
Synergy of Small Antiviral Molecules on a Black-Phosphorus Nanocarrier: Machine Learning and Quantum Chemical Simulation Insights

 , , ,
, , ,
Abstract
:1. Introduction
2. Outcomes and Discussion
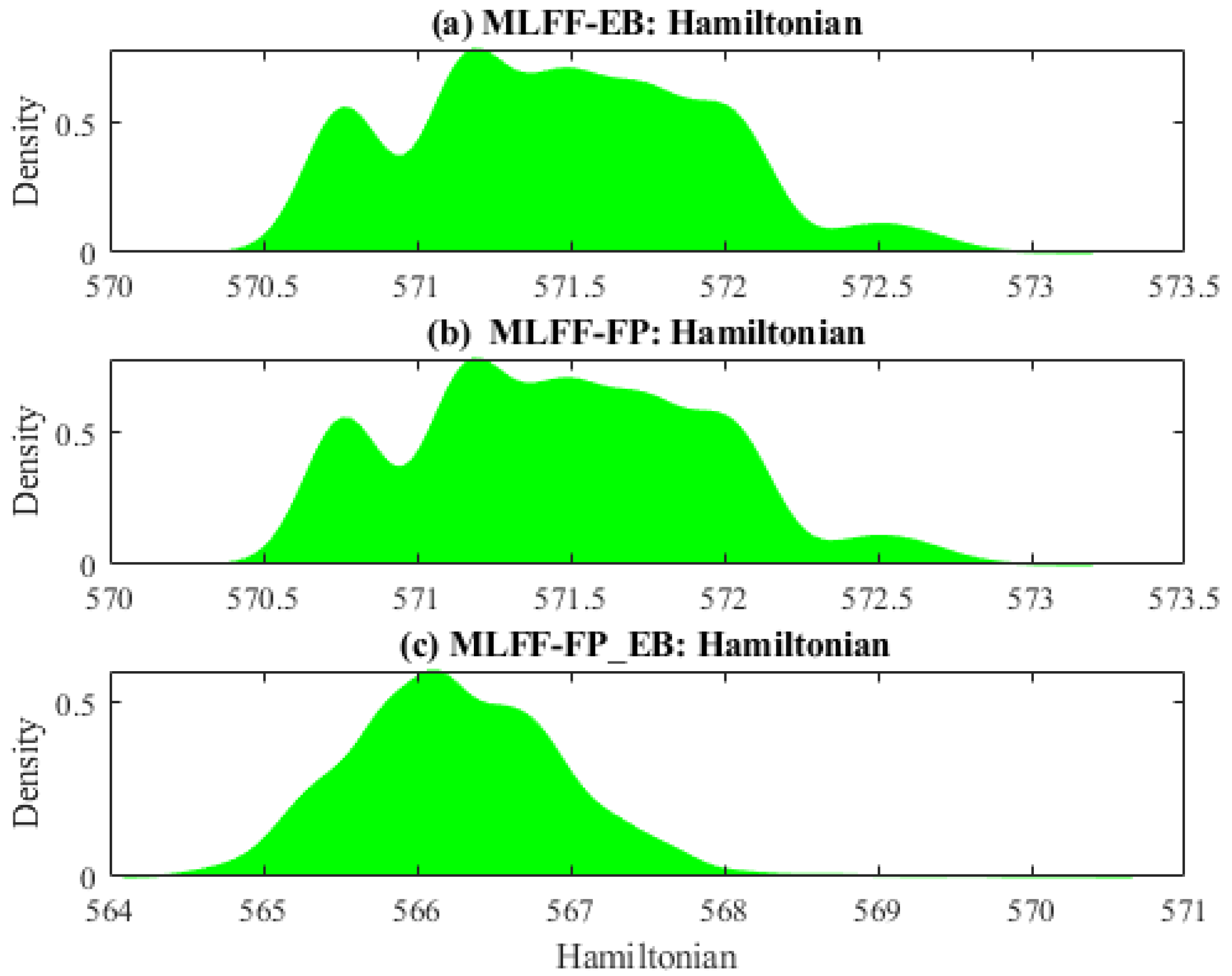
