
Design and Synthesis of Novel Indole Ethylamine Derivatives as a Lipid Metabolism Regulator Targeting PPARα/CPT1 in AML12 Cells
 , , ,
, , ,
Abstract
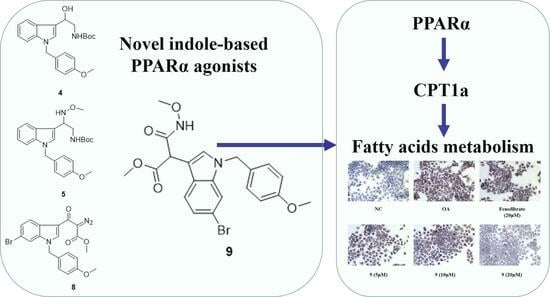
:
1. Introduction
2. Results
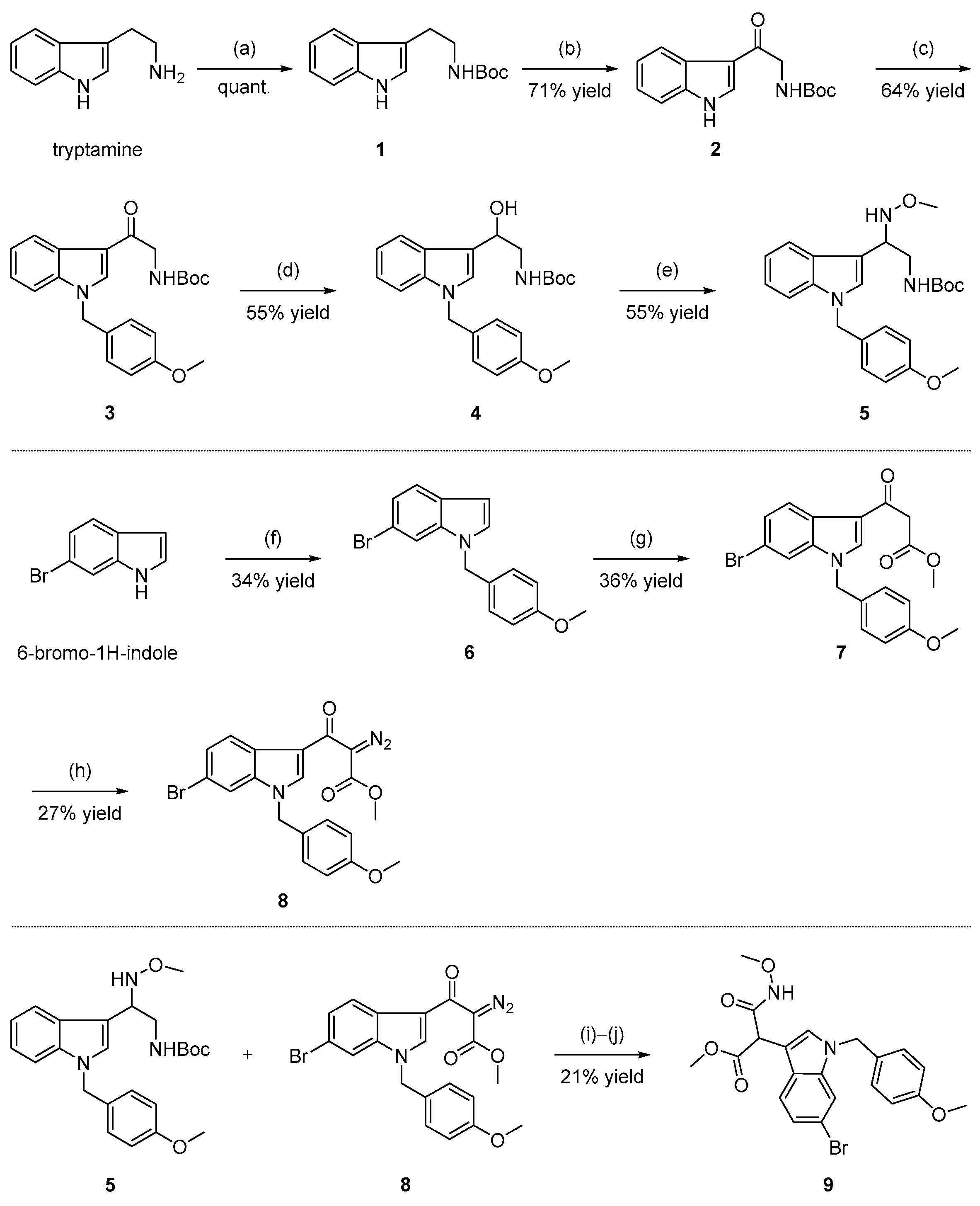
2.1. Chemistry
2.2. Biological Evaluations
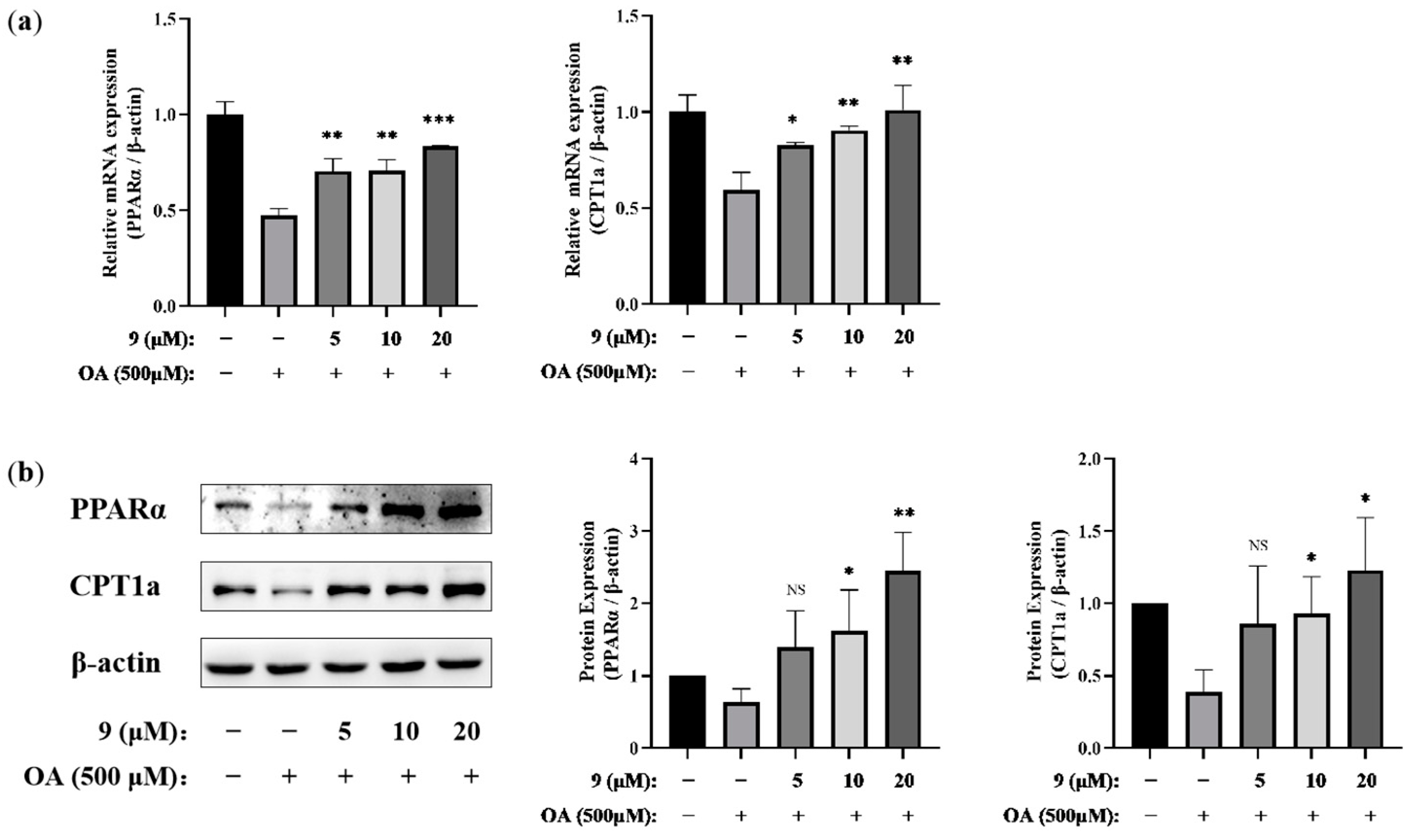
2.2.1. PPARα Agonist Activity
2.2.2. Lipid-Modifying Activity
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Synthesis of tert-butyl (2-(1H-indol-3-yl) ethyl) carbamate (1)
3.1.3. Synthesis of tert-butyl (2-(1H-indol-3-yl)-2-oxoethyl) carbamate (2)
3.1.4. Synthesis of tert-butyl (2-(1-(4-methoxybenzyl)-1H-indol-3-yl)-2-oxoethyl) carbamate (3)
3.1.5. Synthesis of tert-butyl (2-hydroxy-2-(1-(4-methoxybenzyl)-1H-indol-3-yl)ethyl)carbamate (4)
3.1.6. Synthesis of tert-butyl (2-(methoxyamino)-2-(1-(4-methoxybenzyl)-1H-indol-3-yl)ethyl) carbamate (5)
3.1.7. Synthesis of 6-bromo-1-(4-methoxybenzyl)-1H-indole (6)
3.1.8. Synthesis of methyl 3-(6-bromo-1-(4-methoxybenzyl)-1H-indol-3-yl)-3-oxopropanoate (7)
3.1.9. Synthesis of methyl 3-(6-bromo-1-(4-methoxybenzyl)-1H-indol-3-yl)-2-diazo-3-oxopropanoate (8)
3.1.10. Synthesis of methyl-2-(6-bromo-1-(4-methoxybenzyl)-1H-indol-3-yl)-3-(methoxyamino)-3-oxopropanoate (9)
3.2. Biological Evaluations
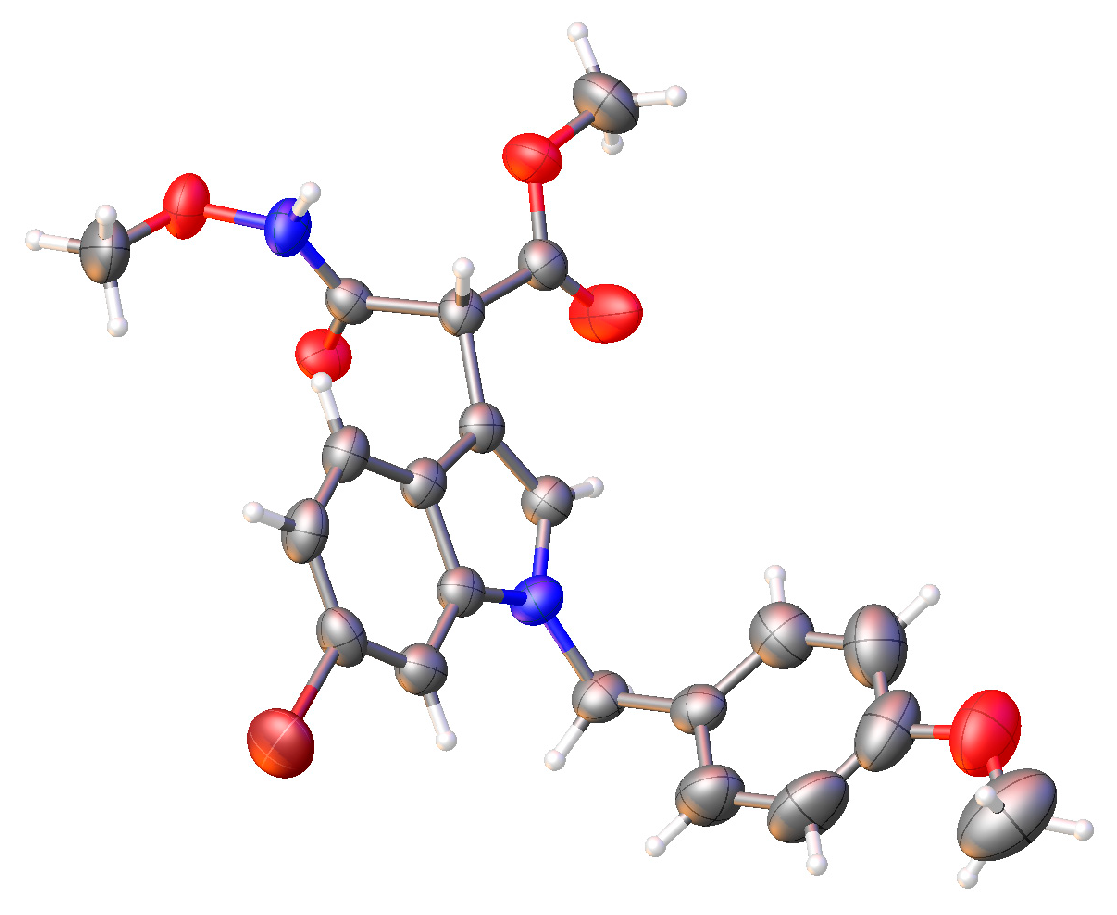
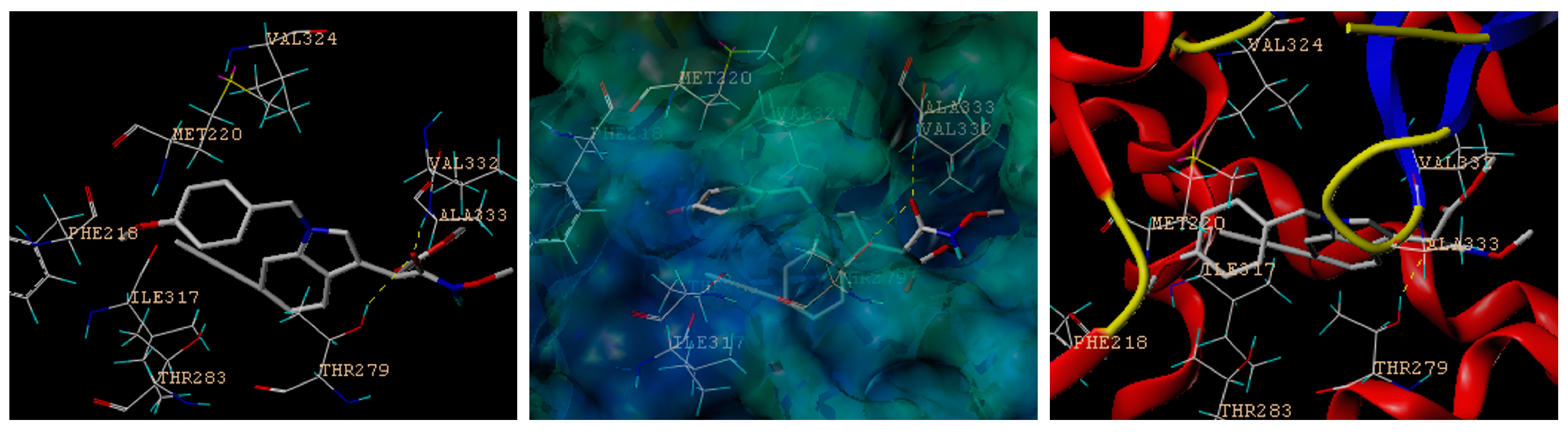
3.2.1. Molecular Modeling
3.2.2. Cell Cultivation and Treatment
3.2.3. Enzyme-Linked Immunosorbent Assay
3.2.4. Cell Viability Assay
3.2.5. Triglyceride Assay
3.2.6. Oil Red O Staining
3.2.7. BODIPY Staining
3.2.8. Quantitative Real-Time PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Primer Target | Sequence 5′ to 3′ |
|---|---|---|
| Mus | PPARα-F | AGAGCCCCATCTGTCCTCTC |
| PPARα-R | ACTGGTAGTCTGCAAAACCAAA | |
| Mus | ATGL-F | CAACGCCACTCACATCTACGG |
| ATGL-R | GGACACCTCAATAATGTTGGCAC | |
