
Sc@B28−, Ti@B28, V@B28+, and V@B292−: Spherically Aromatic Endohedral Seashell-like Metallo-Borospherenes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussions
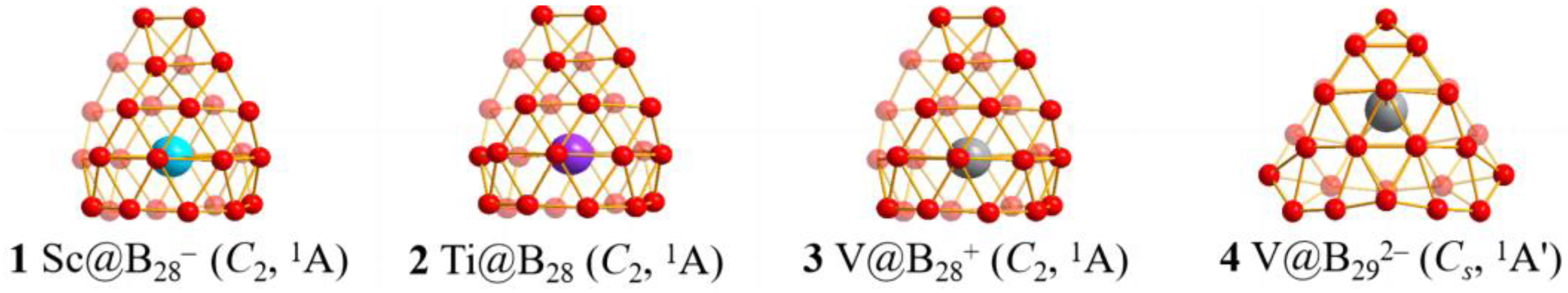
2.1. Structures and Stabilities
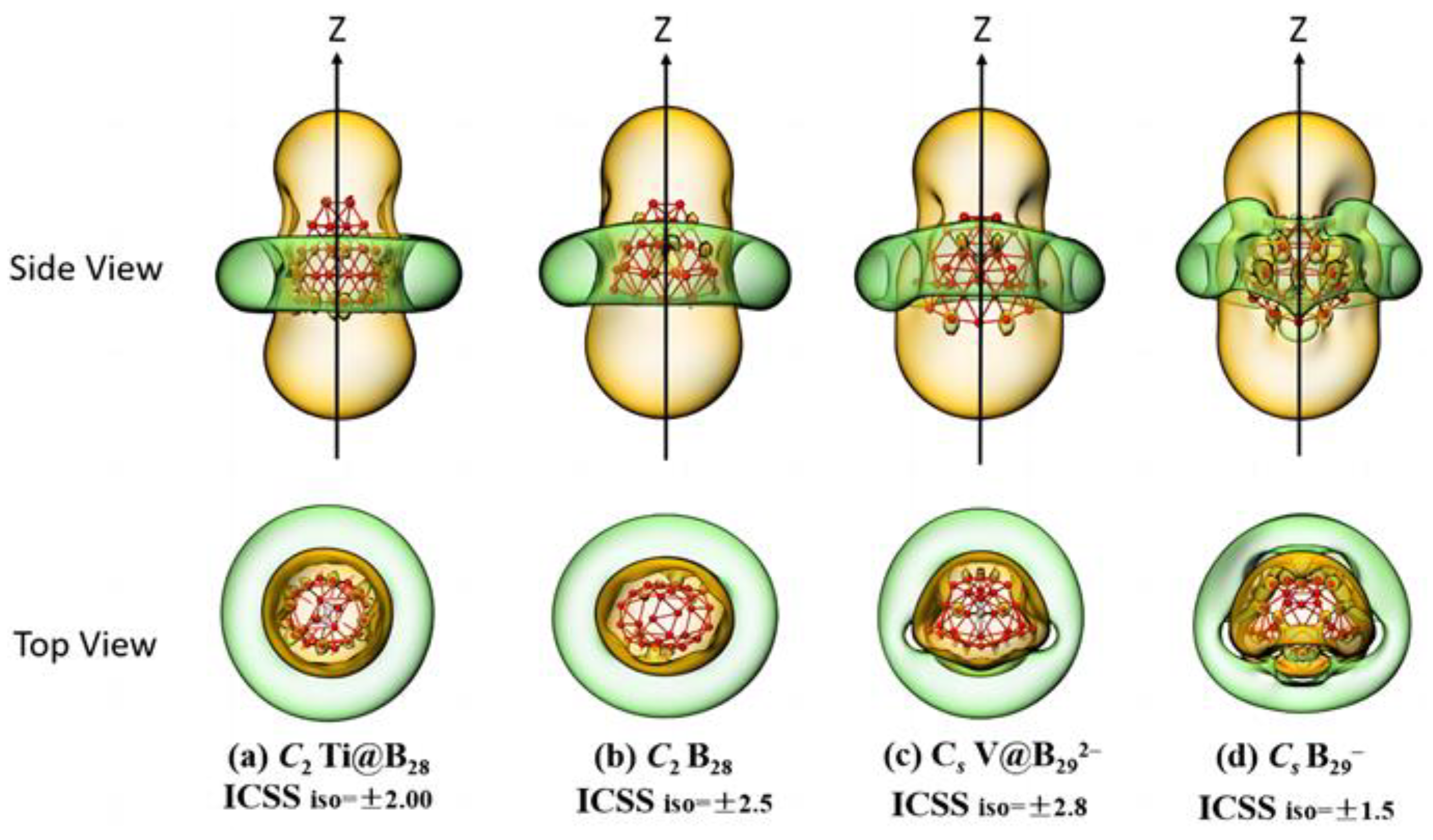
2.2. Bonding Pattern Analyses
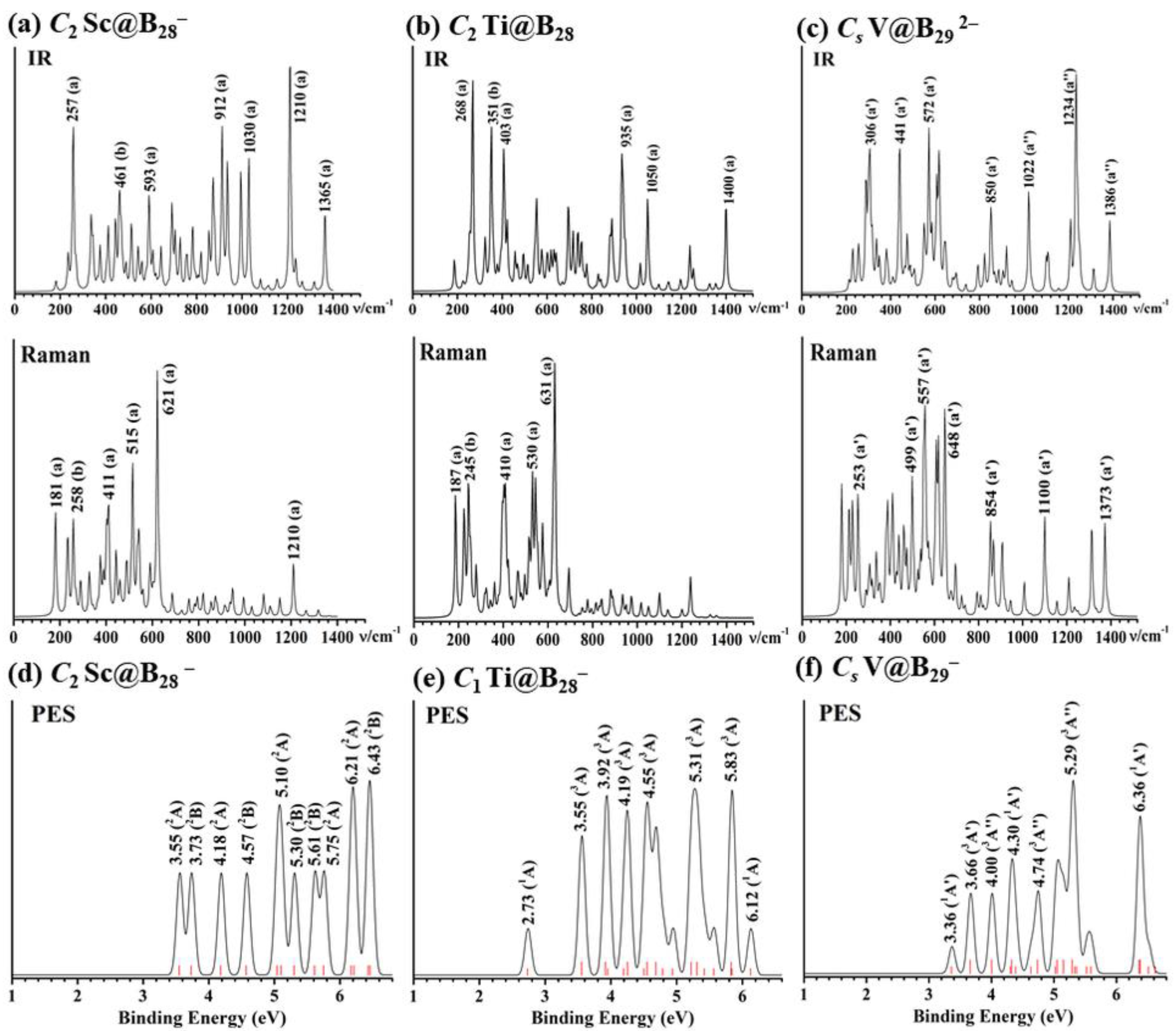
2.3. IR, Raman, and PE Spectral Simulations
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Jian, T.; Chen, X.-N.; Li, S.-D.; Boldyrev, A.-I.; Li, J.; Wang, L.-S. Probing the structures and bonding of size-selected boron and doped-boron clusters. Chem. Soc. Rev. 2019, 48, 3550–3591. [Google Scholar] [CrossRef]
- Wang, L.-S. Photoelectron spectroscopy of size-selected boron clusters: From planar structures to borophenes and borospherenes. Int. Rev. Phys. Chem. 2016, 35, 69–142. [Google Scholar] [CrossRef]
- Bai, H.; Chen, T.-T.; Chen, Q.; Zhao, X. -Y.; Zhang, Y.-Y.; Chen, W.-J.; Li, W.-L.; Cheung, L.-F.; Bai, B.; Cavanagh, J.; Huang, W.; et al. Planar B41− and B42− clusters with double-hexagonal vacancies. Nanoscale 2019, 11, 23286–23295. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Zhao, Y.-F.; Li, W.-L.; Chen, Q.; Bai, H.; Hu, H.-S.; Piazza, Z.-A.; Tian, W.-J.; Lu, H.-G.; Wu, Y.-B.; et al. Observation of an all-boron fullerene. Nat. Chem. 2014, 6, 727–731. [Google Scholar] [CrossRef]
- Chen, Q.; Li, W.-L.; Zhao, Y.-F.; Zhang, S.-Y.; Hu, H.-S.; Bai, H.; Li, H.-R.; Tian, W.-J.; Lu, H.-G.; Zhai, H.-J.; et al. Experimental and theoretical evidence of anaxially chiral borospherene. ACS Nano. 2015, 9, 754–760. [Google Scholar] [CrossRef]
- Chen, W.-J.; Ma, Y.-Y.; Chen, T.-T.; Ao, M.-Z.; Yuan, D.-F.; Chen, Q.; Tian, X.-X.; Mu, Y.-W.; Li, S.-D.; Wang, L.-S. B48−: A bilayer boron cluster. Nanoscale 2021, 13, 3868–3876. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Zhao, Y.-F.; Li, W.-L.; Jian, T.; Chen, Q.; You, X.R.; Ou, T.; Zhao, X.-Y.; Zhai, H.-J.; Li, S.-D.; et al. Observation and characterization of the smallest borospherene, B28− and B28. J. Chem. Phys. 2016, 144, 064307. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-R.; Jian, T.; Li, W.-L.; Miao, C.-Q.; Wang, Y.-J.; Chen, Q.; Luo, X.-M.; Wang, K.; Zhai, H.-J.; Li, S.-D.; et al. Competition between quasi-planar and cage-like structures in the B29− cluster: Photoelectron spectroscopy and ab initio calculations. Phys. Chem. Chem. Phys. 2016, 18, 29147–29155. [Google Scholar] [CrossRef]
