
Iridium-Catalyzed Synthesis of Chiral 1,2,3-Triazoles Units and Precise Construction of Stereocontrolled Oligomers


Abstract
:1. Introduction
2. Results and Discussion
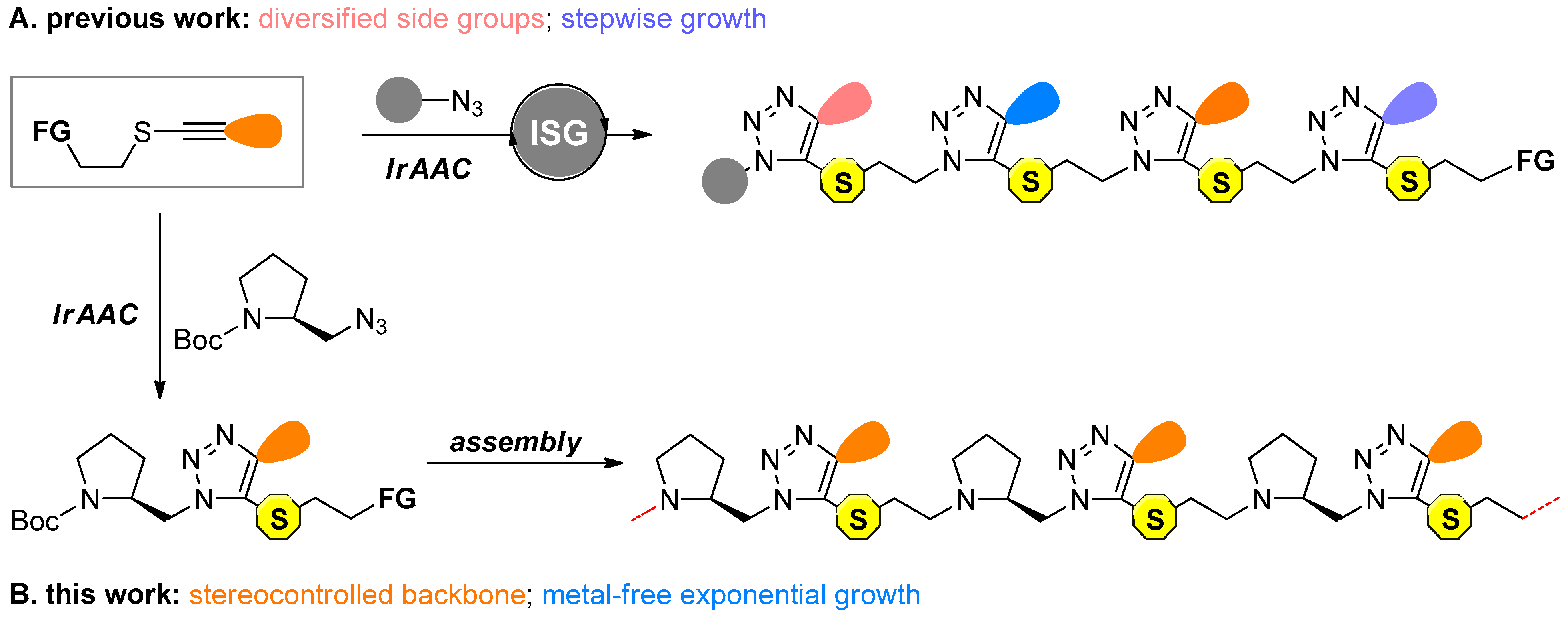
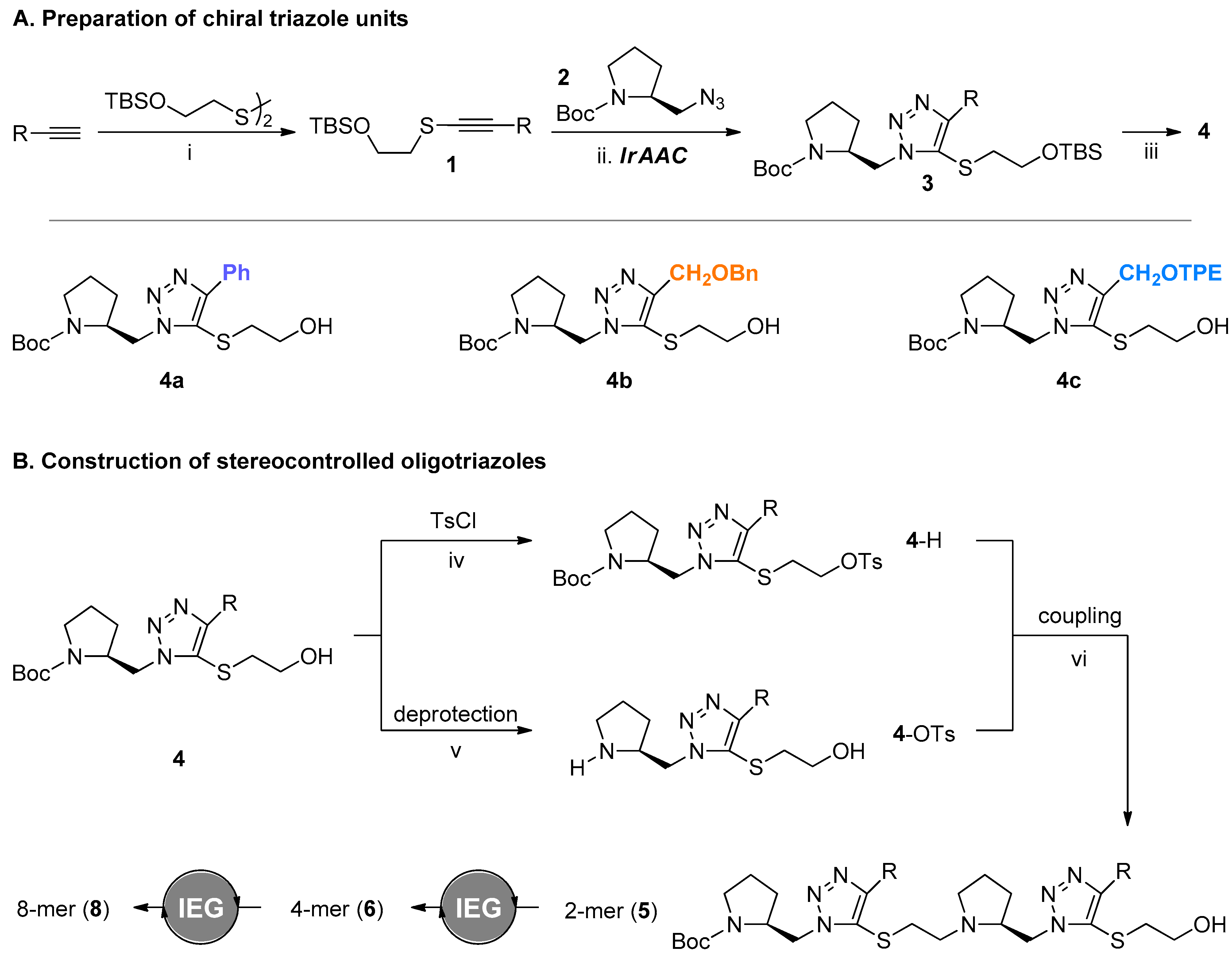
2.1. Preparation of Chiral Triazole Monomers
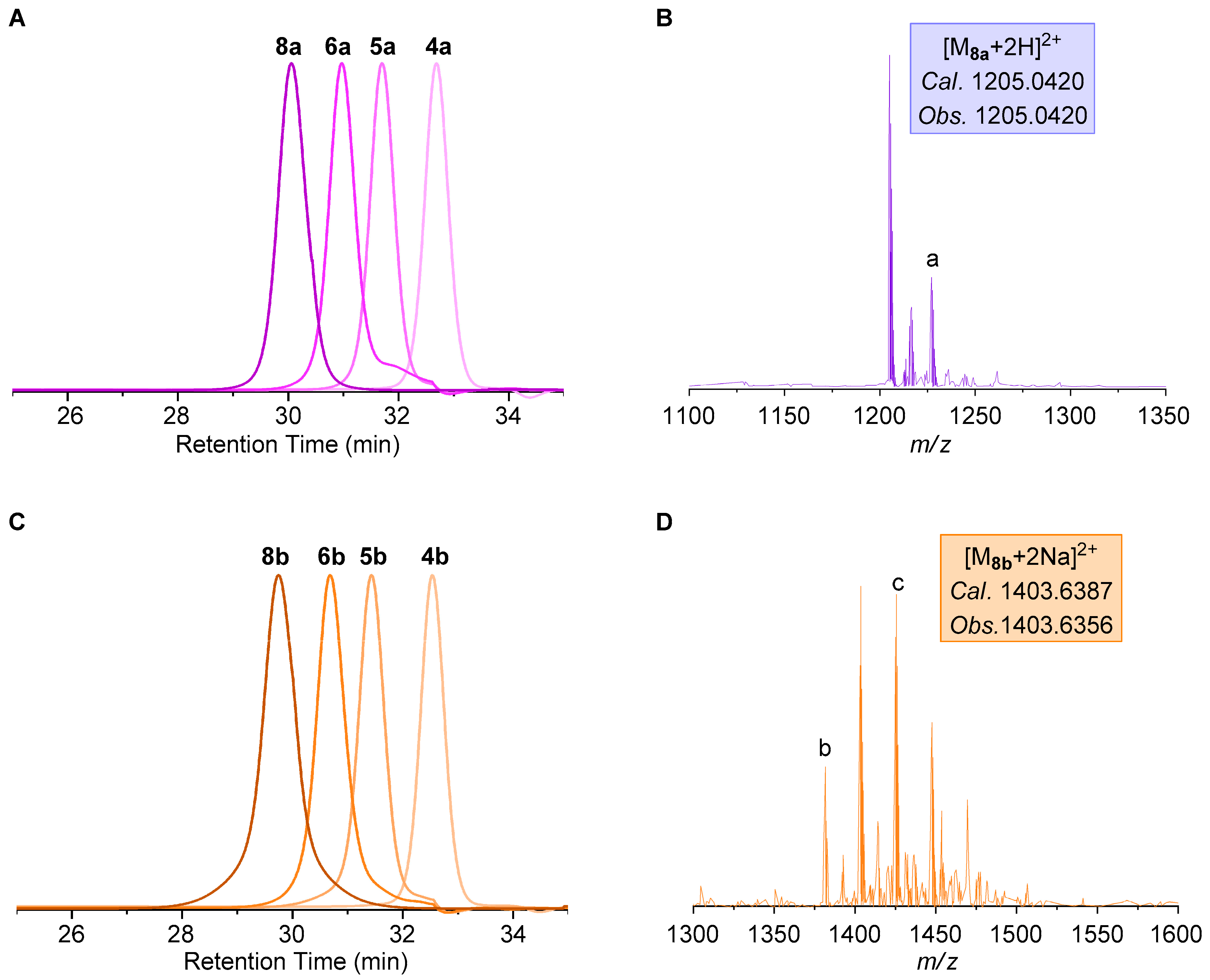
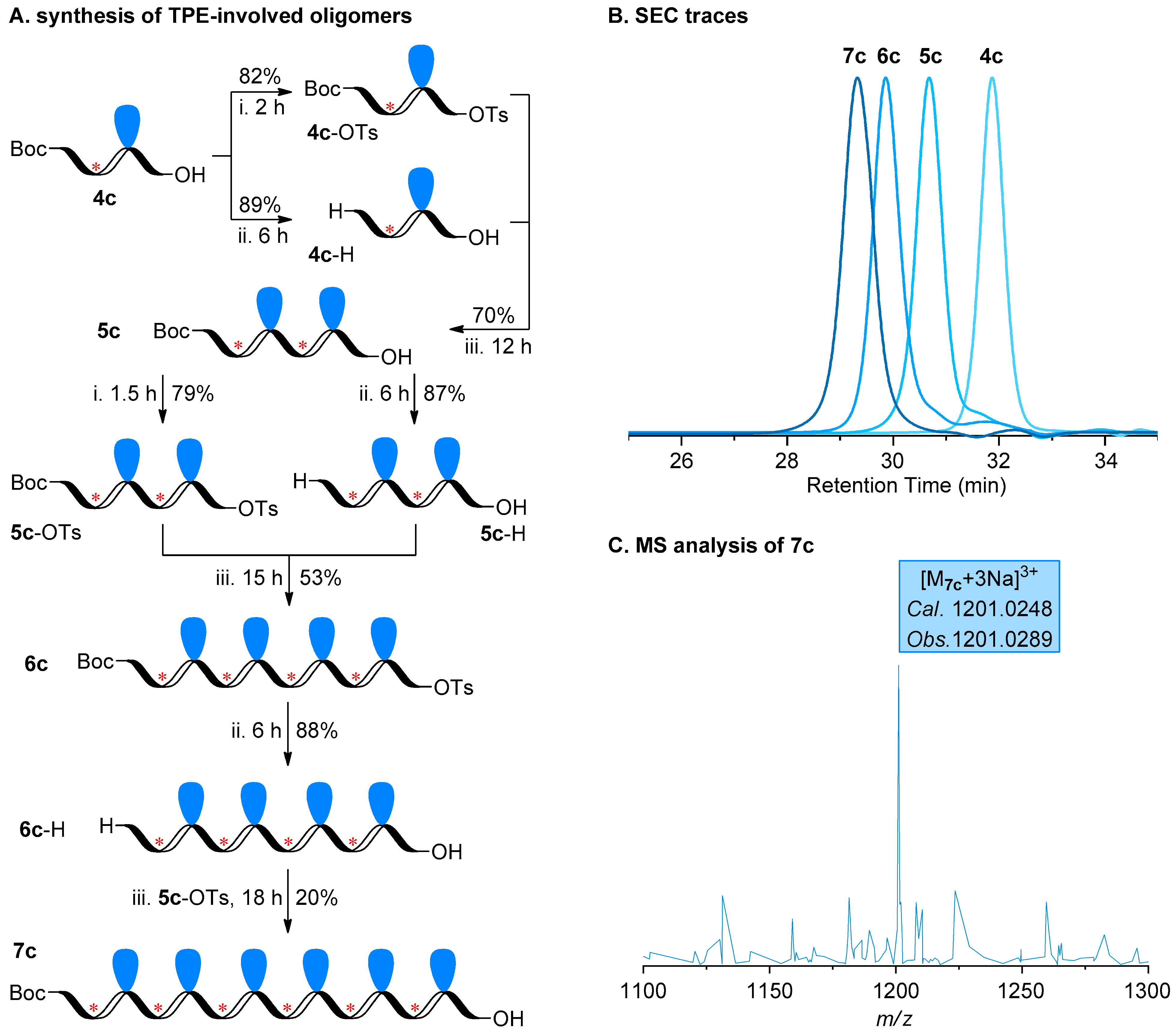
2.2. Construction of Stereoregular Oligotriazoles
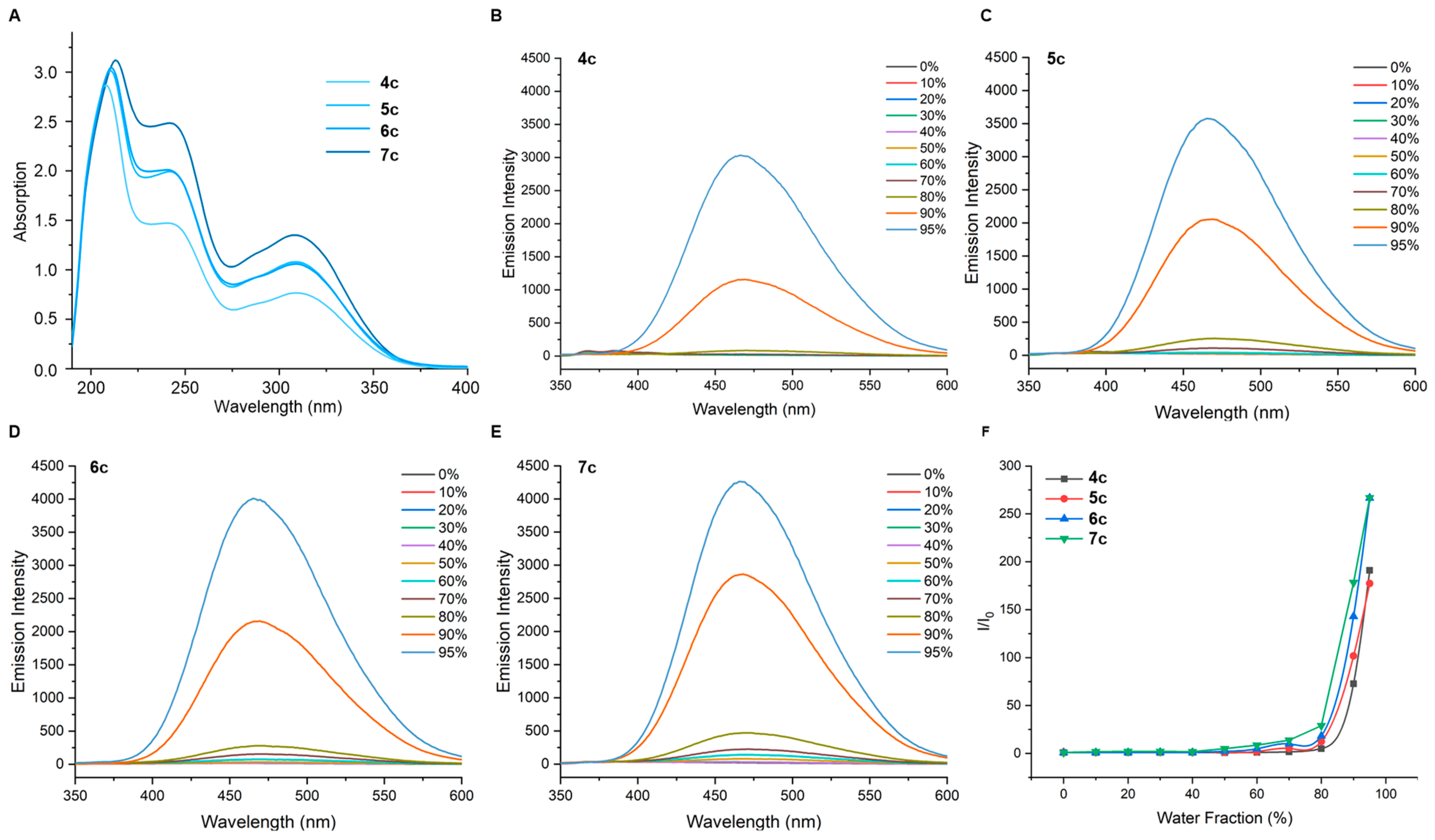
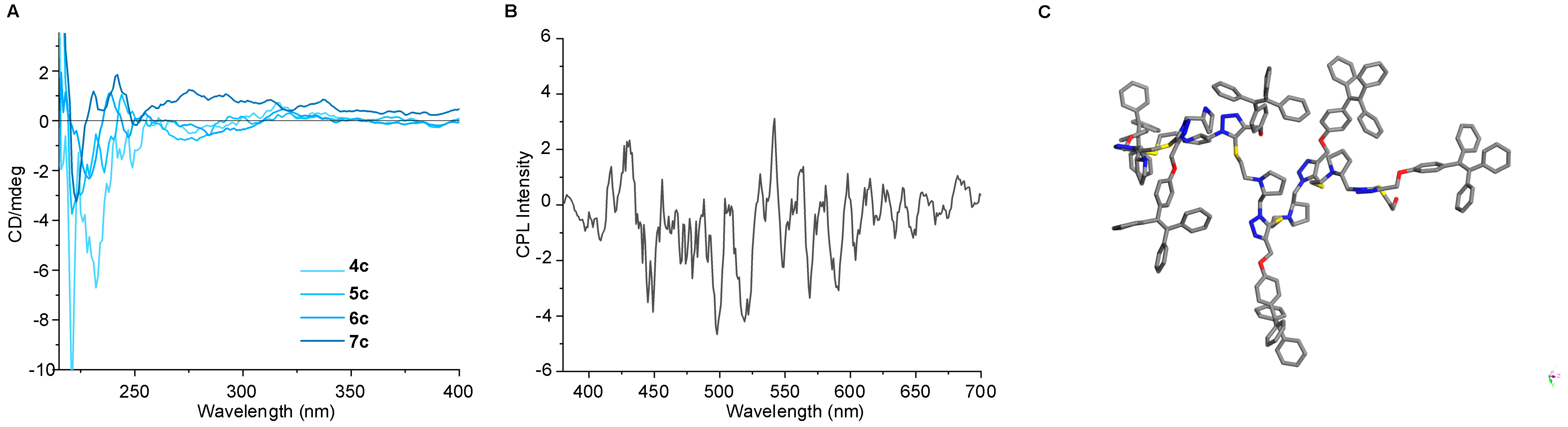
2.3. Photophysical Behaviors of TPE-Involved Oligotriazoles
3. Material and Methods
3.1. General
3.2. Preparation of 1-Thioalkynes
3.3. Synthesis of Triazole Units
3.4. Precise Construction of Oligotriazoles
3.5. Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Oro, L.A.; Claver, C. Iridium Complexes in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Oro, L.A.; Claver, C. Iridium Catalysts for Organic Reactions; Springer: Cham, Switzerland, 2021. [Google Scholar]
- Shibata, T. Iridium-catalyzed cycloadditions. In Iridium Complexes in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2009; pp. 277–298. [Google