
β-Carboline-α-aminophosphonate Derivative: A Promising Antitumor Agent for Breast Cancer Treatment
 ,
,  , and
, and
Abstract
:
1. Introduction
2. Results
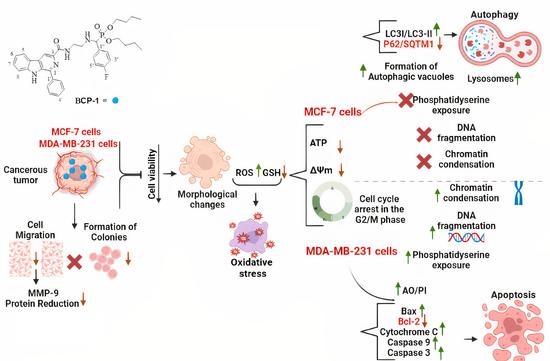
2.1. Evaluation of the Cytotoxic Activity of BCP-1 in Breast Cancer and Healthy Breast Cell Lines
2.2. Morphological Changes Induced by BCP-1 in Breast Cancer Cells
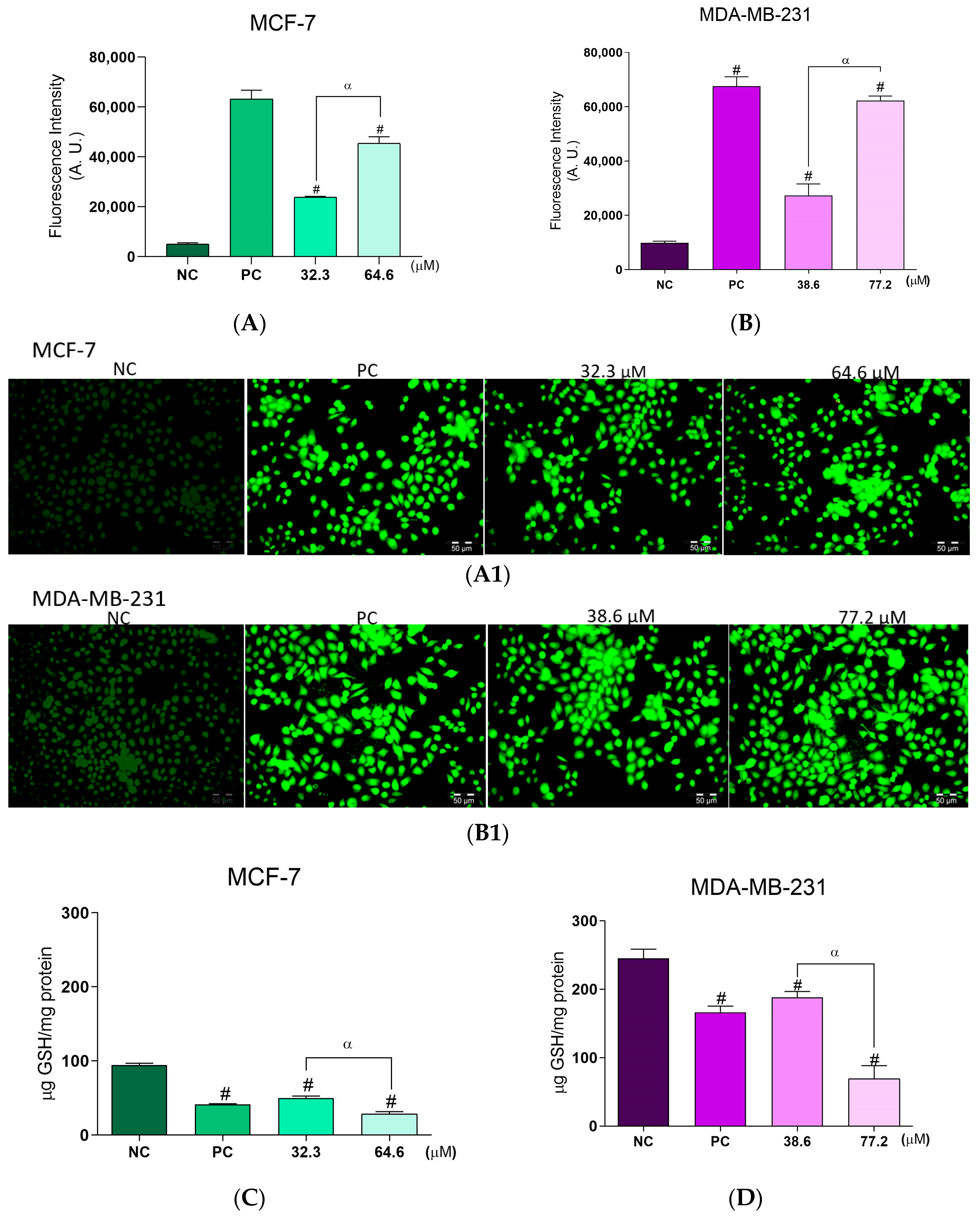
2.3. Effects of BCP-1 on ROS Production and GSH Levels in Breast Cancer Cells
2.4. Effects of BCP-1 on ΔΨm and Intracellular ATP Levels in Breast Cancer Cells
2.5. Effect of BCP-1 on Lipid Peroxidation and Membrane Integrity in Breast Cancer Cells
2.6. Effect of BCP-1 on the Cell Cycle Progression of Breast Cancer Cells