- Tian, W.-J.; Chen, Q.; Li, H.-R.; Yan, M.; Mu, Y.-W.; Lu, H.-G.; Zhai, H.-J.; Li, S.-D. Saturn-like charge-transfer complexes Li4&B36, Li5&B36+, and Li6&B362+: Exohedral metalloborospherenes with a perfect cage-like B364− core. Phys. Chem. Chem. Phys. 2016, 18, 9922–9926. [Google Scholar] [CrossRef]
- Liu, H.; Mu, Y.-W.; Li, S.-D. Axially Chiral Cage-like B38+ and B382+: New aromatic members of the borospherene family. J. Cluster Sci. 2022, 33, 81–87. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, S.-Y.; Bai, H.; Tian, W.-J.; Gao, T.; Li, H.-R.; Miao, C.-Q.; Mu, Y.-W.; Lu, H.-G.; Zhai, H.-J.; et al. Cage-like B41+ and B422+: New chiral members of the borospherene family. Angew. Chem. Int. Ed. 2015, 54, 8160–8164. [Google Scholar] [CrossRef]
- Si, L.; Tang, C.-M. The reversible hydrogen storage abilities of metal Na (Li, K, Ca, Mg, Sc, Ti, Y) decorated all-boron cage B28. Int. J. Hydrogen Energ. 2017, 42, 16611–16619. [Google Scholar] [CrossRef]
- Szwacki, N.-G.; Sadrzadeh, A.; Yakobson, B.-I. Erratum: B80 Fullerene: An ab initio prediction of geometry, stability, and electronic structure. Phys. Rev. Lett. 2007, 98, 166804. [Google Scholar] [CrossRef]
- Research highlights. Nature 2007, 447, 4–5. [CrossRef]
- Pei, L.; Yan, Q.-Q.; Li, S.-D. Predicting the structural transition in medium-sized boron nanoclusters: From bilayer B64, B66, B68, B70, and B72 to Core-Shell B74. Eur. J. Inorg. Chem. 2021, 2021, 2618–2624. [Google Scholar] [CrossRef]
- Yan, Q.-Q.; Pei, L.; Li, S.-D. Predicting bilayer B50, B52, B56 and B58: Structural evolution in bilayer B48–B72 clusters. J. Mol. Model. 2021, 27, 364. [Google Scholar] [CrossRef]
- Yan, Q.-Q.; Zhang, T.; Ma, Y.-Y.; Chen, Q.; Mu, Y.-W.; Li, S.-D. A bottom-up approach from medium-sized bilayer boron nanoclusters to bilayer borophene nanomaterials. Nanoscale 2022, 14, 11443–11451. [Google Scholar] [CrossRef]