| Mus | HSL-F | GCTCATCTCCTATGACCTACGG |
| HSL-R | TCCGTGGATGTGAACAACCAGG | |
| Mus | CPT1a-F | CTATGCGCTACTCGCTGAAGG |
| CPT1a-R | GGCTTTCGACCCGAGAAGA | |
| Mus | β-actin-F | TTCGTTGCCGGTCCACACCC |
| β-actin-R | GCTTTGCACATGCCGGAGCC |
3.2.9. Western Blotting
3.2.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chiriac, S.; Stanciu, C.; Girleanu, I.; Cojocariu, C.; Sfarti, C.; Singeap, A.M.; Cuciureanu, T.; Huiban, L.; Muzica, C.M.; Zenovia, S.; et al. Nonalcoholic Fatty Liver Disease and Cardiovascular Diseases: The Heart of the Matter. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6696857. [Google Scholar] [CrossRef]
- Castera, L.; Friedrich-Rust, M.; Loomba, R. Noninvasive Assessment of Liver Disease in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 156, 1264–1281.e4. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef]
- Wong, V.W.; Chitturi, S.; Wong, G.L.; Yu, J.; Chan, H.L.; Farrell, G.C. Pathogenesis and novel treatment options for non-alcoholic steatohepatitis. Lancet Gastroenterol. Hepatol. 2016, 1, 56–67. [Google Scholar] [CrossRef]
- McCullough, A.J. Pathophysiology of nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2006, 40 (Suppl. S1), S17–S29. [Google Scholar]
- Vergani, L. Fatty Acids and Effects on In Vitro and In Vivo Models of Liver Steatosis. Curr. Med. Chem. 2019, 26, 3439–3456. [Google Scholar] [CrossRef]
- Lian, C.Y.; Zhai, Z.Z.; Li, Z.F.; Wang, L. High fat diet-triggered non-alcoholic fatty liver disease: A review of proposed mechanisms. Chem. Biol. Interact. 2020, 330, 109199. [Google Scholar] [CrossRef]
- Kumar, V.; Xin, X.; Ma, J.; Tan, C.; Osna, N.; Mahato, R.I. Therapeutic targets, novel drugs, and delivery systems for diabetes associated NAFLD and liver fibrosis. Adv. Drug Deliv. Rev. 2021, 176, 113888. [Google Scholar] [CrossRef] [PubMed]
