, 6569–6573. [Google Scholar] [CrossRef] [PubMed]
- Minami, M.; Taguchi, B. Effects of Substituents on the Indoline Ring on the Negative Photochromic Properties of Spirobenzopyran Derivatives. Chem. Lett. 1996, 25, 429–430. [Google Scholar] [CrossRef]
- Nikolaeva, O.G.; Karlutova, O.Y.; Gaeva, E.B.; Dubonosov, A.D.; Metelitsa, A.V.; Bren, V.A.; Minkin, V.I. Synthesis and Photochromic Properties of Bis–Spirocyclic Compounds Based on 1.3–Dihydrocy–6–oxo–6H–benzo[c]chromene–2,4–dicarbaldehyde. Russ. J. Gen. Chem. 2021, 91, 626–630. [Google Scholar] [CrossRef]
- Keum, S.-R.; Hur, M.-S.; Kazmaier, P.M.; Buncel, E. Thermo– and Photochromic Dyes: Indolino–Benzospiropyrans. Part 1. UV–VIS Spectroscopic Studies of 1,3,3–Spiro(2H–1–Benzopyran–2,2′–Indolines) and the Open–Chain Merocyanine Forms; Solvatochromism and Medium Effects on Spiro Ring Formation. Can. J. Chem. 1991, 69, 1940–1947. [Google Scholar] [CrossRef]
- Görner, H. Photochromism of Nitrospiropyrans: Effects of Structure, Solvent and Temperature. Phys. Chem. Chem. Phys. 2001, 3, 416–423. [Google Scholar] [CrossRef]
- Shimizu, I.; Kokado, H.; Inoue, E. Photoreversible Photographic Systems. VI. Reverse Photochromism of 1,3,3–Trimethylspiro[indoline–2,2′–Benzopyran]–8′–Carboxylic Acid. Bull. Chem. Soc. Jpn. 1969, 42, 1730–1734. [Google Scholar] [CrossRef]
- Inoue, E.; Kokado, H.; Shimizu, I.; Kobayashi, H.; Takahashi, Y. Photo–decoloration Process of the Reverse Photochromic Spirans. Bull. Chem. Soc. Jpn. 1972, 45, 1951–1956. [Google Scholar] [CrossRef]
- Sunamoto, J.; Iwamoto, K.; Akutagawa, M.; Nagase, M.; Kondo, H. Rate Control by Restricting Mobility of Substrate in Specific Reaction Field. Negative Photochromism of Water–Soluble Spiropyran in AOT Reversed Micelle. J. Am. Chem. Soc. 1982, 104, 4904–4907. [Google Scholar] [CrossRef]
- Metelitsa, A.V.; Chernyshev, A.V.; Voloshin, N.A.; Solov’eva, E.V.; Dorogan, I.V. Chromogenic properties of hererocyclic compounds: Barochromic effect of indoline spiropyrans in the gas phase. J. Photochem. Photobiol. A Chem. 2022, 430, 113982. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Chen, J.; Tian, Y.; Xiang, C.; Liu, W.; Zhou, Z.; Cui, J.; Chen, X. Visible–Light–Driven Photoswitchable Fluorescent Polymers for Photorewritable Pattern, Anti–Counterfeiting, and Information Encryption. Adv. Funct. Mater. 2023, 33, 2303765. [Google Scholar] [CrossRef]
- Taylor, L.D.; Nicholso, J.; Davis, R.B. Photochromic Chelating Agents. Tetrahedron Lett. 1967, 8, 1585–1588. [Google Scholar] [CrossRef]
- Kawanishi, Y.; Seki, K.; Tamaki, T.; Sakuragi, M.; Suzuki, Y. Tuning Reverse Ring Closure in the Photochromic and Thermochromic Transformation of 1′,3′,3′–Trimethyl–6–Nitrospiro[2H–1–Benzopyran–2,2′–Indoline] Analogues by Ionic Moieties. J. Photochem. Photobiol. A Chem. 1997, 109, 237–242. [Google Scholar] [CrossRef]