- Chen, Q.; Wei, G.-F.; Tian, W.-J.; Bai, H.; Liu, Z.-P.; Zhai, H.-J.; Li, S.-D. Quasi-planar aromatic B36 and B36− clusters: All-boron analogues of coronene. Phys. Chem. Chem. Phys. 2014, 16, 18282–18287. [Google Scholar] [CrossRef]
- Ma, Y.-Y.; Zhao, X.-Y.; Zan, W.-Y.; Mu, Y.-W.; Zhang, Z.-H.; Li, S.-D. Prediction of freestanding semiconducting bilayer borophenes. Nano Res. 2022, 15, 5752–5757. [Google Scholar] [CrossRef]
- Prasad, D.-L.; Jemmis, E.-D. Stuffing improves the stability of fullerenelike boron clusters. Phys. Rev. Lett. 2008, 100, 165504. [Google Scholar] [CrossRef]
- Li, H.; Shao, N.; Shang, B.; Yuan, L.-F.; Yang, J.; Zeng, X.C. Icosahedral B12-containing core–shell structures of B80. Chem. Commun. 2010, 46, 3878–3880. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-J.; Wang, L.; Li, F.-Y.; Chen, Z.-F. B80 and other medium-sized boron clusters: Core−shell structures, not hollow cages. J. Phys. Chem. A 2010, 114, 9969–9972. [Google Scholar] [CrossRef] [PubMed]
- Sai, L.-W.; Wu, X.; Yu, F.-Y. B96: A complete core–shell structure with high symmetry. Phys. Chem. Chem. Phys. 2022, 24, 15687–15690. [Google Scholar] [CrossRef]
- Li, F.-Y.; Jin, P.; Jiang, D.-E.; Wang, L.; Zhang, S.-B.; Zhao, J.-J.; Chen, Z.-F. B80 and B101–103 clusters: Remarkable stability of the core-shell structures established by validated density functionals. J. Chem. Phys. 2012, 136, 074302. [Google Scholar] [CrossRef]
- Sai, L.-W.; Wu, X.; Gao, N.; Zhao, J.-J.; King, R.-B. Boron clusters with 46, 48, and 50 atoms: Competition among the core–shell, bilayer and quasi-planar structures. Nanoscale 2017, 9, 13905–13909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lu, H.-G.; Li, S.-D. B111, B112, B113, and B114: The most stable core-shell borospherenes with an icosahedral B12 core at the center exhibiting superatomic behaviors. Nano Res. 2021, 14, 4719–4724. [Google Scholar] [CrossRef]
- Zhang, M.; Jia, W.-P.; Zhang, T.; Pei, B.-B.; Xu, J.; Tian, X.-X.; Lu, H.-G.; Li, S.-D. Superatomic icosahedral-CnB12-n (n = 0, 1, 2) stuffed mononuclear and binuclear borafullerene and borospherene nanoclusters with spherical aromaticity. Sci. Rep. 2022, 12, 19741. [Google Scholar] [CrossRef]
- Romanescu, C.; Galeev, T.-R.; Li, W.-L.; Boldyrev, A.-I.; Wang, L.-S. Aromatic metal-centered monocyclic boron rings: Co©B8− and Ru©B9−. Angew. Chem. 2011, 50, 9334–9337. [Google Scholar] [CrossRef]