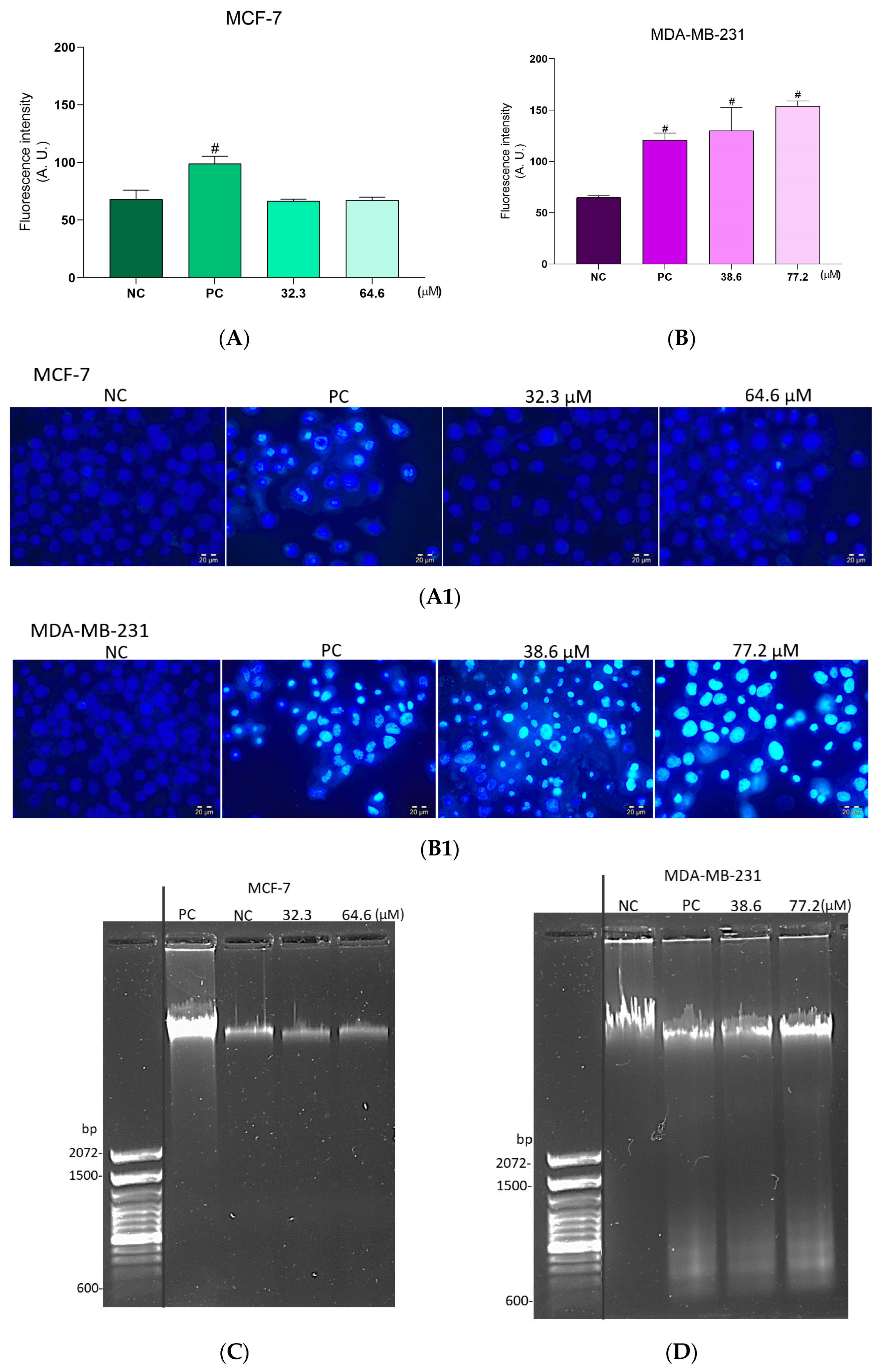
2.7. Effect of the BCP-1 on DNA Fragmentation in Breast Cancer Cells
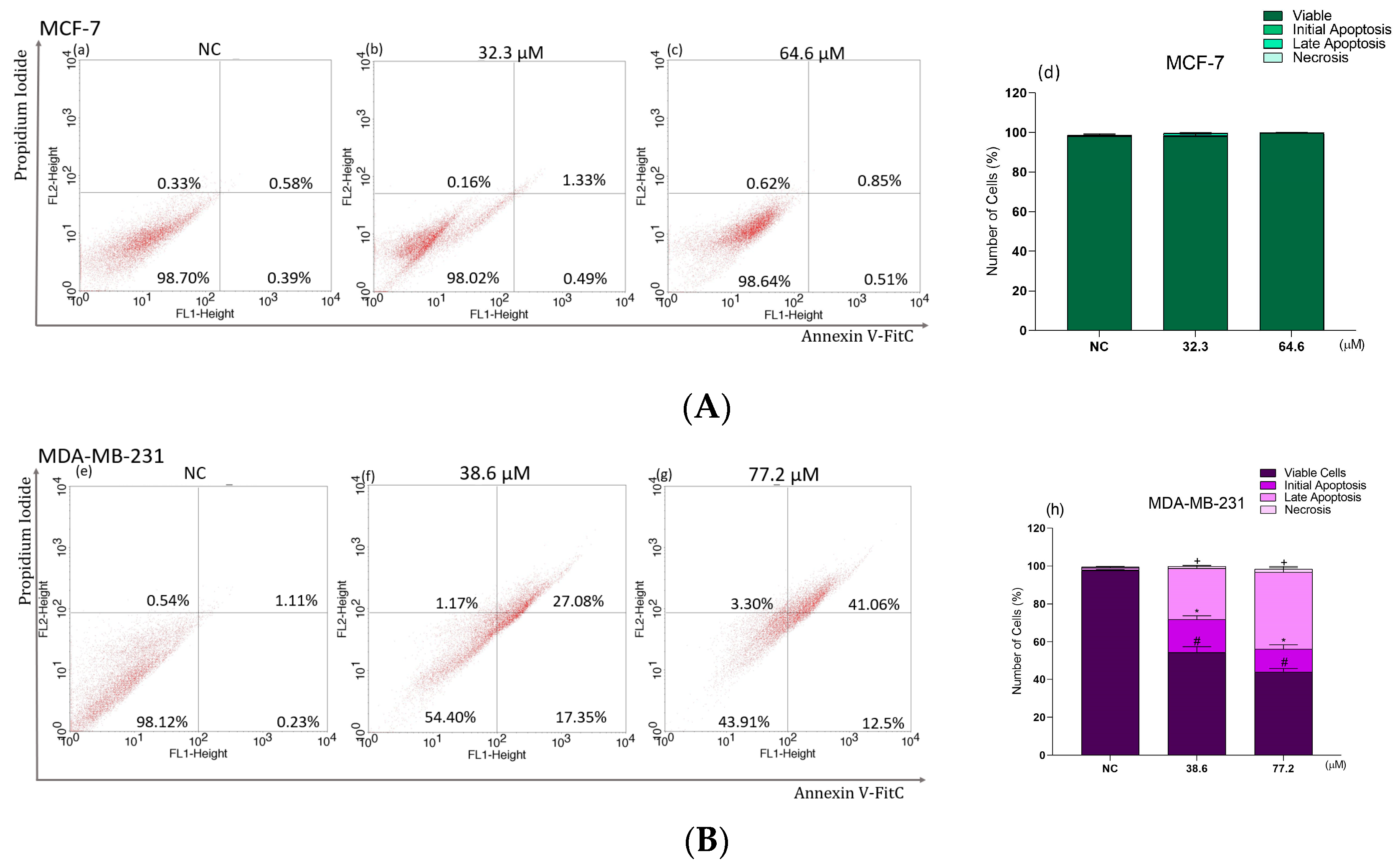
2.8. Phosphatidylserine Exposure Analysis in Breast Cancer Cells Treated with BCP-1
2.9. Effects of BCP-1 on Cell Death and Expression of Apoptosis-Related Proteins in MDA-MB-231 Breast Cancer Cells
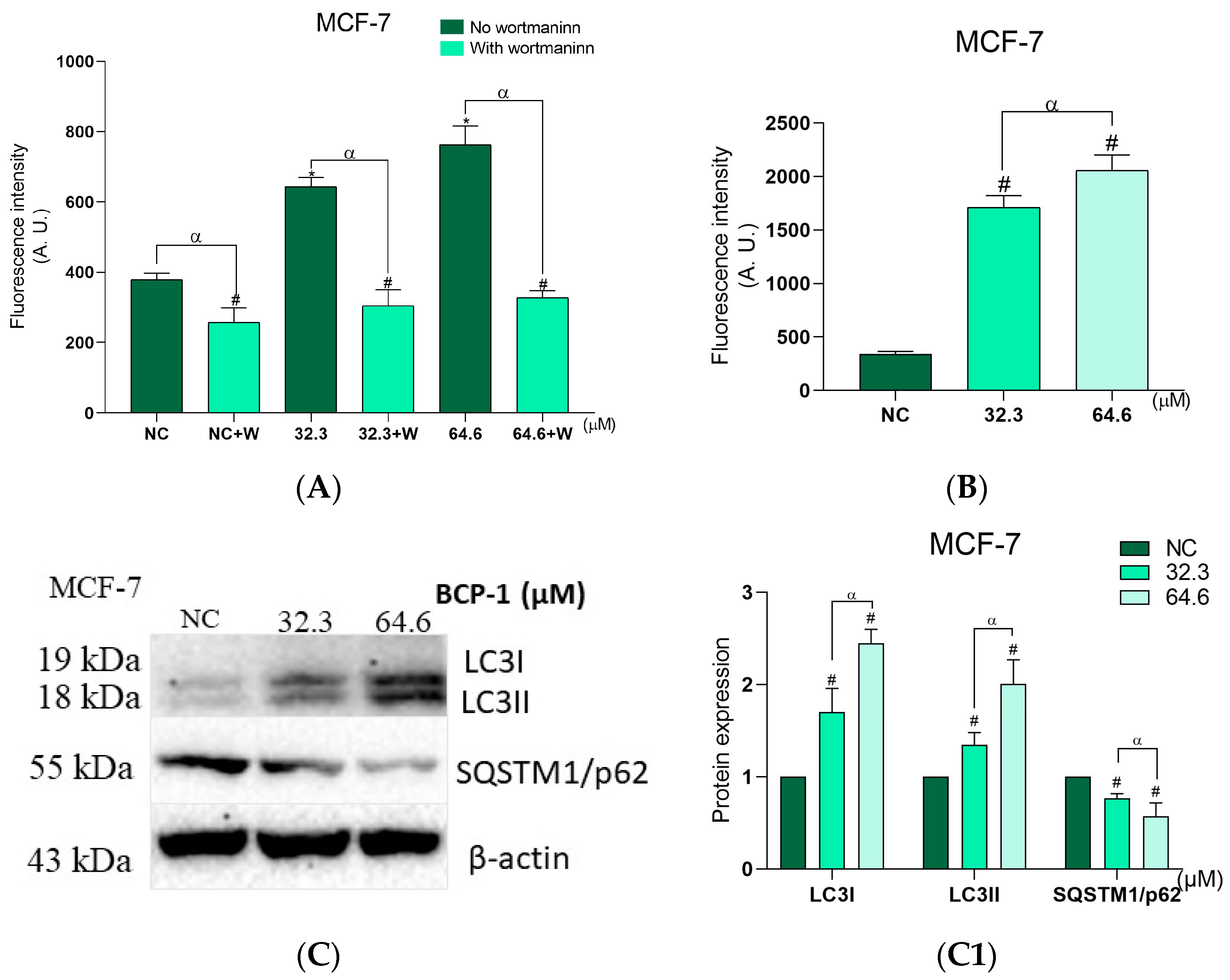
2.10. Effects of BCP-1 on the Number of Autophagic Vacuoles and Acidic Organelles, and the Expression of Proteins Related to Autophagy in MCF-7 Breast Cancer Cells
2.11. Effects of the Antimetastatic Potential of BCP-1 on Breast Cancer Cells
2.12. Effects of BCP-1 on Breast Cancer Cell Colony Formation