- Tanaka, M.; Nakamura, M.; Salhin, M.A.A.; Ikeda, T.; Kamada, K.; Ando, H.; Shibutani, Y.; Kimura, K. Synthesis and Photochromism of Spirobenzopyran Derivatives Bearing an Oxymethylcrown Ether Moiety: Metal Ion–Induced Switching between Positive and Negative Photochromisms. J. Org. Chem. 2001, 66, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, M.I.; Coudret, C.; Pimienta, V.; Micheau, J.C.; Delbaere, S.; Vermeersch, G.; Metelitsa, A.V.; Voloshin, N.; Minkin, V.I. Quantitative Investigations of Cation Complexation of Photochromic 8–Benzothiazole–Substituted Benzopyran: Towards Metal–Ion Sensors. Photochem. Photobiol. Sci. 2010, 9, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Chernyshev, A.V.; Voloshin, N.A.; Raskita, I.M.; Metelitsa, A.V.; Minkin, V.I. Photo– and Ionochromism of 5’–(4,5–Diphenyl–1,3–Oxazol–2–yl) Substituted Spiro[Indoline–Naphthopyrans]. J. Photochem. Photobiol. A Chem. 2006, 184, 289–297. [Google Scholar] [CrossRef]
- Isamu, S.; Hiroshi, K.; Eiichi, I. Photoreversible Photographic Systems. V. Reverse Photochromism of (Photospiran/Acid) System in Acetone. Bull. Chem. Soc. Jpn. 1969, 42, 1726–1729. [Google Scholar]
- Yamaguchi, T.; Leelaphattharaphan, N.N.; Shin, H.; Ogawa, M. Acceleration of Photochromism and Negative Photochromism by the Interactions with Mesoporous Silicas. Photochem. Photobiol. Sci. 2019, 18, 1742–1749. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Imwiset, K.J.; Ogawa, M. Efficient Negative Photochromism by the Photoinduced Migration of Photochromic Merocyanine/Spiropyran in the Solid State. Langmuir 2021, 37, 3702–3708. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Zheng, W.; Zhao, Y.; Li, C.-H. Visible light responsive spiropyran derivatives based on dynamic coordination bonds. Chin. Chem. Lett. 2023, 34, 107457. [Google Scholar] [CrossRef]
- Peng, P.; Strohecker, D.; Liao, Y. Negative Photochromism of a TCF Chromophore. Chem. Commun. 2011, 47, 8575–8577. [Google Scholar] [CrossRef] [PubMed]
- Johns, V.K.; Peng, P.; DeJesus, J.; Wang, Z.; Liao, Y. Visible–Light–Responsive Reversible Photoacid Based on a Metastable Carbanion. Chem. Eur. J. 2014, 20, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.Y.; Lee, J.H.; Park, S.B.; Choi, S. –H.; Lee, J.–S.; Hong, J.–I. Development of tricyanofuran–based activity probes for sulfatase assay in live cells. Dyes Pigm. 2022, 205, 110517. [Google Scholar] [CrossRef]
- Liang, M.; Mu, X.; Li, Y.; Tan, Y.; Hao, X.; Tang, Y.; Wang, Z.; Feng, W.; Lu, Y.; Zhou, X. Heptamethine Cyanine–Based Nanotheranostics with Catalase–Like Activity for Synergistic Phototherapy of Cancer. Adv. Funct. Mater. 2023, 33, 2302112. [Google Scholar] [CrossRef]
- Li, Z.; Zeng, Z.; Wu, S.; Liu, J.; Luo, T.; Liao, J.; Yang, R.; Liu, F. Synthesis and characterization of Y–shaped optical nonlinear chromophores with strong acceptors. New J. Chem. 2023, 47, 9203–9211. [Google Scholar] [CrossRef]
- Liu, T.; Huo, F.; Ge, C.; Li, Y.; He, J.; Zheng, H.; He, Q.; Zhao, Y.; Chen, Z.; Bo, S. Systematic Study on Nonlinear Optical Chromophores with Improved Electro–Optic Activity by Introducing 3,5–Bis(trifluoromethyl)benzene Derivative Isolation Groups into the Bridge. Molecules 2023, 28, 488. [Google Scholar] [CrossRef]
- Belikov, M.Y.; Ievlev, M.Y.; Fedoseev, S.V.; Ershov, O.V. Tuning the Photochromic Properties of Chromophores Containing a Nitrile–Rich Acceptor: A Novel Branch in the Investigation of Negative Photochromes. New J. Chem. 2019, 43, 8414–8417. [Google Scholar] [CrossRef]
- Belikov, M.Y.; Ievlev, M.Y.; Fedoseev, S.V.; Ershov, O.V. Novel Group of Negative Photochromes Containing a Nitrile–Rich Acceptor: Synthesis and Photochromic Properties. Res. Chem. Intermed. 2019, 45, 4625–4636. [Google Scholar] [CrossRef]