- Chen, T.-T.; Li, W.-L.; Bai, H.; Chen, W.-J.; Dong, X.-R.; Li, J.; Wang, L.-S. Re©B8− and Re©B9−: New members of the transition-metal-centered borometallic molecular wheel family. J.Phys. Chem. A 2019, 123, 5317–5324. [Google Scholar] [CrossRef]
- Romanescu, C.; Galeev, T.-R.; Li, W.-L.; Boldyrev, A.-I.; Wang, L.-S. Transition-metal-centered monocyclic boron wheel clusters (M©Bn): A new class of aromatic borometallic compounds. Acc. Chem. Res. 2013, 46, 350–358. [Google Scholar] [CrossRef]
- Popov, I.-A.; Li, W.-L.; Piazza, Z.-A.; Boldyrev, A.-I.; Wang, L.-S. Complexes between planar boron clusters and transition metals: A photoelectron spectroscopy and ab initio study of CoB12− and RhB12−. J. Phys. Chem. A 2014, 118, 8098–8105. [Google Scholar] [CrossRef] [PubMed]
- Popov, I.-A.; Jian, T.; Lopez, G.-V.; Boldyrev, A.-I.; Wang, L.-S. Cobalt-centred boron molecular drums with the highest coordination number in the CoB16− cluster. Nat. Commun. 2015, 6, 8654. [Google Scholar] [CrossRef] [PubMed]
- Jian, T.; Li, W.-L.; Popo, I.-A.; Lope, G.-V.; Chen, X.; Boldyrev, A.-I.; Li, J.; Wang, L.-S. Manganese-centered tubular boron cluster–MnB16−: A new class of transition-metal molecules. J. Chem. Phys. 2016, 144, 154310. [Google Scholar] [CrossRef]
- Jian, T.; Li, W.-L.; Chen, X.; Chen, T.-T.; Lopez, G.-V.; Li, J.; Wang, L.S. Competition between drum and quasi-planar structures in RhB18−: Motifs for metallo-boronanotubes and metallo-borophenes. Chem. Sci. 2016, 7, 7020–7027. [Google Scholar] [CrossRef]
- Li, W.-L.; Jian, T.; Chen, X.; Li, H.-R.; Chen, T.-T.; Luo, X.-M.; Li, S.-D.; Li, J.; Wang, L.-S. Observation of a metal-centered B2-Ta@B18− tubular molecular rotor and a perfect Ta@B20− boron drum with the record coordination number of twenty. Chem. Commun. 2017, 53, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-T.; Li, W.-L.; Li, J.; Wang, L.-S. [La(ηx-Bx)La]− (x = 7–9): A new class of inverse sandwich complexes. Chem. Sci. 2019, 10, 2534–2542. [Google Scholar] [CrossRef]