Scholar]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(i)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Xue, P.; Sun, H.H.Y.; Williams, I.D.; Sharpless, K.B.; Fokin, V.V.; Jia, G. Ruthenium-catalyzed cycloaddition of alkynes and organic azides. J. Am. Chem. Soc. 2005, 127, 15998–15999. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ding, S. Transition metal-catalyzed cycloaddition of azides with internal alkynes. Asian J. Org. Chem. 2020, 9, 1872–1888. [Google Scholar] [CrossRef]
- Ding, S.; Jia, G.; Sun, J. Iridium-catalyzed intermolecular azide−alkyne cycloaddition of internal thioalkynes under mild conditions. Angew. Chem. Int. Ed. 2014, 53, 1877–1880. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.P.; Gellman, S.H.; DeGrado, W.F. β-Peptides: From structure to function. Chem. Rev. 2001, 101, 3219–3232. [Google Scholar] [CrossRef]
- Martinek, T.A.; Fülöp, F. Peptidic foldamers: Ramping up diversity. Chem. Soc. Rev. 2012, 41, 687–702. [Google Scholar] [CrossRef]
- George, K.L.; Horne, W.S. Foldamer tertiary structure through sequence-guided protein backbone alteration. Acc. Chem. Res. 2018, 51, 1220–1228. [Google Scholar] [CrossRef]
- Shi, Y.; Teng, P.; Sang, P.; She, F.; Wei, L.; Cai, J. γ-AA-peptides: Design, structure, and applications. Acc. Chem. Res. 2016, 49, 428–441. [Google Scholar] [CrossRef]