- Willson, T.M.; Brown, P.J.; Sternbach, D.D.; Henke, B.R. The PPARs: From orphan receptors to drug discovery. J. Med. Chem. 2000, 43, 527–550. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Gupta, P.; Singh Saini, A.; Kaushal, C.; Sharma, S. The peroxisome proliferator activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Tech. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer. 2012, 12, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Haczeyni, F.; Wang, H.; Barn, V.; Mridha, A.R.; Yeh, M.M.; Haigh, W.G.; Ioannou, G.N.; Choi, Y.J.; McWherter, C.A.; Teoh, N.C.; et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol. Commun. 2017, 1, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.R.; Giri, S.R.; Bhoi, B.; Trivedi, C.; Rath, A.; Rathod, R.; Ranvir, R.; Kadam, S.; Patel, H.; Swain, P.; et al. Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 2018, 38, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.C.; Staels, B. Sorting out the roles of PPAR in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Zambon, A.; Gervois, P.; Pauletto, P.; Fruchart, J.C.; Staels, B. Modulation of hepatic inflammatory risk markers of cardiovascular diseases by PPAR-α activators: Clinical and experimental evidence. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Foxworthy, P.S.; Perry, D.N.; Hoover, D.M.; Eacho, P.I. Changes in hepatic lipid metabolism associated with lipid accumulation and its reversal in rats given the peroxisome proliferator LY171883. Toxicol. Appl. Pharmacol. 1990, 106, 375–383. [Google Scholar] [CrossRef]
- Dai, J.; Liang, K.; Zhao, S.; Jia, W.; Liu, Y.; Wu, H.; Lv, J.; Cao, C.; Chen, T.; Zhuang, S.; et al. Chemoproteomics reveals baicalin activates hepatic CPT1 to ameliorate diet-induced obesity and hepatic steatosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5896–E5905. [Google Scholar] [CrossRef]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef]
