
Design and Construction of a Synthetic Nanobody Library: Testing Its Potential with a Single Selection Round Strategy

 , , , , and
, , , , and 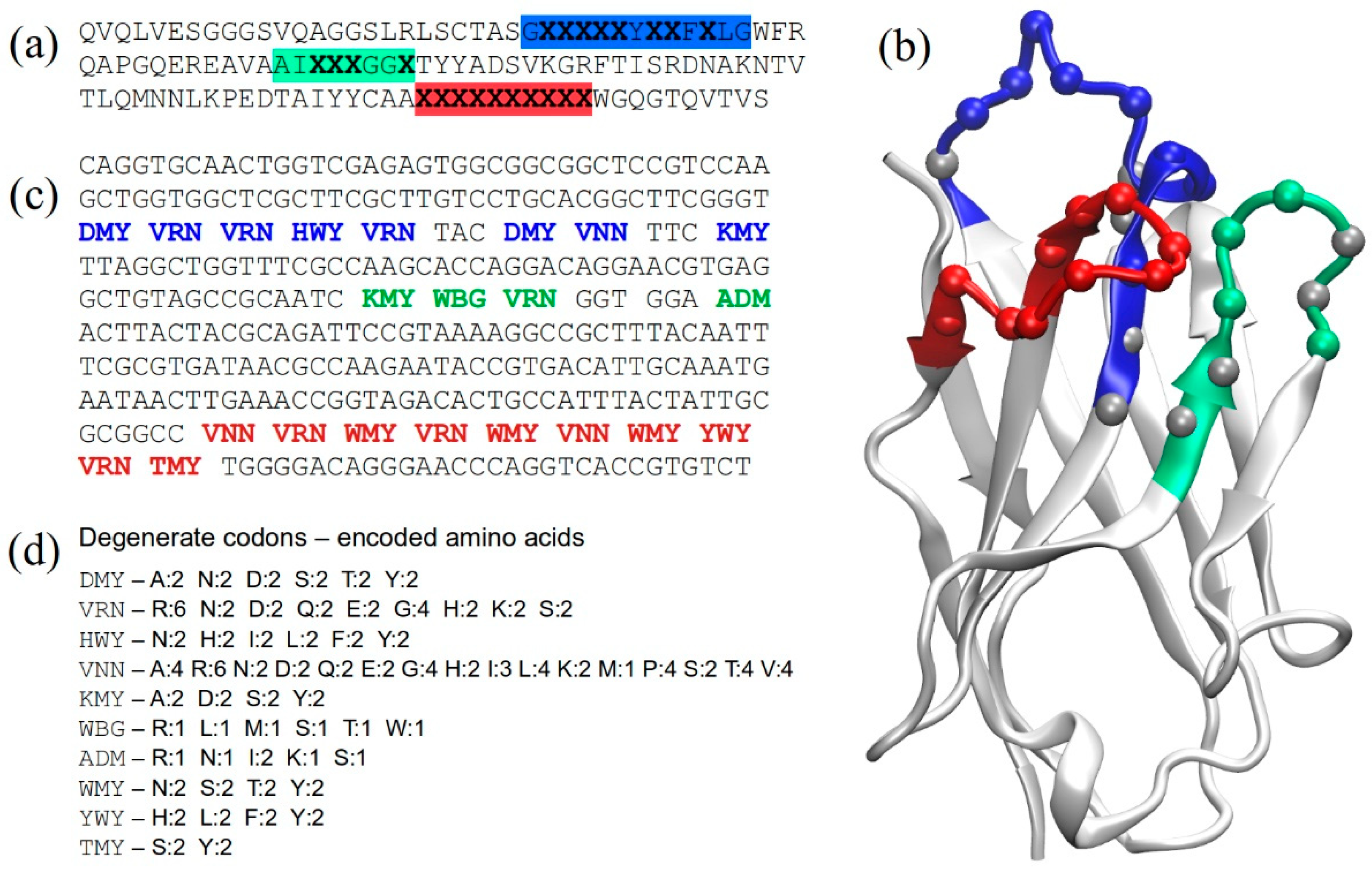{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Structure-Based Library Design
2.2. Library Construction
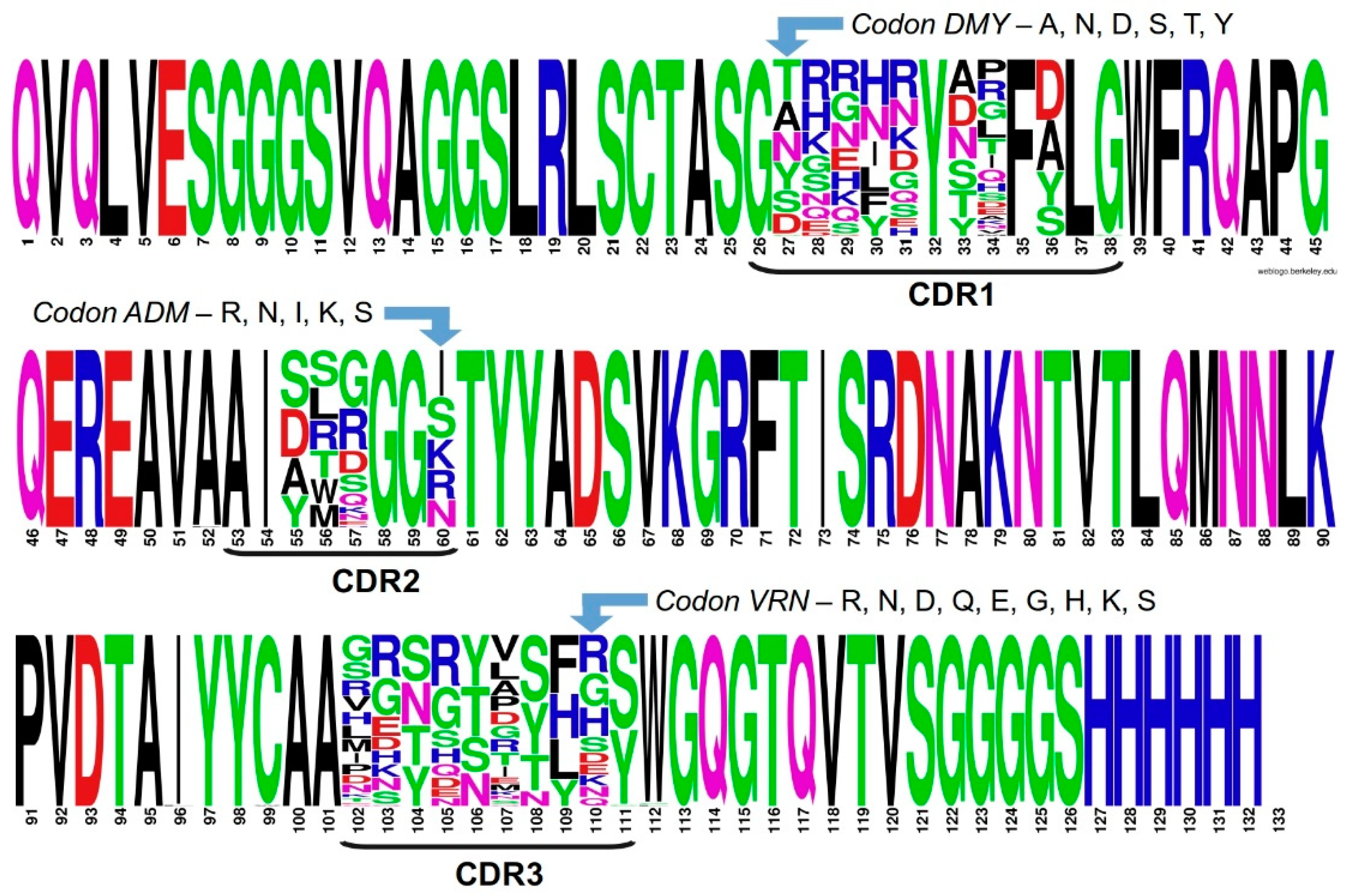
2.3. Assessing Library Quality and Diversity
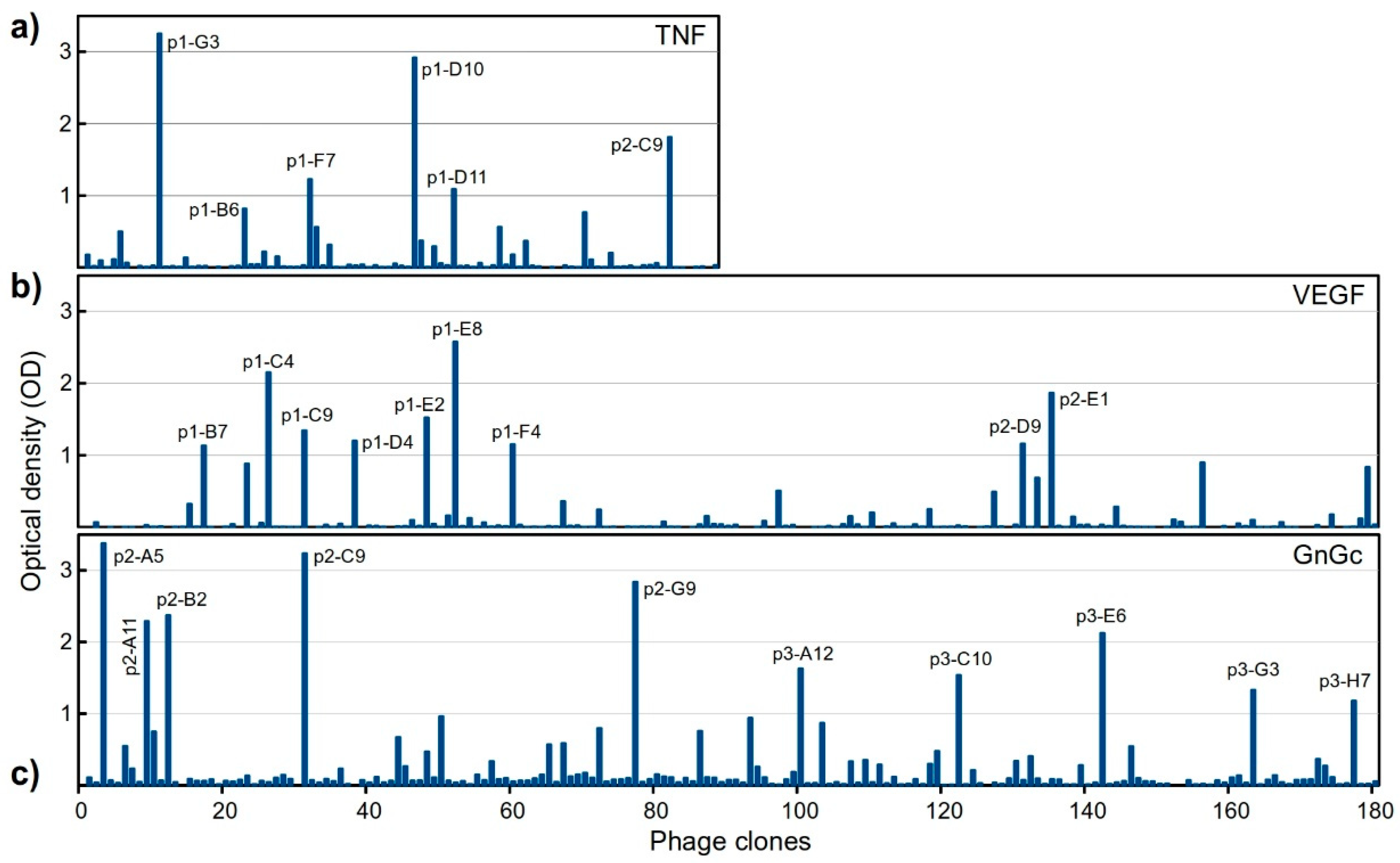
2.4. Library Screening
2.4.1. Selection of Antigen-Specific Binders in a Single Round
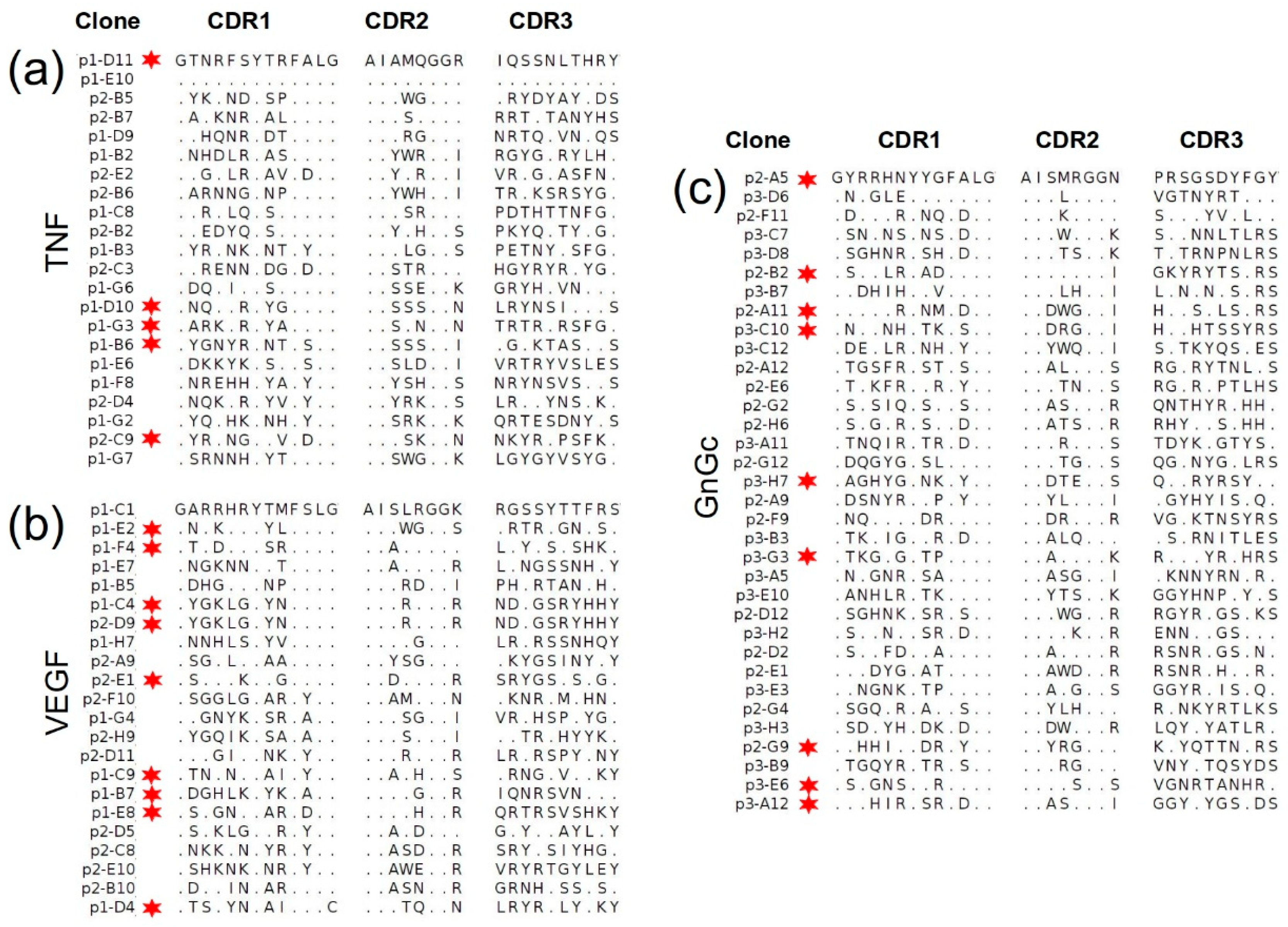
2.4.2. Sequencing of Selected Groups of Phage Clones
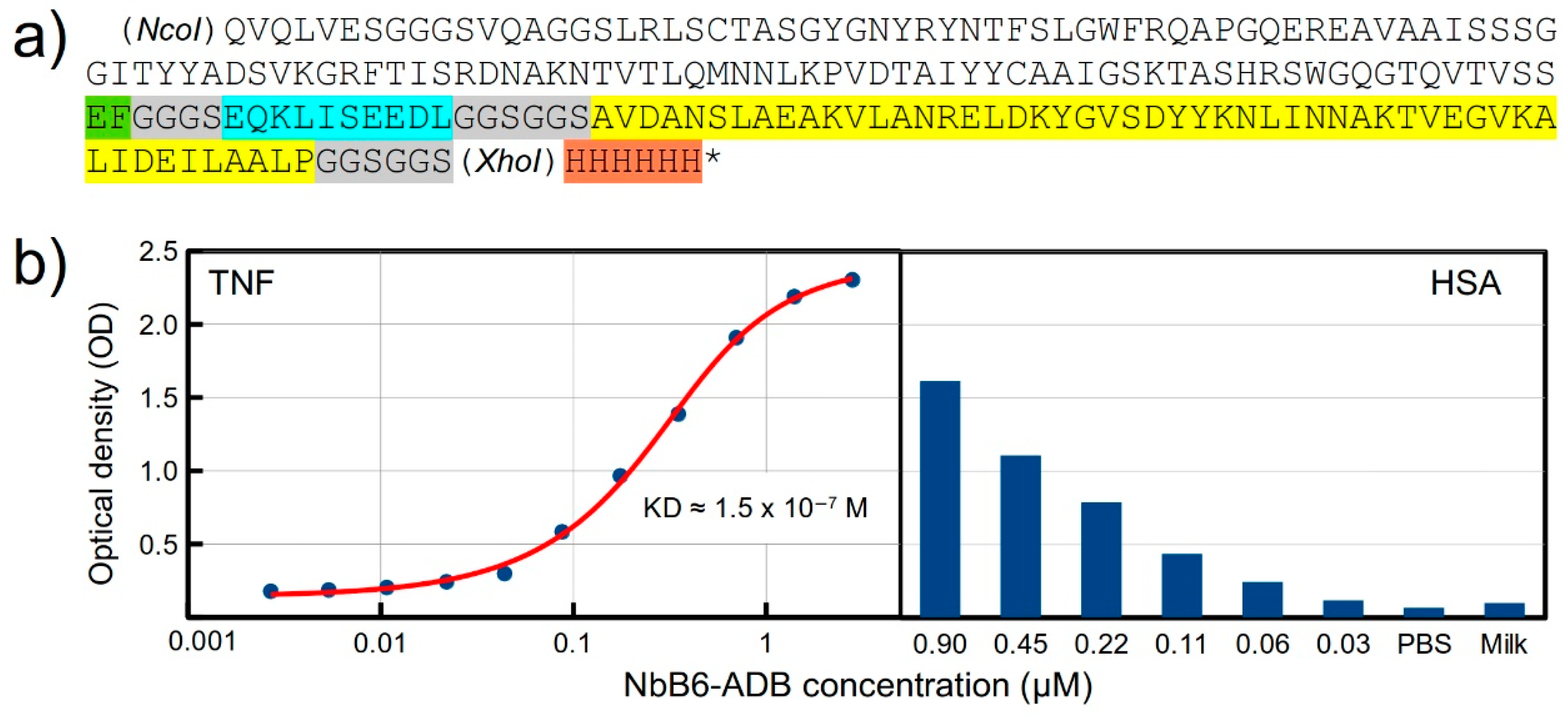
2.5. Design and Expression of a Recombinant Fusion Protein with an Anti-TNF Nb
3. Discussion
4. Materials and Methods
4.1. In Silico Design and Analyses
4.2. Library Construction
4.3. Library Screening
4.4. Binding Assays to Detect Positive Phage Clones
4.5. Sanger Sequencing
4.6. Production of Recombinant Fusion Protein
4.7. Binding Assay for the Fusion Protein
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally Occurring Antibodies Devoid of Light Chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Molina-Abad, E.; Moreno, E. NbThermo: A New Thermostability Database for Nanobodies. Database 2023, baad021. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh-Ghassabeh, G.; Devoogdt, N.; De Pauw, P.; Vincke, C.; Muyldermans, S. Nanobodies and Their Potential Applications. Nanomedicine 2013, 8, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C. Nanobody Approval Gives Domain Antibodies a Boost. Nat. Rev. Drug. Discov. 2019, 18, 485–487. [Google Scholar] [CrossRef]