3. Discussion
4. Material and Methods
4.1. Reagents
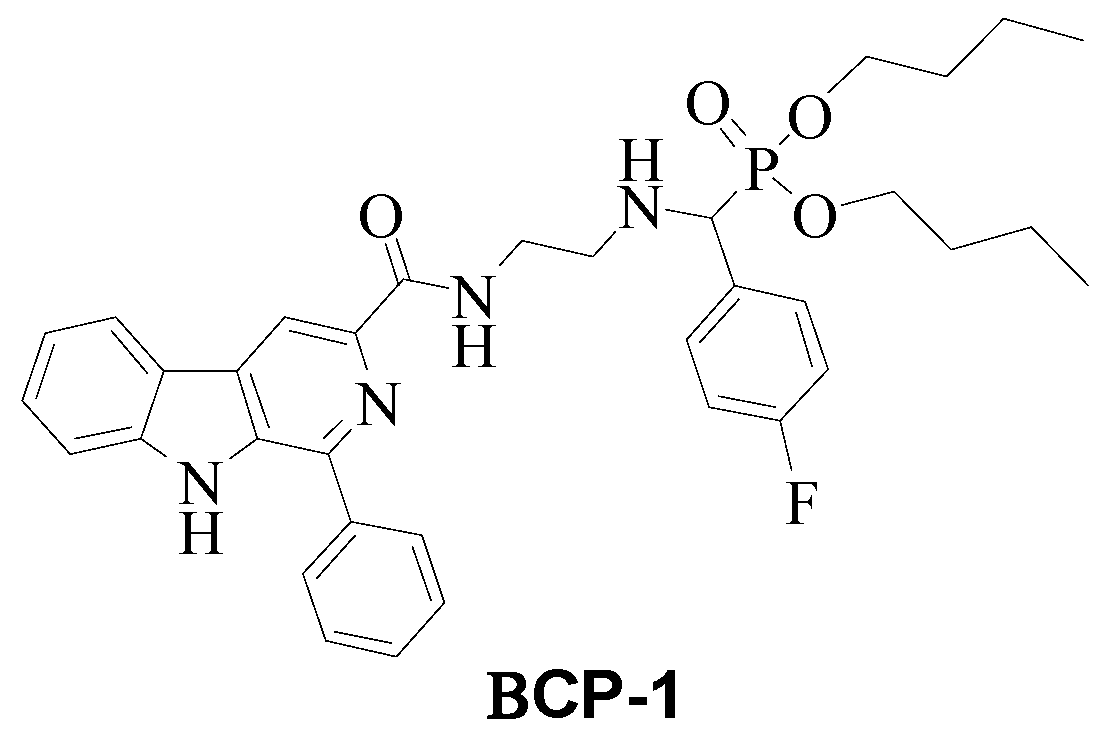
4.2. Synthesis and Characterization of β-Carboline-α-Aminophosphonate BCP-1
4.3. Cell Lines and Cell Culture
4.4. Evaluation of Cell Viability
4.5. Evaluation of the Cell Morphology by Scanning Electron Microscopy (SEM)
4.6. Evaluation of the Production of Reactive Oxygen Species (ROS)
4.7. Evaluation of the Endogenous Antioxidant Defense System through GSH Levels
4.8. Mitochondrial Membrane Potential Assessment (ΔΨm)
4.9. Quantification of Intracellular Adenosine Triphosphate (ATP) Levels
4.10. Cell Membrane Assessment
4.10.1. Evaluation of Lipid Peroxidation
4.10.2. Assessment of Cell Membrane Integrity
4.11. Cell cycle Analysis
4.12. Cell Chromatin Analysis
4.13. Evaluation of DNA Fragmentation by Agarose Gel Electrophoresis
4.14. Detection of Phosphatidylserine Exposure
4.15. Evaluation of the Type of Cell Death by Double Labeling with AO/PI
4.16. Western Blotting
4.17. Assessment of Autophagic Vacuoles
4.18. Evaluation of Acidic Organelles
4.19. Cell Migration Assay
4.20. Clonogenic Assay
4.21. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wu, H.; Chen, L.; Zhu, F.; Han, X.; Sun, L.; Chen, K. Toxins the cytotoxicity effect of Resveratrol: Cell cycle arrest and induced apoptosis of breast cancer 4T1 cells. Toxins 2019, 11, 731. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 1 December 2022).