- Schreurs, M.; Kuipers, F.; van der Leij, F.R. Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes. Rev. 2010, 11, 380–388. [Google Scholar] [CrossRef]
- Shearer, B.G.; Hoekstra, W.J. Recent advances in peroxisome proliferator-activated receptor science. Curr. Med. Chem. 2003, 10, 267–280. [Google Scholar] [CrossRef]
- Campbell, I.W. The clinical significance of PPAR gamma agonism. Curr. Mol. Med. 2005, 5, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Stuart, K.; Hartland, A.; Bhartia, M.; Ramachandran, S. Drugs and the peroxisome proliferator activated receptors. Front. Clin. Drug Res. Diabetes Obes. 2014, 1, 149–210. [Google Scholar]
- Wright, M.B.; Bortolini, M.; Tadayyon, M.; Bopst, M. Challenges and opportunities in development of PPAR agonists. Mol. Endocrinol. 2014, 28, 1756–1768. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Kim, E.S.; Koh, M.; Lee, S.J.; Lim, D.; Yang, Y.R.; Jang, H.J.; Seo, K.A.; Min, S.H.; Lee, I.H.; et al. A novel non-agonist peroxisome proliferator-activated receptor γ (PPARγ) ligand UHC1 blocks PPARγ phosphorylation by cyclin-dependent kinase 5 (CDK5) and improves insulin sensitivity. J. Biol. Chem. 2014, 289, 26618–26629. [Google Scholar] [CrossRef]
- Choi, J.H.; Banks, A.S.; Kamenecka, T.M.; Busby, S.A.; Chalmers, M.J.; Kumar, N.; Kuruvilla, D.S.; Shin, Y.; He, Y.; Bruning, J.B.; et al. Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature 2011, 477, 477–481. [Google Scholar] [CrossRef]
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L.; Jacquier, E.; Gauthier, E.; Lepais, V.; Chatar, M.; et al. Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) α/γ/δ Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J. Med. Chem. 2018, 61, 2246–2265. [Google Scholar] [CrossRef] [PubMed]
- Eeda, V.; Wu, D.; Lim, H.Y.; Wang, W. Design, synthesis, and evaluation of potent novel peroxisome proliferator-activated receptor γ indole partial agonists. Bioorg. Med. Chem. Lett. 2019, 29, 126664. [Google Scholar] [CrossRef]