- Sang, P.; Shi, Y.; Huang, B.; Xue, S.; Odom, T.; Cai, J. Sulfono-γ-AApeptides as helical mimetics: Crystal structures and applications. Acc. Chem. Res. 2020, 53, 2425–2442. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.F.; Meyer, T.Y.; Ouchi, M.; Sawamoto, M. Sequence Controlled Polymers: Synthesis, Self-Assembly, and Properties; American Chemical Society: Washington, DC, USA, 2014. [Google Scholar]
- Lutz, J.-F. Sequence-Controlled Polymers; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Solleder, S.C.; Schneider, R.V.; Wetzel, K.S.; Boukis, A.C.; Meier, M.A.R. Recent progress in the design of monodisperse, sequence-defined macromolecules. Macromol. Rapid Commun. 2017, 38, 1600711. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.A.R.; Barner-Kowollik, C. A new class of materials: Sequence-defined macromolecules and their emerging applications. Adv. Mater. 2019, 31, 1806027. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.L.; Sing, C.E. 100th Anniversary of macromolecular science viewpoint: Opportunities in the physics of sequence-defined polymers. ACS Macro Lett. 2020, 9, 216–225. [Google Scholar] [CrossRef]
- Yang, C.; Wu, K.B.; Deng, Y.; Yuan, J.; Niu, J. Geared toward applications: A perspective on functional sequence-controlled polymers. ACS Macro Lett. 2021, 10, 243–257. [Google Scholar] [CrossRef]
- Takizawa, K.; Nulwala, H.; Hu, J.; Yoshinaga, K.; Hawker, C.J. Molecularly defined (L)-lactic acid oligomers and polymers: Synthesis and characterization. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 5977–5990. [Google Scholar] [CrossRef]
- Mosca, S.; Wojcik, F.; Hartmann, L. Precise positioning of chiral building blocks in monodisperse, sequence-defined polyamides. Macromol. Rapid Commun. 2011, 32, 197–202. [Google Scholar] [CrossRef]
- Kanasty, R.L.; Vegas, A.J.; Ceo, L.M.; Maier, M.; Charisse, K.; Nair, J.K.; Langer, R.; Anderson, D.G. Sequence-defined oligomers from hydroxyproline building blocks for parallel synthesis applications. Angew. Chem. Int. Ed. 2016, 55, 9529–9533. [Google Scholar] [CrossRef]
- Núñez-Villanueva, D.; Hunter, C.A. Homochiral oligomers with highly flexible backbones form stable H-bonded duplexes. Chem. Sci. 2017, 8, 206–213. [Google Scholar] [CrossRef]