- Chen, T.-T.; Li, W.-L.; Chen, W.-J.; Yu, X.-H.; Dong, X.-R.; Li, J.; Wang, L.-S. Spherical trihedral metallo-borospherenes. Nat. Commun. 2020, 11, 2766. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-Y.; Yan, M.; Wei, Z.-H.; Li, S.-D. Donor–acceptor duality of the transition-metal-like B2 core in core–shell-like metallo-borospherenes La3&[B2@B17]− and La3&[B2@B18]−. RSC Adv. 2020, 10, 34225–34230. [Google Scholar] [CrossRef]
- Lu, X.-Q.; Gao, C.-Y.; Wei, Z.-H.; Li, S.-D. Cage-like La4B24 and Core-Shell La4B290/+/−: Perfect spherically aromatic tetrahedral metallo-borospherenes. J. Mol. Model. 2021, 27, 130. [Google Scholar] [CrossRef]
- Lu, X.-Q.; Ao, M.-Z.; Tian, X.-X.; Zan, W.-Y.; Mu, Y.-W.; Li, S.-D. Perfect cubic La-doped boron clusters La6&[La@B24]+/0 as the embryos of low-dimensional lanthanide boride nanomaterials. RSC Adv. 2020, 10, 12469–12474. [Google Scholar] [CrossRef]
- Ao, M.-Z.; Lu, X.-Q.; Mu, Y.-W.; Zan, W.-Y.; Li, S.-D. La@[La5&B30]0/−/2−: Endohedral trihedral metallo-borospherenes with spherical aromaticity. Phys. Chem. Chem. Phys. 2022, 24, 3918–3923. [Google Scholar] [CrossRef]
- Yan, L.-J. Large B7 triangles in hollow spherical trihedral metallo-borospherenes and their endohedral complexes of B20TMn (TM = Sc, Y; n = 3, 4): A theoretical characterization. Inorg. Chem. 2022, 61, 10652–10660. [Google Scholar] [CrossRef]
- Li, H.-R.; Liu, H.; Lu, X.-Q.; Zan, W.-Y.; Tian, X.-X.; Lu, H.-G.; Wu, Y.-B.; Mu, Y.-W.; Li, S.-D. Cage-like Ta@Bnq complexes (n = 23–28, q = −1– +3) in 18-electron configurations with the highest coordination number of twenty-eight. Nanoscale 2018, 10, 7451–7456. [Google Scholar] [CrossRef]
- Li, H.-R.; Liu, H.; Tian, X.-X.; Zan, W.-Y.; Mu, Y.-W.; Lu, H.-G.; Li, J.; Wang, Y.-K.; Li, S.-D. Structural transition in metal-centered boron clusters: From tubular molecular rotors Ta@B21 and Ta@B22+ to cage-like endohedral metalloborospherene Ta@B22−. Phys. Chem. Chem. Phys. 2017, 19, 27025–27030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, X.-Y.; Yan, M.; Li, S.-D. From inverse sandwich Ta2B7+ and Ta2B8 to spherical trihedral Ta3B12−: Prediction of the smallest metallo-borospherene. RSC. Adv. 2020, 10, 29320–29325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, X.-Q.; Yan, M.; Li, S.-D. Perfect spherical tetrahedral metallo-borospherene Ta4B18 as a superatom following the 18-electron rule. ACS Omega 2021, 6, 10991–10996. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Chen, Q.; Zhai, H.-J.; Li, S.-D. Endohedral and exohedral metalloborospherenes: M@B40 (M=Ca, Sr) and M&B40 (M=Be, Mg). Angew Chem Int Ed. 2015, 54, 941–945. [Google Scholar] [CrossRef]