- Keam, S.J. Ozoralizumab: First Approval. Drugs. 2023, 83, 87–92. [Google Scholar] [CrossRef]
- Muyldermans, S. A Guide to: Generation and Design of Nanobodies. FEBS. J. 2021, 288, 2084–2102. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Molina-Zapata, A.; Pose, A.G.; Moreno, E. Structural Insights into the Design of Synthetic Nanobody Libraries. Molecules 2022, 27, 2198. [Google Scholar] [CrossRef]
- Moutel, S.; Bery, N.; Bernard, V.; Keller, L.; Lemesre, E.; de Marco, A.; Ligat, L.; Rain, J.-C.; Favre, G.; Olichon, A.; et al. NaLi-H1: A Universal Synthetic Library of Humanized Nanobodies Providing Highly Functional Antibodies and Intrabodies. Elife 2016, 5, e16228. [Google Scholar] [CrossRef]
- McMahon, C.; Baier, A.S.; Pascolutti, R.; Wegrecki, M.; Zheng, S.; Ong, J.X.; Erlandson, S.C.; Hilger, D.; Rasmussen, S.G.F.; Ring, A.M.; et al. Yeast Surface Display Platform for Rapid Discovery of Conformationally Selective Nanobodies. Nat. Struct. Mol. Biol. 2018, 25, 289–296. [Google Scholar] [CrossRef]
- Zimmermann, I.; Egloff, P.; Hutter, C.A.; Arnold, F.M.; Stohler, P.; Bocquet, N.; Hug, M.N.; Huber, S.; Siegrist, M.; Hetemann, L.; et al. Synthetic Single Domain Antibodies for the Conformational Trapping of Membrane Proteins. Elife 2018, 7, e34317. [Google Scholar] [CrossRef]
- Sevy, A.M.; Chen, M.-T.; Castor, M.; Sylvia, T.; Krishnamurthy, H.; Ishchenko, A.; Hsieh, C.-M. Structure- and Sequence-Based Design of Synthetic Single-Domain Antibody Libraries. Protein. Eng. Des. Selection. 2020, 33, gzaa028. [Google Scholar] [CrossRef]