- Lamotte, Y.; Martres, P.; Faucher, N.; Laroze, A.; Grillot, D.; Ancellin, N.; Saintillan, Y.; Beneton, V.; Gampe, R.T., Jr. Synthesis and biological activities of novel indole derivatives as potent and selective PPARgamma modulators. Bioorg. Med. Chem. Lett. 2010, 20, 1399–1404. [Google Scholar] [CrossRef]
- Mahindroo, N.; Huang, C.F.; Peng, Y.H.; Wang, C.C.; Liao, C.C.; Lien, T.W.; Chittimalla, S.K.; Huang, W.J.; Chai, C.H.; Prakash, E.; et al. Novel indole-based peroxisome proliferator-activated receptor agonists: Design, SAR, structural biology, and biological activities. J. Med. Chem. 2005, 48, 8194–8208. [Google Scholar] [CrossRef]
- Wang, L.P.; Mei, X.G.; Wang, C.; Zhu, W.M. Biomimetic semi-synthesis of fradcarbazole A and its analogues. Tetrahedron 2015, 71, 7990–7997. [Google Scholar] [CrossRef]
- de la Fuente, M.C.; Dominguez, D. Normal electron demand Diels-Alder cycloaddition of indoles to 2,3-dimethyl-1,3-butadiene. Tetrahedron 2011, 67, 3997–4001. [Google Scholar] [CrossRef]
- Jin, J.; Qiu, F.G. Total Synthesis of (±)-1-Acetylaspidoalbidine and (±)-1-Methylaspidospermidine. Adv. Synth. Catal. 2014, 356, 340–346. [Google Scholar] [CrossRef]
- Wen, S.J.; Zhong, H.W.; Yao, Z.J. Synthesis of a fully protected (2S,3R)-N-(1′,1′-dimethyl-2′-propenyl)-3-hydroxytryptophan from tryptophan. Tetrahedron Lett. 2002, 43, 5291–5294. [Google Scholar] [CrossRef]
- Zhang, N.; Song, D.; Chen, W.; Zhang, S.; Zhang, P.; Zhang, N.; Ma, S. Modification of 5-methylphenanthridium from benzothiazoles to indoles as potent FtsZ inhibitors: Broadening the antibacterial spectrum toward vancomycin-resistant enterococci. Eur. J. Med. Chem. 2021, 224, 113723. [Google Scholar] [CrossRef] [PubMed]