- Yan, W.; Ma, X.; Zhao, X.; Zhang, S. Baicalein induces apoptosis and autophagy of breast cancer cells via inhibiting PI3K/AKT pathway in vivo and vitro. Drug Des. Dev. Ther. 2018, 2018, 3961–3972. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, I.; Farid, M.; Javidfar, M.; Zabet, N.; Shokoohian, B.; Arki, M.K.; Shpichka, A.; Noorbazargan, H.; Aghdaei, H.A. The optimized formulation of tamoxifen-loaded niosomes efficiently induced apoptosis and cell cycle arrest in breast cancer cells. AAPS PharmSciTech 2022, 23, 57. [Google Scholar] [CrossRef] [PubMed]
- Nigjeh, E.K.; Yeap, S.K.; Nordin, N.; Kamalideghan, B.; Ky, H. Citral induced apoptosis in mda-mb-231 spheroid cells. BMC Complement. Altern. Med. 2018, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-rad, J.; Ozleyen, A.; Tumer, T.B.; Adetunji, C.O. Natural products and synthetic analogs as a source of antitumor drugs. Biomolecules 2019, 9, 679. [Google Scholar] [CrossRef]
- Soni, J.P.; Yeole, Y.; Shankaraiah, N. β-Carboline-Based molecular hybrids as anticancer agents: A brief sketch. RSC Med. Chem. 2021, 10, 730–750. [Google Scholar] [CrossRef]
- Mizerska-Kowalska, M.; Sowa, M.; Donarska, B.; Płaziński, W.; Sławińska-Brych, A.; Tomasik, A.; Ziarkowska, A.; Łączkowski, K.Z.; Zdzisińska, B. New Borane-Protected Derivatives of α-Aminophosphonous Acid as Anti-Osteosarcoma Agents: ADME Analysis and Molecular Modeling, In Vitro Studies on Anti-Cancer Activities, and NEP Inhibition as a Possible Mechanism of Anti-Proliferative Activity. Int. J. Mol. Sci. 2022, 23, 6716. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Wang, C.; Yi, X.; Li, M.; Xiangjiu, H. Anticancer activities of harmine by inducing a pro-death autophagy and apoptosis in human gastric cancer cells. Phytomedicine 2017, 17, 10–18. [Google Scholar] [CrossRef]
- Farouk, L.; Laroubi, A.; Aboufatima, R.; Benharref, A.; Chait, A. Evaluation of the analgesic effect of alkaloid extract of Peganum harmala L.: Possible Mechanisms Involved. J. Ethnopharmacol. 2008, 115, 449–454. [Google Scholar] [CrossRef]
- Cui, G.; Shu, B.; Veeran, S.; Yuan, H.; Yi, X.; Zhong, G. Natural β-Carboline alkaloids regulate the PI3K/Akt/MTOR pathway and induce autophagy in insect Sf9 cells. Pestic. Biochem. Phys. 2018, 154, 67–77. [Google Scholar] [CrossRef]
- Abe, A.; Yamada, H.; Moriya, S.; Iyazawa, K.M. The β -Carboline alkaloid harmol induces cell death via autophagy but not apoptosis in human non-small cell lung cancer A549 cells. Biol. Pharm. Bull. 2011, 34, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Luo, Y.; Zou, J. Autophagy and its role in gastric cancer. Clin. Chim. Acta 2019, 489, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Nassan, M.A.; Aldhahrani, A.; Amer, H.H.; Elhenawy, A.; Swelum, A.A.; Ali, O.M.; Zaki, Y.H. Investigation of the Anticancer Effect of α-Aminophosphonates and Arylidine Derivatives of 3-Acetyl-1-aminoquinolin-2(1H)-one on the DMBA Model of Breast Cancer in Albino Rats with In Silico Prediction of Their Thymidylate Synthase Inhibitory Effect. Molecules 2022, 27, 756. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.V.; Sharif, N.S.; Gajare, R.I.; Vazquez, J.A.S.; Sangshetti, J.N.; Damale, M.D.; Nikalje, A.P.G. New 2-Oxoindolin Phosphonates as novel agents to treat cancer: A green synthesis and molecular modeling. Molecules 2018, 23, 1981. [Google Scholar] [CrossRef] [PubMed]