- Zimmermann, I.; Egloff, P.; Hutter, C.A.J.; Kuhn, B.T.; Bräuer, P.; Newstead, S.; Dawson, R.J.P.; Geertsma, E.R.; Seeger, M.A. Generation of Synthetic Nanobodies against Delicate Proteins. Nat. Protoc. 2020, 15, 1707–1741. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Y.; Su, W.; Li, S. Construction of Synthetic Nanobody Library in Mammalian Cells by DsDNA-Based Strategies. Chem. BioChem. 2021, 22, 2957–2965. [Google Scholar] [CrossRef]
- Chen, X.; Gentili, M.; Hacohen, N.; Regev, A. A Cell-Free Nanobody Engineering Platform Rapidly Generates SARS-CoV-2 Neutralizing Nanobodies. Nat. Commun. 2021, 12, 5506. [Google Scholar] [CrossRef]
- Moreno, E.; Valdés-Tresanco, M.S.; Molina-Zapata, A.; Sánchez-Ramos, O. Structure-Based Design and Construction of a Synthetic Phage Display Nanobody Library. BMC. Res. Notes. 2022, 15, 124. [Google Scholar] [CrossRef]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular Basis for the Preferential Cleft Recognition by Dromedary Heavy-Chain Antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef]
- Uchański, T.; Pardon, E.; Steyaert, J. Nanobodies to Study Protein Conformational States. Curr. Opin. Struct. Biol. 2020, 60, 117–123. [Google Scholar] [CrossRef]
- Shi, Z.; Li, X.; Wang, L.; Sun, Z.; Zhang, H.; Chen, X.; Cui, Q.; Qiao, H.; Lan, Z.; Zhang, X.; et al. Structural Basis of Nanobodies Neutralizing SARS-CoV-2 Variants. Structure 2022, 30, 707–720.e5. [Google Scholar] [CrossRef]
- Conrath, K.E.; Lauwereys, M.; Galleni, M.; Matagne, A.; Frère, J.-M.; Kinne, J.; Wyns, L.; Muyldermans, S. β-Lactamase Inhibitors Derived from Single-Domain Antibody Fragments Elicited in the Camelidae. Antimicrob. Agents. Chemother. 2001, 45, 2807–2812. [Google Scholar] [CrossRef]