2.1. Data Description
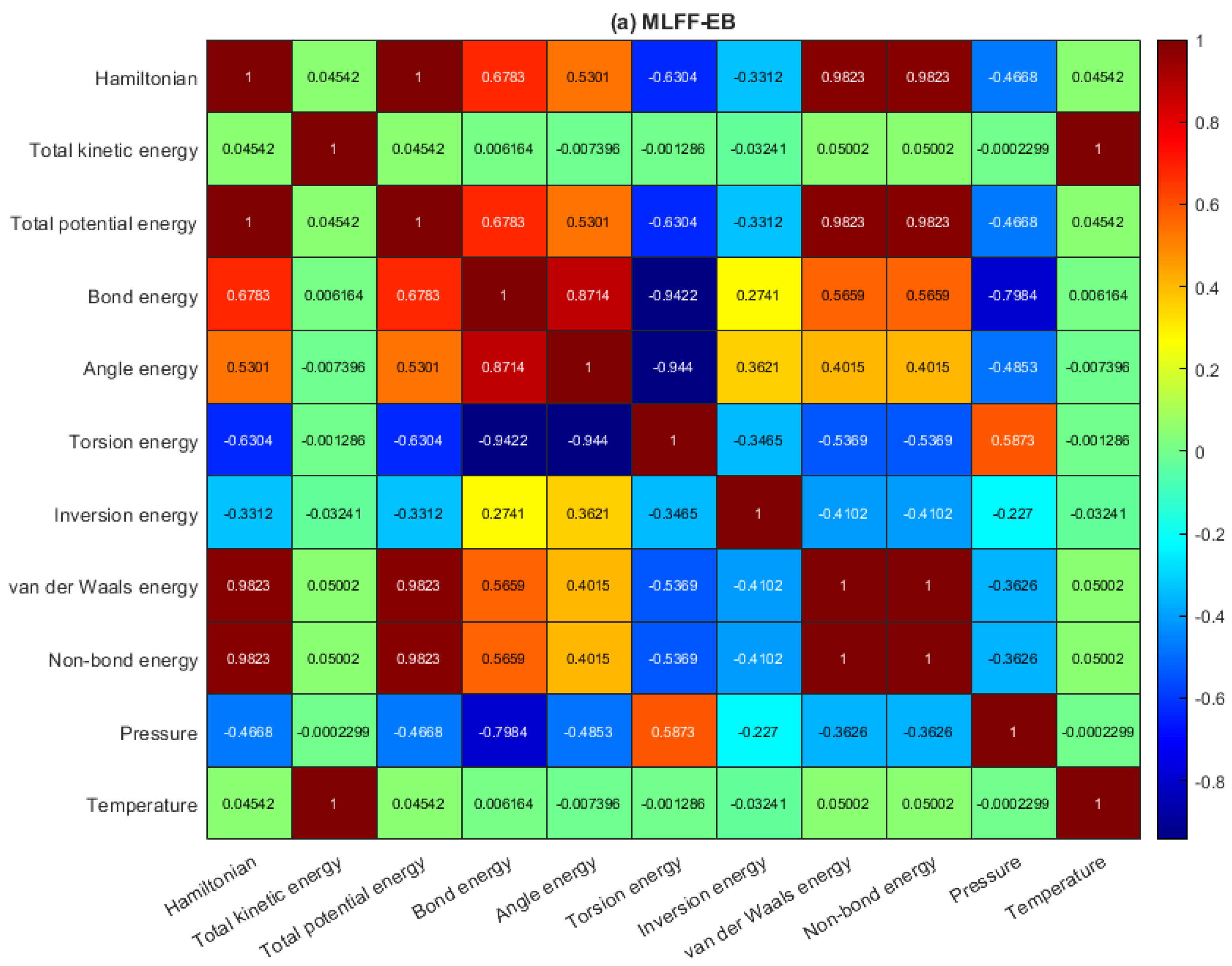
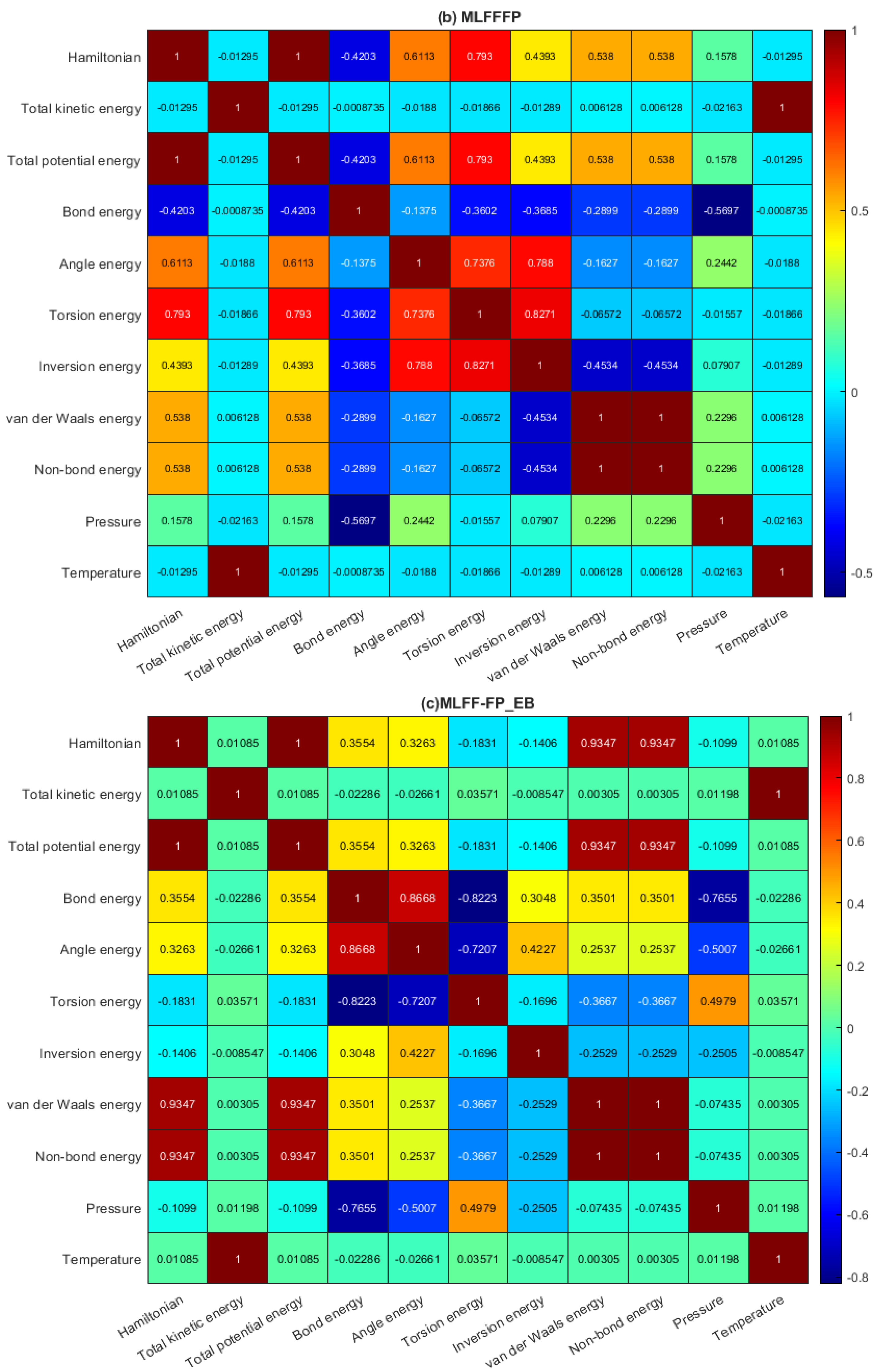
2.2. Data Analysis
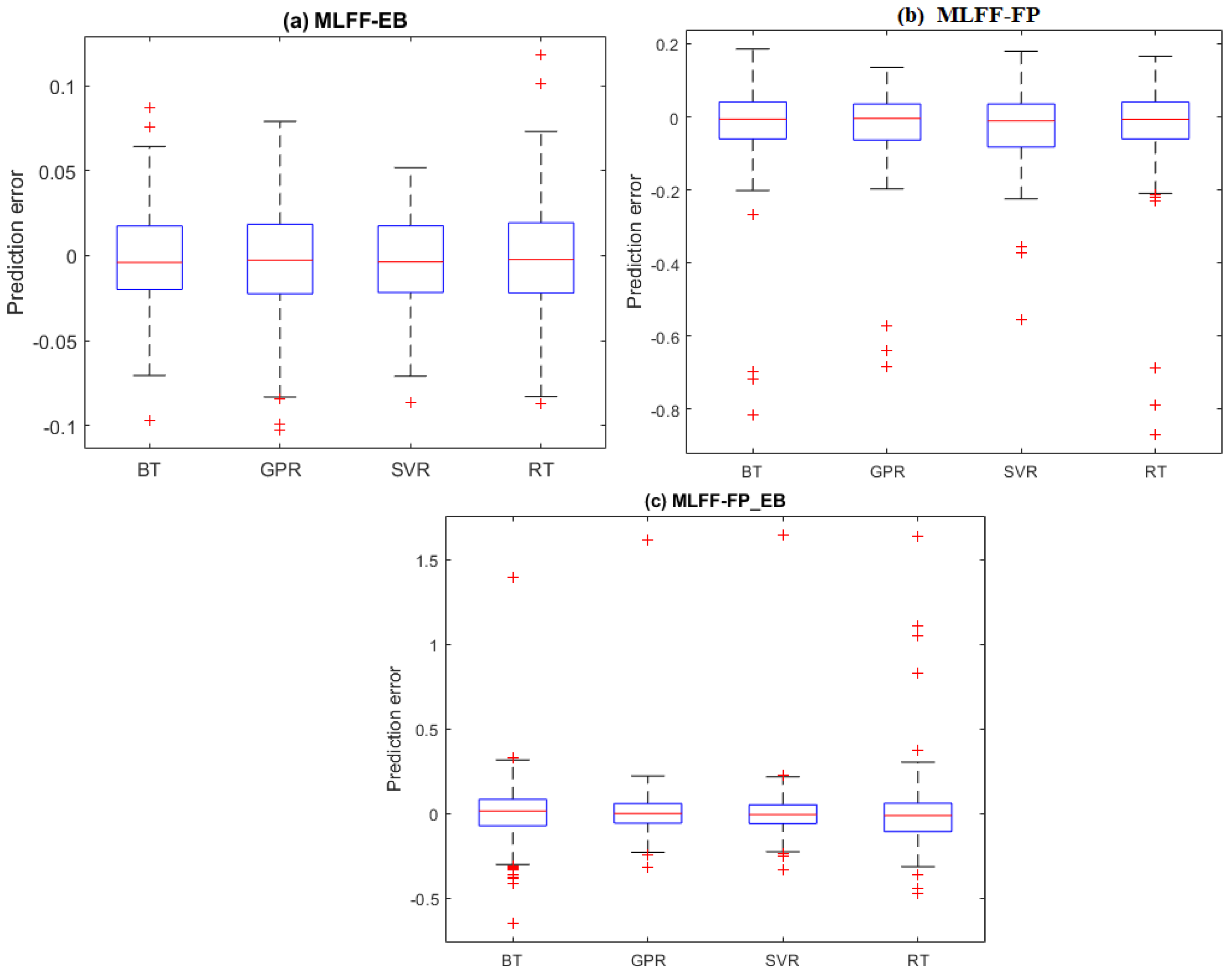
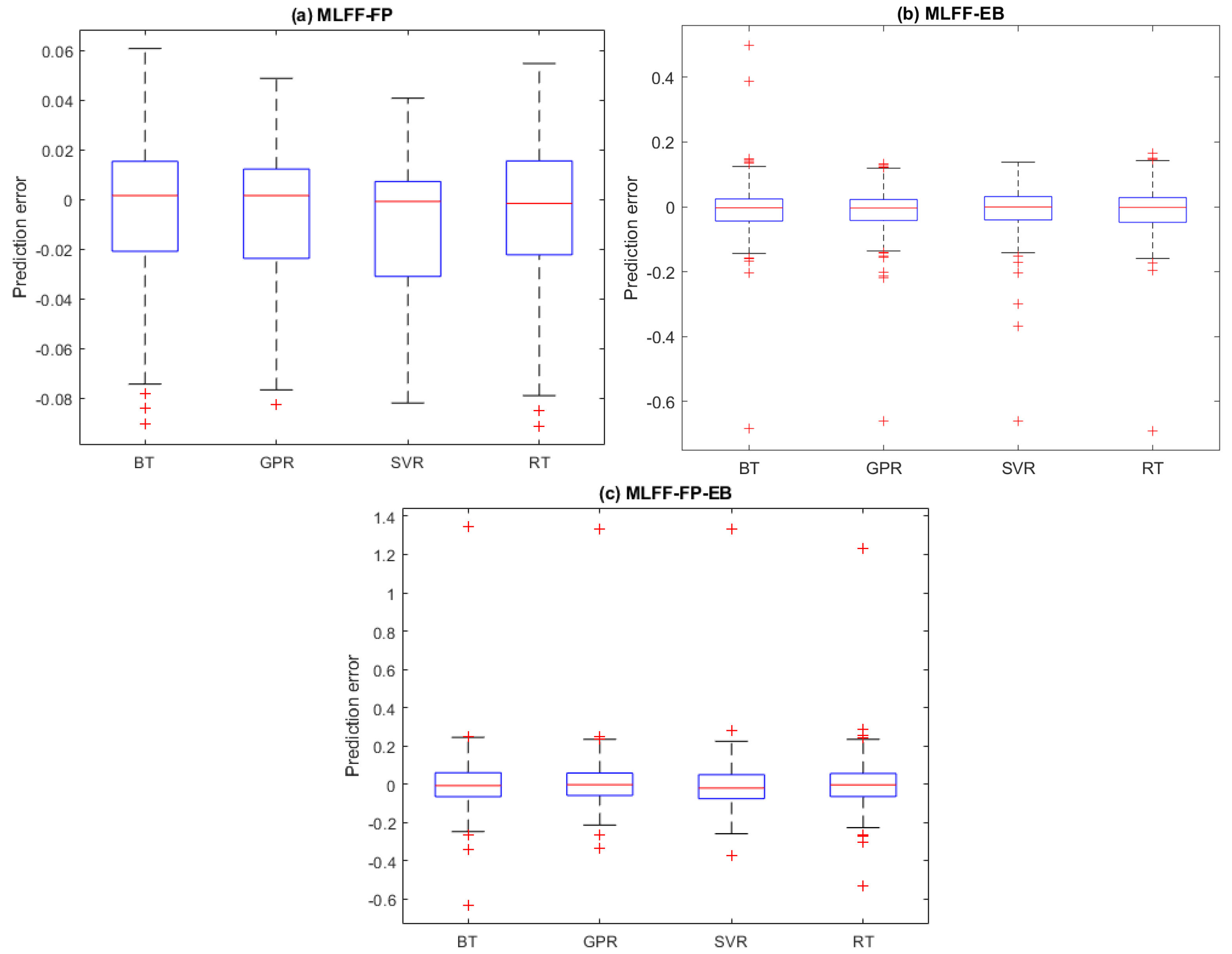
2.3. Prediction Results
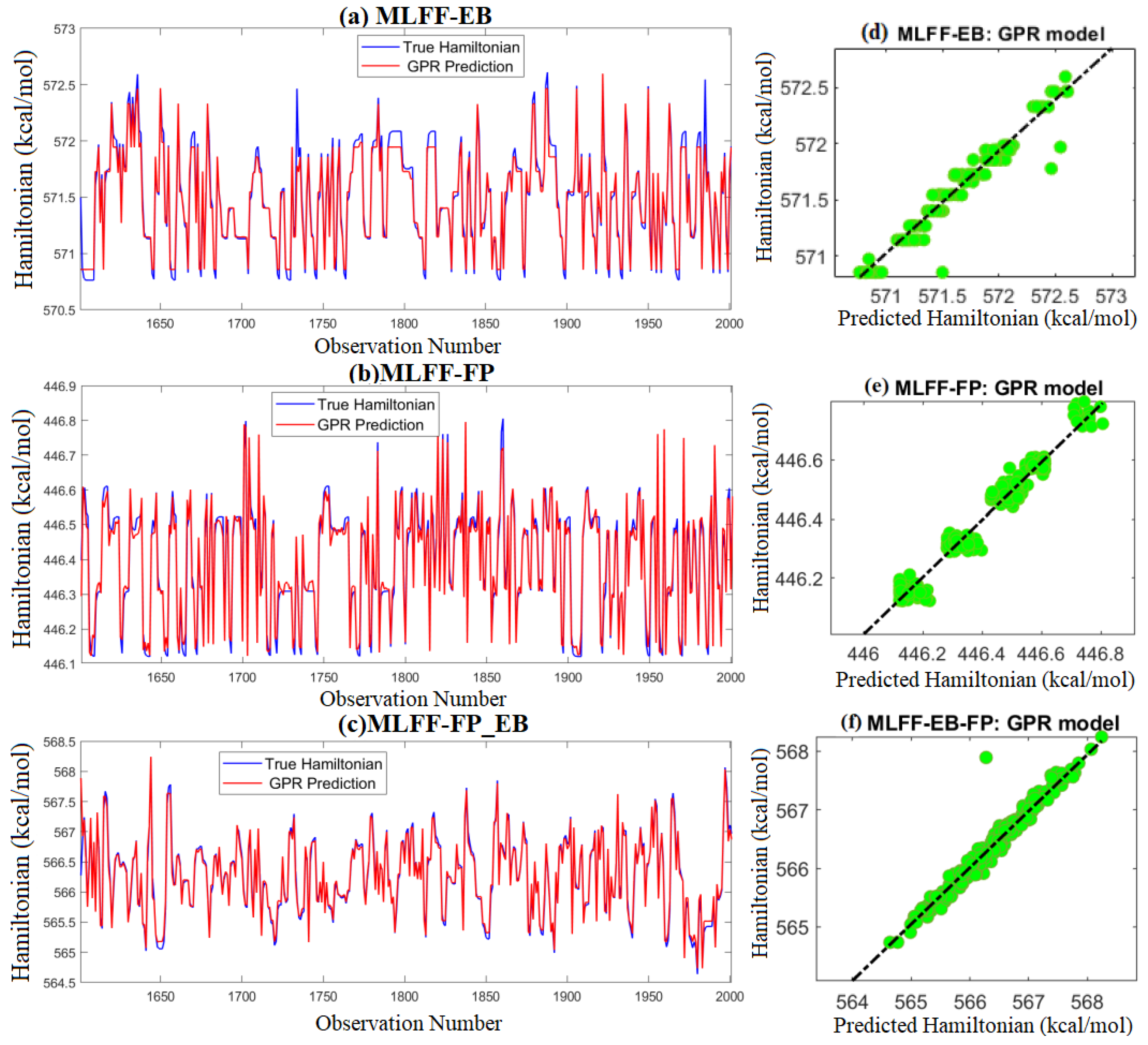