- Tomoo, T.; Nakatsuka, T.; Katayama, T.; Hayashi, Y.; Fujieda, Y.; Terakawa, M.; Nagahira, K. Design, Synthesis, and Biological Evaluation of 3-(1-Aryl-1H-indol-5-yl)propanoic Acids as New Indole-Based Cytosolic Phospholipase A2α Inhibitors. J. Med. Chem. 2014, 57, 7244–7262. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Park, J.; Yu, E.; Sim, J.; Park, C.M. Electrosynthesis of Dihydropyrano [4,3-b] indoles Based on a Double Oxidative [3+3] Cycloaddition. Angew. Chem. Int. Ed. Engl. 2020, 59, 11886–11891. [Google Scholar] [CrossRef]
- Hansen, S.R.; Spangler, J.E.; Hansen, J.H.; Davies, H.M. Metal-free N-H insertions of donor/acceptor carbenes. Org. Lett. 2012, 14, 4626–4629. [Google Scholar] [CrossRef]
- McKeage, K.; Keating, G.M. Fenofibrate: A review of its use in dyslipidaemia. Drugs 2011, 71, 1917–1946. [Google Scholar] [CrossRef]
- Kamata, S.; Oyama, T.; Saito, K.; Honda, A.; Yamamoto, Y.; Suda, K.; Ishikawa, R.; Itoh, T.; Watanabe, Y.; Shibata, T.; et al. PPARα Ligand-Binding Domain Structures with Endogenous Fatty Acids and Fibrates. Iscience 2020, 23, 101727. [Google Scholar] [CrossRef]
- Yamashita, S.; Rizzo, M.; Su, T.C.; Masuda, D. Novel Selective PPARα Modulator Pemafibrate for Dyslipidemia, Nonalcoholic Fatty Liver Disease (NAFLD), and Atherosclerosis. Metabolites 2023, 13, 626. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Leatherman, G.F.; Foster, D.W. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonylCoA. J. Biol. Chem. 1978, 253, 4128–4136. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Pan, J.; Qu, N.; Lei, Y.; Han, J.; Zhang, J.; Han, D. The AMPK pathway in fatty liver disease. Front. Physiol. 2022, 13, 970292. [Google Scholar] [CrossRef] [PubMed]
- Lampidonis, A.D.; Rogdakis, E.; Voutsinas, G.E.; Stravopodis, D.J. The resurgence of Hormone-Sensitive Lipase (HSL) in mammalian lipolysis. Gene 2011, 477, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sperry, J.; Moody, C.J. Diazonamide studies. A direct synthesis of the indole bis-oxazole fragment from tri- and tetra-peptides using biomimetic oxidative cyclizations. Tetrahedron 2010, 66, 6483–6495. [Google Scholar] [CrossRef]