- Yu, T.-R.; Gao, Y.; Xu, D.-X.; Wang, Z.-G. Actinide endohedral boron clusters: A closed-shell electronic structure of U@B40. Nano Res. 2018, 11, 354–359. [Google Scholar] [CrossRef]
- Czekner, J.; Cheung, L.-F.; Wang, L.-S. Probing the structures of neutral B11 and B12 using high-resolutionphotoelectron imaging of B11− and B12−. J. Phys. Chem. C. 2017, 121, 10752–10759. [Google Scholar] [CrossRef]
- Yuan, Y.; Cheng, L.-J. Ferrocene analogues of sandwich B12·Cr·B12: A theoretical study. J. Chem. Phys. 2013, 138, 024301. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Staroverov, V.-N.; Scuseria, G.-E.; Tao, J.; Perdew, J.-P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.-S.; Seeger, R.-J.; Pople, A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Purvis, G.-D., III; Bartlett, R.-J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.-W.; Pople, J.-A.; Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Glendening, E.-D.; Landis, C.-R.; Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Vondele, J.-V.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.-S.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Zubarev, D.-Y.; Boldyrev, A.-I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, N.-V.; Boldyrev, A.-I. Chemical bonding analysis of excited states using the adaptive natural density partitioning method. Phys. Chem. Chem. Phys. 2019, 21, 9590–9596. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-Y.; Nan, Z.-A.; Wang, Q.-M. Superatomic orbital splitting in coinage metal nanoclusters. J. Phys. Chem. Lett. 2022, 13, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Kleinpeter, E.; Klod, S.; Koch, A. Visualization of through space NMR shieldings of aromatic and anti-aromatic molecules and a simple means to compare and estimate aromaticity. J. Mol. Struc: Theochem. 2007, 811, 45–60. [Google Scholar] [CrossRef]