2.3.1. Hamiltonian Energy Prediction
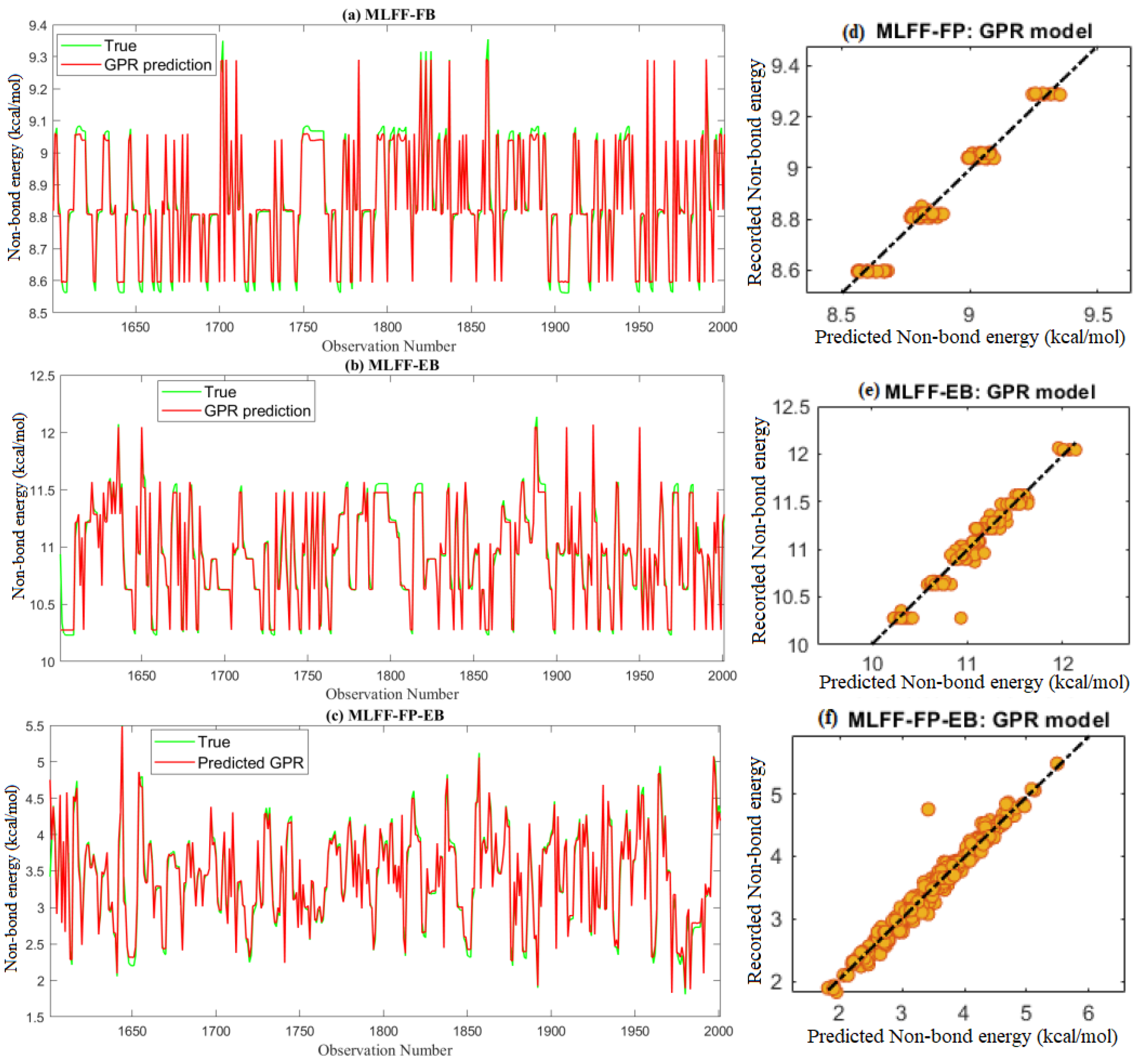
2.3.2. Non-Bond Energy Prediction
3. Pharmachemical Implications
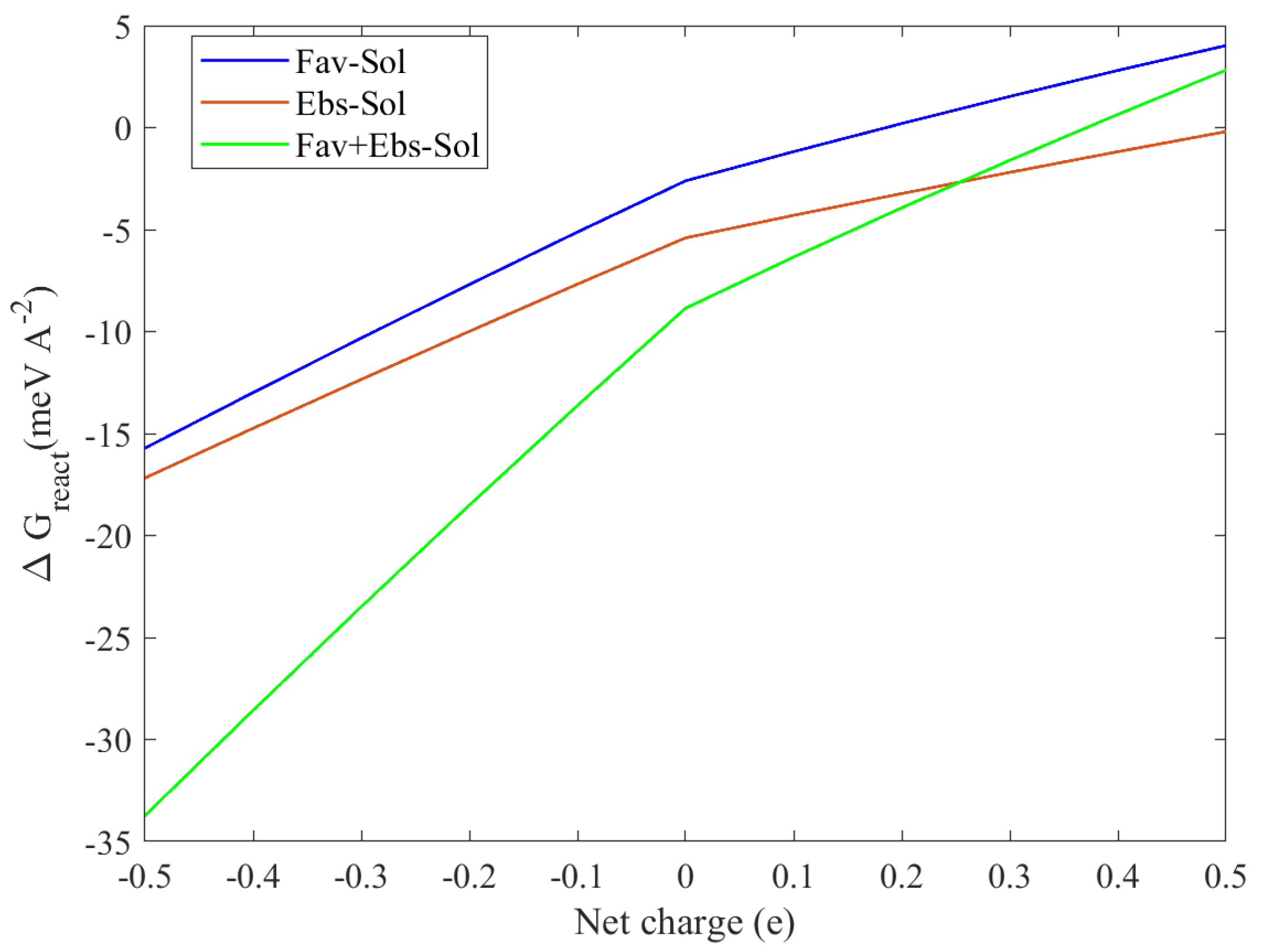
3.1. Drug Release
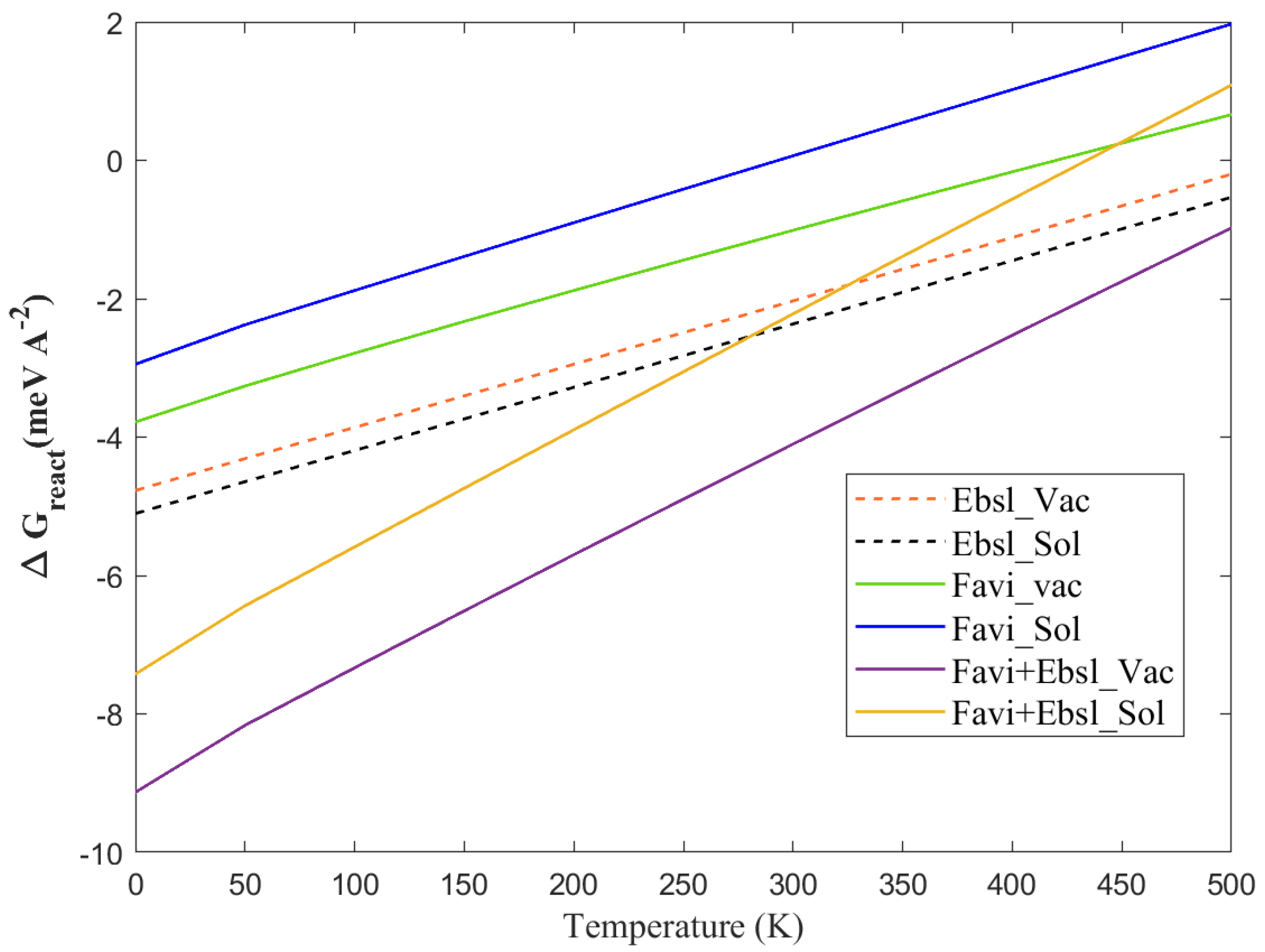