- Kim, D.; Wang, C.Y.; Hu, R.; Lee, J.Y.; Luu, T.T.; Park, H.J.; Lee, S.K. Antitumor Activity of Vanicoside B Isolated from Persicaria dissitiflora by Targeting CDK8 in Triple-Negative Breast Cancer Cells. J. Nat. Prod. 2019, 82, 3140–3149. [Google Scholar] [CrossRef]
- Qiu, J.; Chen, L.; Zhang, L.; Xu, F.; Zhang, C.; Ren, G.; Dou, X.; Xie, Z.; Tiao, Z. Formula modulates intestinal microbiota and liver purine metabolism to suppress hepatic steatosis and pyroptosis in NAFLD therapy. J. Phytomed. 2023, 121, 155111. [Google Scholar] [CrossRef]
- Li, S.; Li, J.; Shen, C.; Zhang, X.; Sun, S.; Cho, M.; Sun, C.; Song, Z. tert-Butylhydroquinone (tBHQ) Protects Hepatocytes against Lipotoxicity via Inducing Autophagy Independently of Nrf2 Activation. J. Biochim. Biophys. Act. 2014, 1841, 22–33. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.-C.; Wei, G.; Liao, Z.-Q.; Wang, F.-X.; Zong, C.; Qiu, J.; Le, Y.; Yu, Z.-L.; Yang, S.Y.; Wang, H.-S.; et al. Design and Synthesis of Novel Indole Ethylamine Derivatives as a Lipid Metabolism Regulator Targeting PPARα/CPT1 in AML12 Cells. Molecules 2024, 29, 12. https://doi.org/10.3390/molecules29010012
Liu Y-C, Wei G, Liao Z-Q, Wang F-X, Zong C, Qiu J, Le Y, Yu Z-L, Yang SY, Wang H-S, et al. Design and Synthesis of Novel Indole Ethylamine Derivatives as a Lipid Metabolism Regulator Targeting PPARα/CPT1 in AML12 Cells. Molecules. 2024; 29(1):12. https://doi.org/10.3390/molecules29010012
Chicago/Turabian StyleLiu, Yu-Chen, Gang Wei, Zhi-Qiang Liao, Fang-Xin Wang, Chunxiao Zong, Jiannan Qiu, Yifei Le, Zhi-Ling Yu, Seo Young Yang, Heng-Shan Wang, and et al. 2024. "Design and Synthesis of Novel Indole Ethylamine Derivatives as a Lipid Metabolism Regulator Targeting PPARα/CPT1 in AML12 Cells" Molecules 29, no. 1: 12. https://doi.org/10.3390/molecules29010012