- Pawez Kafarski, A.M.; Berlicki, L. Remarkable Potential of the r-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Liu, J.; Liao, P.; Hu, J.; Zhu, H.; Wang, Y.; Li, Y.; Li, Y.; He, B. Synthesis and antitumor activities of chiral dipeptide thioureas containing an α-aminophosphonate Moiety. Molecules 2017, 22, 238. [Google Scholar] [CrossRef]
- Ye, M.Y.; Yao, G.Y.; Pan, Y.M.; Liao, Z.X.; Zhang, Y. Synthesis and antitumor activities of novel α-Aminophosphonate derivatives containing an alizarin moiety. Eur. J. Med. Chem. 2014, 83, 116–128. [Google Scholar] [CrossRef]
- Souza, W.F.; Araújo, W.M.; Freitas-Junior, J.C.M.; Morgado-Díaz, J.A. Sinalização celular em câncer. Ciência E Cult. 2014, 6, 30–33. [Google Scholar] [CrossRef]
- Cao, M.R.; Li, Q.; Liu, Z.L.; Liu, H.; Wang, W.; Liao, X.L.; Pan, Y.P.; Jiang, J.W. Harmine induces apoptosis in hepg2 cells via mitochondrial signaling pathway. Hepatobiliary Pancreat. Dis. Int. 2011, 10, 599–604. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, B.; Bian, B.; Zhao, H.; Kiyomi, A.; Hayashi, H. Cytotoxic effects of hellebrigenin and arenobufagin against human breast cancer cells. Front. Oncol. 2021, 11, 711220. [Google Scholar] [CrossRef]
- Badisa, R.B.; Darling-reed, S.F.; Joseph, P.; John, S.; Latinwo, L.M.; Goodman, C.B. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 Cells. Anticancer Res. 2009, 29, 2993–2996. [Google Scholar] [PubMed]
- Badisa, R.B.; Mina, D.A.; Latinwo, L.M.; Soliman, K.F.A. Selective anticancer activity of neurotoxin 1-methyl-4- phenylpyridinium on non-small cell lung adenocarcinoma A549 cells. Anticancer Res. 2014, 34, 5447–5452. [Google Scholar] [PubMed]
- Awang, N.; Kamaludin, N.F.; Chan, K.M. Cytotoxicity and mode of cell death induced by triphenyltin (IV) compounds in vitro. J. Biol. Sci. 2014, 14, 84–93. [Google Scholar] [CrossRef]
- Instituto Nacional de Câncer José Alencar Gomes da Silva. Detecção Precoce do Câncer. Rio de Janeiro: INCA, 2021. Available online: https://www.inca.gov.br/app/mortalidade (accessed on 8 December 2022).
- Nafie, E.; Lolarga, J.; Lam, B.; Guo, J.; Abdollahzadeh, E.; Rodriguez, S.; Glackin, C.; Liu, J. Harmine inhibits breast cancer cell migration and invasion by inducing the degradation of twist1. PLoS ONE 2021, 16, e0247652. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.T.; Tsai, Y.S.; Chen, M.S.; Li, Y.Z.; Lin, W.C.; Lee, R.; Tseng, Y.S.; Sheu, S.M. Flavopereirine induces cell cycle arrest and apoptosis via the AKT/P38 MAPK/ERK1/2 signaling pathway in human breast cancer cells. Eur. J. Pharmacol. 2019, 863, 172658. [Google Scholar] [CrossRef]
- Razak, N.A.; Abu, N.; Ho, W.Y.; Zamberi, N.R.; Tan, S.W. Cytotoxicity of eupatorin in MCF-7 and MDA-MB-231 human breast cancer cells via cell cycle arrest, anti-angiogenesis and induction of apoptosis. Sci. Rep. 2019, 9, 1514. [Google Scholar] [CrossRef]
- Dasari, S.; Bakthavachalam, V.; Chinnapaka, S.; Samy, A.L.P.A.; Munirathiknam, G. Neferine, an alkaloid from lotus seed embryo target HeLa and SiHa cervical cancer cells via pro-oxidant anticancer mechanism. Phytother. Res. 2020, 34, 2366–2384. [Google Scholar] [CrossRef]
- Jelic, M.D.; Mandic, A.D.; Maricic, M.S.; Srdjenovic, B.U. Oxidative stress and its role in cancer. J. Cancer Res. Ther. 2021, 17, 22–28. [Google Scholar] [CrossRef]