- Wang, G.-J.; Zhou, M.-F.; Goettel, J.-T.; Schrobilgen, G.-J.; Su, J.; Li, J.; Schloeder, T.; Riedel, S. Identification of an iridium-containing compound with a formal oxidation state of IX. Nature 2014, 514, 475–477. [Google Scholar] [CrossRef]
- Ciuparu, D.; Klie, R.-F.; Zhu, Y.-M.; Pfefferle, L. Synthesis of pure boron single-wall nanotubes. J. Phys. Chem. B. 2004, 108, 3967–3969. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Casida, M.-E.; Jamorski, C.; Casida, K.-C.; Salahub, D.-R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Zhao, Y.-F.; Chen, X.; Li, J. TGMin: A global-minimum structure search program based on a constrained basin-hopping algorithm. Nano Res. 2017, 10, 3407–3420. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, Y.-F.; Zhang, Y.-Y.; Li, J. TGMin: An efficient global minimum searching program for free and surface-supported clusters. J. Comput. Chem. 2019, 40, 1105–1112. [Google Scholar] [CrossRef]
- Goedecker, S. Minima hopping: An efficient search method for the global minimum of the potential energy surface of complex molecular systems. J. Chem. Phys. 2004, 120, 9911–9917. [Google Scholar] [CrossRef]
- Goedecker, S.; Hellmann, W.; Lenosky, T. Global Minimum Determination of the Born-Oppenheimer Surface within Density Functional Theory. Phys. Rev. Lett. 2005, 95, 055501. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.-W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graphics. 1996, 14, 33. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Zhang, M.; Lu, X.-Q.; Yan, Q.-Q.; Zhao, X.-N.; Li, S.-D. Sc@B28−, Ti@B28, V@B28+, and V@B292−: Spherically Aromatic Endohedral Seashell-like Metallo-Borospherenes. Molecules 2023, 28, 3892. https://doi.org/10.3390/molecules28093892
Zhang T, Zhang M, Lu X-Q, Yan Q-Q, Zhao X-N, Li S-D. Sc@B28−, Ti@B28, V@B28+, and V@B292−: Spherically Aromatic Endohedral Seashell-like Metallo-Borospherenes. Molecules. 2023; 28(9):3892. https://doi.org/10.3390/molecules28093892
Chicago/Turabian StyleZhang, Ting, Min Zhang, Xiao-Qin Lu, Qiao-Qiao Yan, Xiao-Ni Zhao, and Si-Dian Li. 2023. "Sc@B28−, Ti@B28, V@B28+, and V@B292−: Spherically Aromatic Endohedral Seashell-like Metallo-Borospherenes" Molecules 28, no. 9: 3892. https://doi.org/10.3390/molecules28093892