- Dumoulin, M.; Conrath, K.; Van Meirhaeghe, A.; Meersman, F.; Heremans, K.; Frenken, L.G.J.; Muyldermans, S.; Wyns, L.; Matagne, A. Single-Domain Antibody Fragments with High Conformational Stability. Protein. Sci. 2002, 11, 500–515. [Google Scholar] [CrossRef]
- Saerens, D.; Pellis, M.; Loris, R.; Pardon, E.; Dumoulin, M.; Matagne, A.; Wyns, L.; Muyldermans, S.; Conrath, K. Identification of a Universal VHH Framework to Graft Non-Canonical Antigen-Binding Loops of Camel Single-Domain Antibodies. J. Mol. Biol. 2005, 352, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Meng, W.; Guo, H.; Pan, W.; Liu, J.; Peng, T.; Chen, L.; Chen, C.-Y. Potent Neutralization of Influenza A Virus by a Single-Domain Antibody Blocking M2 Ion Channel Protein. PLoS ONE 2011, 6, e28309. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, G.; Hu, Y.; Ou, W.; Wan, Y. Construction of a Synthetic Phage-Displayed Nanobody Library with CDR3 Regions Randomized by Trinucleotide Cassettes for Diagnostic Applications. J. Transl. Med. 2014, 12, 343. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Liu, X.; Wang, C.; Zhang, X.; Li, X.; Hou, J.; Ren, L.; Jin, Q.; Wang, J.; Yang, W. Humanized Single Domain Antibodies Neutralize SARS-CoV-2 by Targeting the Spike Receptor Binding Domain. Nat. Commun. 2020, 11, 4528. [Google Scholar] [CrossRef]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General Strategy to Humanize a Camelid Single-Domain Antibody and Identification of a Universal Humanized Nanobody Scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Nomenclature for Incompletely Specified Bases in Nucleic Acid Sequences: Rcommendations 1984. Nucleic Acids Res. 1985, 13, 3021–3030. [Google Scholar] [CrossRef]