- Zou, N.; Wei, Y.; Li, F.; Yang, Y.; Cheng, X.; Wang, C. The Inhibitory Effects of Compound Muniziqi Granule against B16 cells and harmine induced autophagy and apoptosis by inhibiting Akt/MTOR pathway. BMC Complement. Altern. Med. 2017, 17, 517. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Irina, B.; Jankauskas, S.S.; Babenko, A.A. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Jobling, M.; Hol, E.; Tyler-smith, C.; Garland, Y. The cell: A molecular approach. J. Biol. Med. 2014, 87, 603–604. [Google Scholar]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Ewies, E.F.; El-hussieny, M.; El-sayed, N.F.; Fouad, M.A. Design, synthesis and biological evaluation of novel α-Aminophosphonates oxadiazoles via optimized iron triflate catalyzed reaction as apoptotic inducers. Eur. J. Med. Chem. 2019, 20, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Chae, H.D.; Yun, J.; Jung, M.; Kim, Y.; Kim, S.; Han, M.H.; Shin, D.Y. Constitutive activation of cyclin B1-associated cdc2 kinase overrides p53-mediated G2-M arrest. Cancer Res. 2000, 60, 542–545. [Google Scholar]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Sarkar, S.; Bhattacharjee, P.; Bhadra, B. DNA binding and apoptotic induction ability of Harmalol in HepG2: Biophysical and biochemical approaches. Chem. Biol. Interac. 2016, 258, 142–152. [Google Scholar] [CrossRef]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906. [Google Scholar] [CrossRef]
- Yoshida, G.J. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: From pathophysiology to treatment. J. Hematol. Oncol. 2017, 10, 67. [Google Scholar] [CrossRef]
- Lou, C.; Xu, X.; Chen, Y.; Zhao, H. Alisol A suppresses proliferation, migration, and invasion in human breast cancer MDA-MB-231 Cells. Molecules 2019, 24, 3651. [Google Scholar] [CrossRef]
- Ranjan, A.; Srivastava, S.K. Penfluridol suppresses pancreatic tumor growth by autophagy- mediated apoptosis. Nature 2016, 6, 26165. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H. Guidelines for the Use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. P62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 2015, 4672–4678. [Google Scholar] [CrossRef] [PubMed]
- Sanchéz-Martín, P.; Saito, T.; Komatsu, M. P62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef]
- Abe, A.; Kokuba, H. Harmol induces autophagy and subsequent apoptosis in U251MG human glioma cells through the downregulation of survivin. Oncol. Rep. 2013, 29, 1333–1342. [Google Scholar] [CrossRef]
- Tang, X.; Chen, X.; Wan, N.; Chen, Q. Curcumin induces ferroptosis in non-small-cell lung cancer via activating autophagy. Thorac. Cancer 2021, 12, 1219–1230. [Google Scholar] [CrossRef]
- Gao, J.; Zhu, H.; Wan, H.; Zou, X.; Ma, X.; Gao, G. Harmine suppresses the proliferation and migration of human ovarian cancer cells through inhibiting ERK/CREB pathway. Oncol. Rep. 2017, 38, 2927–2934. [Google Scholar] [CrossRef]
- Harbeck, N.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. 2019, 5, 66. [Google Scholar] [CrossRef]
- Zhu, Y.; Lv, Y.X.; Guo, C.; Xiao, Z.M.; Jiang, Q.J.; Kuang, H. Harmine inhibits the proliferation and migration of glioblastoma cells via the FAK/AKT pathway. Life Sci. 2021, 270, 119112. [Google Scholar] [CrossRef]
- Lin, M.L.; Chung, J.G.; Lu, Y.C.; Yang, C.Y.; Chen, S.S. Rhein inhibits invasion and migration of human nasopharyngeal carcinoma cells in vitro by down-regulation of matrix metalloproteinases-9 and vascular endothelial growth factor. Oral Oncol. 2009, 45, 531–537. [Google Scholar] [CrossRef]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; Bree, C.V. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.R.; Katla, R.; Rocha, M.P.D.; Albuquerque, T.B.; Silva, C.D.G.; Kupfer, V.L.; Rinaldi, A.W.; Domingues, N.L.C. Zinc Di (L -Prolinate). Mediated synthesis of α-aminophosphonates under mild conditions. Synthesis 2016, 48, 4489–4494. [Google Scholar] [CrossRef]