- Huang, Z.; Noble, B.B.; Corrigan, N.; Chu, Y.; Satoh, K.; Thomas, D.S.; Hawker, C.J.; Moad, G.; Kamigaito, M.; Coote, M.L.; et al. Discrete and stereospecific oligomers prepared by sequential and alternating single unit monomer insertion. J. Am. Chem. Soc. 2018, 140, 13392–13406. [Google Scholar] [CrossRef]
- Dong, R.; Liu, R.; Gaffney, P.R.J.; Schaepertoens, M.; Marchetti, P.; Williams, C.M.; Chen, R.; Livingston, A.G. Sequence-defined multifunctional polyethers via liquid-phase synthesis with molecular sieving. Nat. Chem. 2019, 11, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Mertens, C.; Soete, M.; Ślęzkowski, M.L.; Palmans, A.R.A.; Meijer, E.W.; Badi, N.; Du Prez, F.E. Stereocontrolled, multi-functional sequence-defined oligomers through automated synthesis. Polym. Chem. 2020, 11, 4271–4280. [Google Scholar] [CrossRef]
- Dobitz, S.; Aronoff, M.R.; Wennemers, H. Oligoprolines as molecular entities for controlling distance in biological and material sciences. Acc. Chem. Res. 2017, 50, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Fiala, T.; Barros, E.P.; Ebert, M.-O.; Ruijsenaars, E.; Riniker, S.; Wennemers, H. Frame shifts affect the stability of collagen triple helices. J. Am. Chem. Soc. 2022, 144, 18642–18649. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Sun, Y.; Ding, S. Stereocontrolled sequence-defined oligotriazoles through metal-free elongation strategies. Macromolecules 2021, 54, 9437–9444. [Google Scholar] [CrossRef]