- Hoogenboom, H.R.; Griffiths, A.D.; Johnson, K.S.; Chiswell, D.J.; Hudson, P.; Winter, G. Multi-Subunit Proteins on the Surface of Filamentous Phage: Methodologies for Displaying Antibody (Fab) Heavy and Light Chains. Nucleic Acids Res. 1991, 19, 4133–4137. [Google Scholar] [CrossRef]
- van Loo, G.; Bertrand, M.J.M. Death by TNF: A Road to Inflammation. Nat. Rev. Immunol. 2022, 15, 1–15. [Google Scholar] [CrossRef]
- Leone, G.M.; Mangano, K.; Petralia, M.C.; Nicoletti, F.; Fagone, P. Past, Present and (Foreseeable) Future of Biological Anti-TNF Alpha Therapy. J. Clin. Med. 2023, 12, 1630. [Google Scholar] [CrossRef]
- Ghalehbandi, S.; Yuzugulen, J.; Pranjol, M.Z.I.; Pourgholami, M.H. The Role of VEGF in Cancer-Induced Angiogenesis and Research Progress of Drugs Targeting VEGF. Eur. J. Pharmacol. 2023, 175586. [Google Scholar] [CrossRef]
- Arezumand, R.; Alibakhshi, A.; Ranjbari, J.; Ramazani, A.; Muyldermans, S. Nanobodies As Novel Agents for Targeting Angiogenesis in Solid Cancers. Front. Immunol. 2017, 8, 1746. [Google Scholar] [CrossRef]
- Dennis, M.S.; Zhang, M.; Meng, Y.G.; Kadkhodayan, M.; Kirchhofer, D.; Combs, D.; Damico, L.A. Albumin Binding as a General Strategy for Improving the Pharmacokinetics of Proteins. J. Biolo. Chem. 2002, 277, 35035–35043. [Google Scholar] [CrossRef]
- Jonsson, A.; Dogan, J.; Herne, N.; Abrahmsen, L.; Nygren, P.-A. Engineering of a Femtomolar Affinity Binding Protein to Human Serum Albumin. Protein Eng. Des. Sel. 2008, 21, 515–527. [Google Scholar] [CrossRef]