- Baréa, P.; Barbosa, V.A.; Bidóia, D.L.; De Paula, J.C.; Stefanello, T.F.; Da Costa, W.F.; Nakamura, C.V.; Sarragiotto, M.H. Synthesis, antileishmanial activity and mechanism of action studies of novel β-carboline-1,3,5-triazine hybrids. Eur. J. Med. Chem. 2018, 150, 579–590. [Google Scholar] [CrossRef]
- Souza, R.P.; Bonfim-mendonça, P.S.; Gimenes, F.; Ratti, B.A.; Kaplum, V.; Bruschi, M.L.; Nakamura, C.V.; Silva, S.O.; Maria-engler, S.S.; Consolaro, M.E.L. Oxidative stress triggered by apigenin induces apoptosis in a comprehensive panel of human cervical cancer-derived cell lines. Oxid. Med. Cell. Longev. 2017, 2017, 1512745. [Google Scholar] [CrossRef] [PubMed]
- Daré, R.G.; Nakamura, C.V.; Ximenes, V.F.; Lautenschlager, S.O. Tannic Acid, a Promising Anti-Photoaging Agent: Evidences of its antioxidant and anti-wrinkle potentials, and its ability to prevent photodamage and MMP-1 expression in L929 fibroblasts exposed to UVB. Free Radic. Biol. Med. 2020, 160, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Kaplum, V.; Ramos, A.C.; Consolaro, M.E.L.; Fernandez, M.A.; Ueda-nakamura, T.; Dias-filho, B.P.; Silva, S.O.; Hurst, S.M. Proanthocyanidin polymer-rich fraction of stryphnodendron adstringens promotes in vitro and in vivo cancer cell death via oxidative stress. Front. Pharmacol. 2018, 9, 694. [Google Scholar] [CrossRef]
- Lazarin-bidóia, D.; Desoti, C.; Martins, C.; Ribeiro, M.; Din, U. Dibenzylideneacetones are potent trypanocidal compounds that affect the trypanosoma cruzi redox system. Antimicrob. Agents Chemother. 2016, 60, 890–903. [Google Scholar] [CrossRef]
- Zani, A.P.; Zani, C.P.; Din, Z.U.; Rodrigues-filho, E.; Ueda-nakamura, T.; Garcia, F.P.; Silva, S.O.; Nakamura, C.V. Dibenzylideneacetone induces apoptosis in cervical cancer cells through Ros-mediated mitochondrial damage. Antioxidants 2023, 12, 317. [Google Scholar] [CrossRef]
- He, Y.Q.; Wang, Y. Plumbagin induces the apoptosis of human tongue carcinoma cells through the mitochondria-mediated pathway. Med. Sci. Monit. Basic Res. 2013, 19, 228–236. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
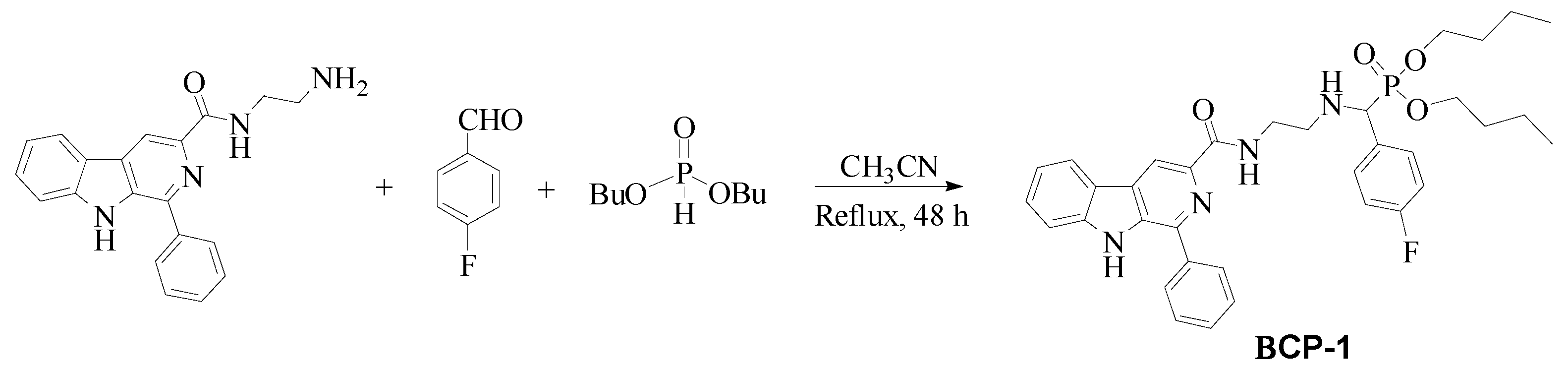{kind=link}
| Cell Line |
IC50 24 h (µM) |
IC50 48 h (µM) | SI 24 h | SI 48 h |
|---|---|---|---|---|
| MCF-7 | 50.2 ± 3.0 | 32.3 ± 1.0 | 2.96 | 3.62 |
| MDA-MB-231 | 66.7 ± 3.2 | 38.6 ± 1.1 | 2.23 | 3.03 |
| MCF-10A | 149.2 ± 4.2 | 117.3 ± 3.4 | ___ | ___ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zani, C.P.; Zani, A.P.; Thomazini, C.M.; Retamiro, K.M.; de Oliveira, A.R.; Gonçalves, D.L.; Sarragiotto, M.H.; Garcia, F.P.; de Oliveira Silva, S.; Nakamura, C.V.; et al. β-Carboline-α-aminophosphonate Derivative: A Promising Antitumor Agent for Breast Cancer Treatment. Molecules 2023, 28, 3949. https://doi.org/10.3390/molecules28093949
Zani CP, Zani AP, Thomazini CM, Retamiro KM, de Oliveira AR, Gonçalves DL, Sarragiotto MH, Garcia FP, de Oliveira Silva S, Nakamura CV, et al. β-Carboline-α-aminophosphonate Derivative: A Promising Antitumor Agent for Breast Cancer Treatment. Molecules. 2023; 28(9):3949. https://doi.org/10.3390/molecules28093949
Chicago/Turabian StyleZani, Caroline Pinto, Aline Pinto Zani, Cristiane Melissa Thomazini, Karina Miyuki Retamiro, Aline Rufino de Oliveira, Débora Laís Gonçalves, Maria Helena Sarragiotto, Francielle Pelegrin Garcia, Sueli de Oliveira Silva, Celso Vataru Nakamura, and et al. 2023. "β-Carboline-α-aminophosphonate Derivative: A Promising Antitumor Agent for Breast Cancer Treatment" Molecules 28, no. 9: 3949. https://doi.org/10.3390/molecules28093949