- Zhang, X.; Gou, F.; Wang, X.; Wang, Y.; Ding, S. Easily functionalized and readable sequence-defined polytriazoles. ACS Macro Lett. 2021, 10, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Meng, C.; Ma, J.; Zhang, X.; Ding, S. Construction of sequence-defined polytriazoles by IrAAC and CuAAC reactions. Chem. Commun. 2020, 56, 3955–3958. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Wang, Y.; Ding, S. IrAAC-based construction of dual sequence-defined polytriazoles. Polym. Chem. 2021, 12, 3825–3831. [Google Scholar] [CrossRef]
- Niu, L.; Zhang, X.; Ding, S. Unsymmetrical growth synthesis of nontraditional dendrimers. Macromolecules 2022, 55, 3469–3475. [Google Scholar] [CrossRef]
- Niu, L.; Song, N.; Wang, X.; Ding, S. Internally functionalized dendrimers based on fully substituted 1,2,3-triazoles. Macromol. Rapid Commun. 2022, 43, 2200375. [Google Scholar] [CrossRef]
- Mei, J.; Leung, N.L.C.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Aggregation-induced emission: Together we shine, united we soar. Chem. Rev. 2015, 115, 11718–11940. [Google Scholar] [CrossRef]
- Shi, Y.; Yin, G.; Yan, Z.; Sang, P.; Wang, M.; Brzozowski, R.; Eswara, P.; Wojtas, L.; Zheng, Y.; Li, X.; et al. Helical sulfono-γ-AApeptides with aggregation-induced emission and circularly polarized luminescence. J. Am. Chem. Soc. 2019, 141, 12697–12706. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sang, P.; Yin, G.; Gao, R.; Liang, X.; Brzozowski, R.; Odom, T.; Eswara, P.; Zheng, Y.; Li, X.; et al. Aggregation-induced emissive and circularly polarized homogeneous sulfono-γ-AApeptide foldamers. Adv. Opt. Mater. 2020, 8, 1902122. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | m/z cal. | m/z obs. | Yield | Time | Overall Yield | |
|---|---|---|---|---|---|---|
| 4a | Boc-Ph-OH | 405.1995 | 405.1999 a | - | - | |
| 4a-H | H-Ph-OH | 96% | 6 h | |||
| 4a-OTs | Boc-Ph-OTs | 87% | 2 h | |||
| 5a | Boc-(Ph)2-OH | 713.3026 | 713.3039 b | 68% | 10 h | |
| 5a-H | H-(Ph)2-OH | 96% | 6 h | |||
| 5a-OTs | Boc-(Ph)2-OTs | 86% | 1.5 h | |||
| 6a | Boc-(Ph)4-OH | 632.2892 | 632.2893 c | 52% | 10 h | |
| 6a-H | H-(Ph)4-OH | 90% | 6 h | |||
| 6a-OTs | Boc-(Ph)4-OTs | 82% | 2 h | |||
| 8a | Boc-(Ph)8-OH | 1205.0420 | 1205.0420 c | 25% | 10 h | 7.3% |