- Johansson, M.U.; Frick, I.-M.; Nilsson, H.; Kraulis, P.J.; Hober, S.; Jonasson, P.; Linhult, M.; Nygren, P.-Å.; Uhlén, M.; Björck, L.; et al. Structure, Specificity, and Mode of Interaction for Bacterial Albumin-Binding Modules. J. Biol. Chem. 2002, 277, 8114–8120. [Google Scholar] [CrossRef]
- Eble, J.A. Titration ELISA as a Method to Determine the Dissociation Constant of Receptor Ligand Interaction. J. Vis. Exp. 2018, 15, 57334. [Google Scholar] [CrossRef]
- Lakzaei, M.; Rasaee, M.J.; Fazaeli, A.A.; Aminian, M. A Comparison of Three Strategies for Biopanning of Phage-scFv Library against Diphtheria Toxin. J. Cell. Physiol. 2019, 234, 9486–9494. [Google Scholar] [CrossRef]
- Jaroszewicz, W.; Morcinek-Orłowska, J.; Pierzynowska, K.; Gaffke, L.; Węgrzyn, G. Phage Display and Other Peptide Display Technologies. FEMS Microbiol. Rev. 2022, 46, fuab052. [Google Scholar] [CrossRef]
- Scarrone, M.; González-Techera, A.; Alvez-Rosado, R.; Delfin-Riela, T.; Modernell, Á.; González-Sapienza, G.; Lassabe, G. Development of Anti-Human IgM Nanobodies as Universal Reagents for General Immunodiagnostics. New Biotechnol. 2021, 64, 9–16. [Google Scholar] [CrossRef]
- Roshan, R.; Naderi, S.; Behdani, M.; Cohan, R.A.; Ghaderi, H.; Shokrgozar, M.A.; Golkar, M.; Kazemi-Lomedasht, F. Isolation and Characterization of Nanobodies against Epithelial Cell Adhesion Molecule as Novel Theranostic Agents for Cancer Therapy. Mol. Immunol. 2021, 129, 70–77. [Google Scholar] [CrossRef]
- Kazemi-Lomedasht, F.; Behdani, M.; Bagheri, K.P.; Habibi-Anbouhi, M.; Abolhassani, M.; Arezumand, R.; Shahbazzadeh, D.; Mirzahoseini, H. Inhibition of Angiogenesis in Human Endothelial Cell Using VEGF Specific Nanobody. Mol. Immunol. 2015, 65, 58–67. [Google Scholar] [CrossRef]