- Belikov, M.Y.; Ievlev, M.Y.; Fedoseev, S.V.; Ershov, O.V. Synthesis and Fine–Tuning of Thermal Stability of the Negative Nitrile–Rich Photochromes of Hydroxytricyanopyrrole (HTCP) Series. Res. Chem. Intermed. 2020, 46, 3477–3490. [Google Scholar] [CrossRef]
- Belikov, M.Y.; Fedoseev, S.V.; Ievlev, M.Y.; Ershov, O.V.; Lipin, K.V.; Tafeenko, V.A. Direct synthesis of variously substituted negative photochromes of hydroxytricyanopyrrole (HTCP) series. Synth. Commun. 2020, 50, 2413–2421. [Google Scholar] [CrossRef]
- Fedoseev, S.V.; Belikov, M.Y. Synthesis and Tunable Fluorescence of N,N–Disubstitute ortho–Aminostyryl D–π–A Chromophores Based on a Hydroxytricyanopyrrole (HTCP) Acceptor. ChemistrySelect 2023, 8, e202300771. [Google Scholar] [CrossRef]
- Fedoseev, S.V.; Belikov, M.Y.; Ievlev, M.Y. Synthesis and optical properties of the first representatives of N, N–disubstituted aminostyryl D–π–A chromophores with tunable hydroxytricyanopyrrole (HTCP) acceptor. Dyes Pigm. 2022, 204, 110455. [Google Scholar] [CrossRef]
- Fedoseev, S.V.; Belikov, M.Y.; Ielev, M.Y.; Ershov, O.V.; Tafeenko, V.A. Tuning solid–state fluorescence of a novel group D–π–A chromophores with a reactive hydroxytricyanopyrrole (HTCP) acceptor. Dyes Pigm. 2019, 165, 451–457. [Google Scholar] [CrossRef]
- Zeng, C.; Chen, Z.; Yang, M.; Lv, J.; Li, H.; Gao, J.; Yuan, Z. A Hydroxytricyanopyrrole–Based Fluorescent Probe for Sensitive and Selective Detection of Hypochlorous Acid. Molecules 2022, 27, 7237. [Google Scholar] [CrossRef]
- Petermayer, C.; Dube, H. Indigoid Photoswitches: Visible Light Responsive Molecular Tools. Acc. Chem. Res. 2018, 51, 1153–1163. [Google Scholar] [CrossRef]
- Qian, H.; Pramanik, S.; Aprahamian, I. Photochromic Hydrazone Switches with Extremely Long Thermal Half–Lives. J. Am. Chem. Soc. 2017, 139, 9140–9143. [Google Scholar] [CrossRef]
- Bouas–Laurent, H.; Castellan, A.; Desvergne, J.-P.; Lapouyade, R. Photodimerization of Anthracenes in Fluid Solutions: (Part 2) Mechanistic Aspects of the Photocycloaddition and of the Photochemical and Thermal Cleavage. Chem. Soc. Rev. 2001, 30, 248–263. [Google Scholar] [CrossRef]
- Van Dijken, D.J.; Kovaříček, P.; Ihring, S.P.; Hecht, S. Acylhydrazones as Widely Tuable Photoswitches. J. Am. Chem. Soc. 2015, 137, 14982–14991. [Google Scholar] [CrossRef]
- Koibuchi, R.; Omasa, K.; Yoshikawa, I.; Houjou, H. Photoinduced Crystal–Liquid Transition of Acylhydrazone–Based Photoswitching Molecules. J. Phys. Chem. Lett. 2023, 14, 8320–8326. [Google Scholar] [CrossRef] [PubMed]


























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Mou, B.; Yamada, M.; Li, W.; Nakashima, T.; Kawai, T. From Visible to Near–Infrared Light–Triggered Photochromism: Negative Photochromism. Molecules 2024, 29, 155. https://doi.org/10.3390/molecules29010155
Li R, Mou B, Yamada M, Li W, Nakashima T, Kawai T. From Visible to Near–Infrared Light–Triggered Photochromism: Negative Photochromism. Molecules. 2024; 29(1):155. https://doi.org/10.3390/molecules29010155
Chicago/Turabian StyleLi, Ruiji, Bingzhao Mou, Mihoko Yamada, Wei Li, Takuya Nakashima, and Tsuyoshi Kawai. 2024. "From Visible to Near–Infrared Light–Triggered Photochromism: Negative Photochromism" Molecules 29, no. 1: 155. https://doi.org/10.3390/molecules29010155