| 4b | Boc-Bn-OH | 449.2223 | 449.2226 a | |||
| 4b-H | H-Bn-OH | 92% | 6 h | |||
| 4b-OTs | Boc-Bn-OTs | 84% | 1.5 h | |||
| 5b | Boc-(Bn)2-OH | 801.3551 | 801.3547 a | 76% | 10 h | |
| 5b-H | H-(Bn)2-OH | 94% | 6 h | |||
| 5b-OTs | Boc-(Bn)2-OTs | 82% | 1.5 h | |||
| 6b | Boc-(Bn)4-OH | 720.3416 | 720.3419 c | 59% | 10 h | |
| 6b-H | H-(Bn)4-OH | 90% | 6 h | |||
| 6b-OTs | Boc-(Bn)4-OTs | 86% | 2 h | |||
| 8b | Boc-(Bn)8-OH | 1403.6387 | 1403.6356 d | 22% | 15 h | 7.6% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Yu, T.; Ding, S. Iridium-Catalyzed Synthesis of Chiral 1,2,3-Triazoles Units and Precise Construction of Stereocontrolled Oligomers. Molecules 2023, 28, 3726. https://doi.org/10.3390/molecules28093726
Zhang X, Yu T, Ding S. Iridium-Catalyzed Synthesis of Chiral 1,2,3-Triazoles Units and Precise Construction of Stereocontrolled Oligomers. Molecules. 2023; 28(9):3726. https://doi.org/10.3390/molecules28093726
Chicago/Turabian StyleZhang, Xueyan, Tian Yu, and Shengtao Ding. 2023. "Iridium-Catalyzed Synthesis of Chiral 1,2,3-Triazoles Units and Precise Construction of Stereocontrolled Oligomers" Molecules 28, no. 9: 3726. https://doi.org/10.3390/molecules28093726