- Wu, M.; Tu, Z.; Huang, F.; He, Q.; Fu, J.; Li, Y. Panning Anti-LPS Nanobody as a Capture Target to Enrich Vibrio Fluvialis. Biochem. Biophys. Res. Commun. 2019, 512, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Lunder, M.; Bratkovič, T.; Urleb, U.; Kreft, S.; Štrukelj, B. Ultrasound in Phage Display: A New Approach to Nonspecific Elution. Biotechniques 2008, 44, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Tonikian, R.; Zhang, Y.; Boone, C.; Sidhu, S.S. Identifying Specificity Profiles for Peptide Recognition Modules from Phage-Displayed Peptide Libraries. Nat. Protoc. 2007, 2, 1368–1386. [Google Scholar] [CrossRef]
- Chen, G.; Sidhu, S.S. Design and Generation of Synthetic Antibody Libraries for Phage Display. In Monoclonal Antibodies: Methods and Protocols; Ossipow, V., Fischer, N., Eds.; Humana Press: Totowa, NJ, USA, 2014; pp. 113–131. [Google Scholar]
- Contreras, M.A.; Macaya, L.; Neira, P.; Camacho, F.; González, A.; Acosta, J.; Montesino, R.; Toledo, J.R.; Sánchez, O. New Insights on the Interaction Mechanism of RhTNFα with Its Antagonists Adalimumab and Etanercept. Biochem. J. 2020, 477, 3299–3311. [Google Scholar] [CrossRef]
- Parra, N.C.; Mansilla, R.; Aedo, G.; Vispo, N.S.; González-Horta, E.E.; González-Chavarría, I.; Castillo, C.; Camacho, F.; Sánchez, O. Expression and Characterization of Human Vascular Endothelial Growth Factor Produced in SiHa Cells Transduced with Adenoviral Vector. Protein J. 2019, 38, 693–703. [Google Scholar] [CrossRef]
- Beltrán-Ortiz, C.E.; Starck-Mendez, M.F.; Fernández, Y.; Farnós, O.; González, E.E.; Rivas, C.I.; Camacho, F.; Zuñiga, F.A.; Toledo, J.R.; Sánchez, O. Expression and Purification of the Surface Proteins from Andes Virus. Protein. Expr. Purif. 2017, 139, 63–70. [Google Scholar] [CrossRef]
- Baek, H.; Suk, K.; Kim, Y.; Cha, S. An Improved Helper Phage System for Efficient Isolation of Specific Antibody Molecules in Phage Display. Nucleic. Acids. Res. 2002, 30, e18. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contreras, M.A.; Serrano-Rivero, Y.; González-Pose, A.; Salazar-Uribe, J.; Rubio-Carrasquilla, M.; Soares-Alves, M.; Parra, N.C.; Camacho-Casanova, F.; Sánchez-Ramos, O.; Moreno, E. Design and Construction of a Synthetic Nanobody Library: Testing Its Potential with a Single Selection Round Strategy. Molecules 2023, 28, 3708. https://doi.org/10.3390/molecules28093708
Contreras MA, Serrano-Rivero Y, González-Pose A, Salazar-Uribe J, Rubio-Carrasquilla M, Soares-Alves M, Parra NC, Camacho-Casanova F, Sánchez-Ramos O, Moreno E. Design and Construction of a Synthetic Nanobody Library: Testing Its Potential with a Single Selection Round Strategy. Molecules. 2023; 28(9):3708. https://doi.org/10.3390/molecules28093708
Chicago/Turabian StyleContreras, María Angélica, Yunier Serrano-Rivero, Alaín González-Pose, Julieta Salazar-Uribe, Marcela Rubio-Carrasquilla, Matheus Soares-Alves, Natalie C. Parra, Frank Camacho-Casanova, Oliberto Sánchez-Ramos, and Ernesto Moreno. 2023. "Design and Construction of a Synthetic Nanobody Library: Testing Its Potential with a Single Selection Round Strategy" Molecules 28, no. 9: 3708. https://doi.org/10.3390/molecules28093708