3.1.1. The pH Sensitivity
3.1.2. Thermotherapy Properties
4. Materials and Methods
4.1. Molecular Dynamics Simulations
4.2. Gaussian Process Regressor
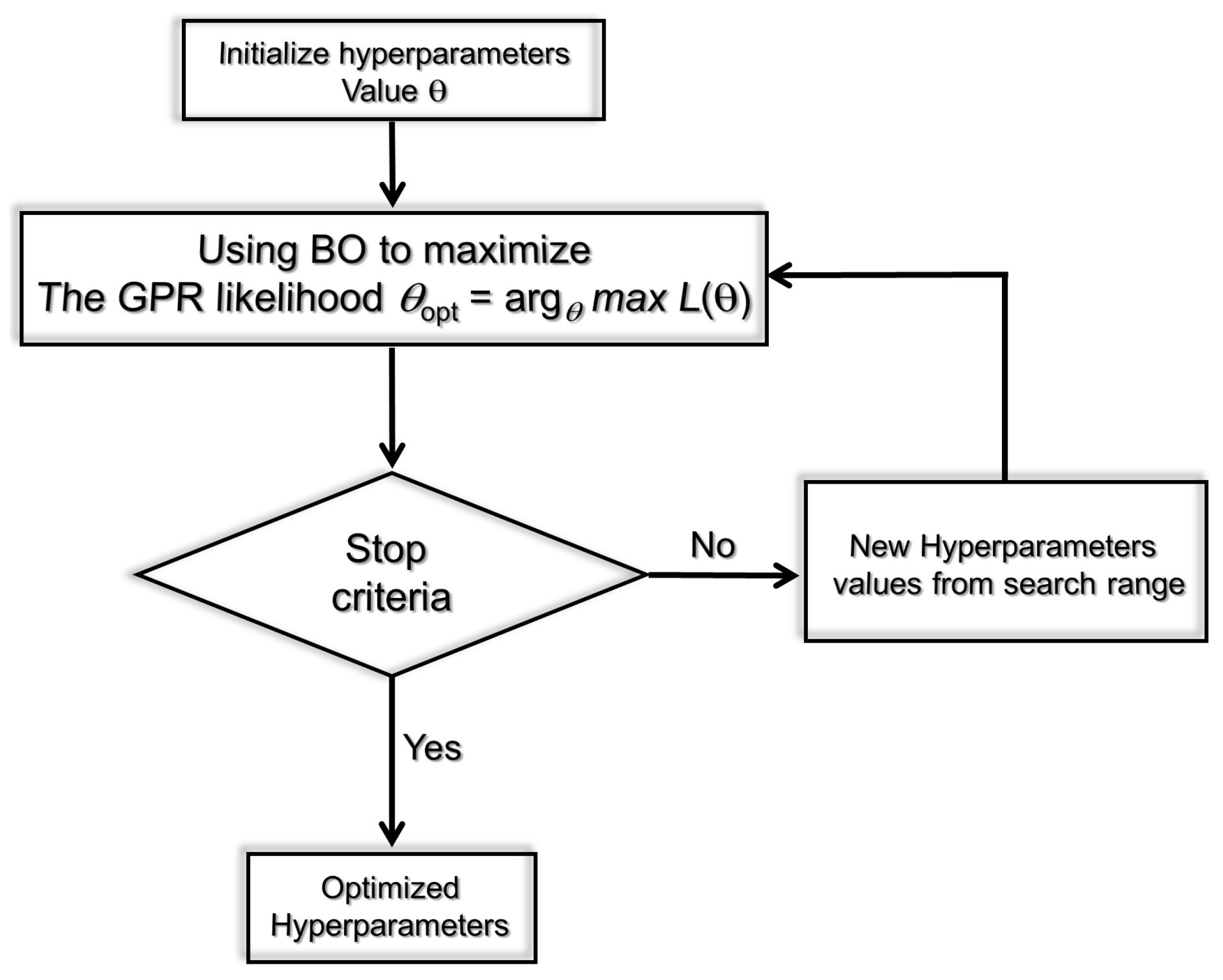
4.3. Bayesian Optimization Procedure
4.4. SVR Models
4.5. Bagged Tree Model
4.6. Evaluation Metrics
- RMSE: Root Mean Squared Error measures the differences between predicted and actual values. It is calculated by taking the square root of the average of the squared differences between the predicted and actual values.
- MAPE: Mean Absolute Percentage Error is a measure of the accuracy of a model in predicting values. It is calculated as the average of the absolute percentage difference between predicted and actual values.
- MAE: Mean Absolute Error is a measure of the average magnitude of the errors in a set of predictions, without considering their direction. It is calculated as the average of the absolute differences between predicted and actual values.
- R-squared: is a statistical measure that represents the proportion of the variance for a dependent variable that is explained by an independent variable or variables in a regression model. It is also known as the coefficient of determination, and its value ranges between 0 and 1.
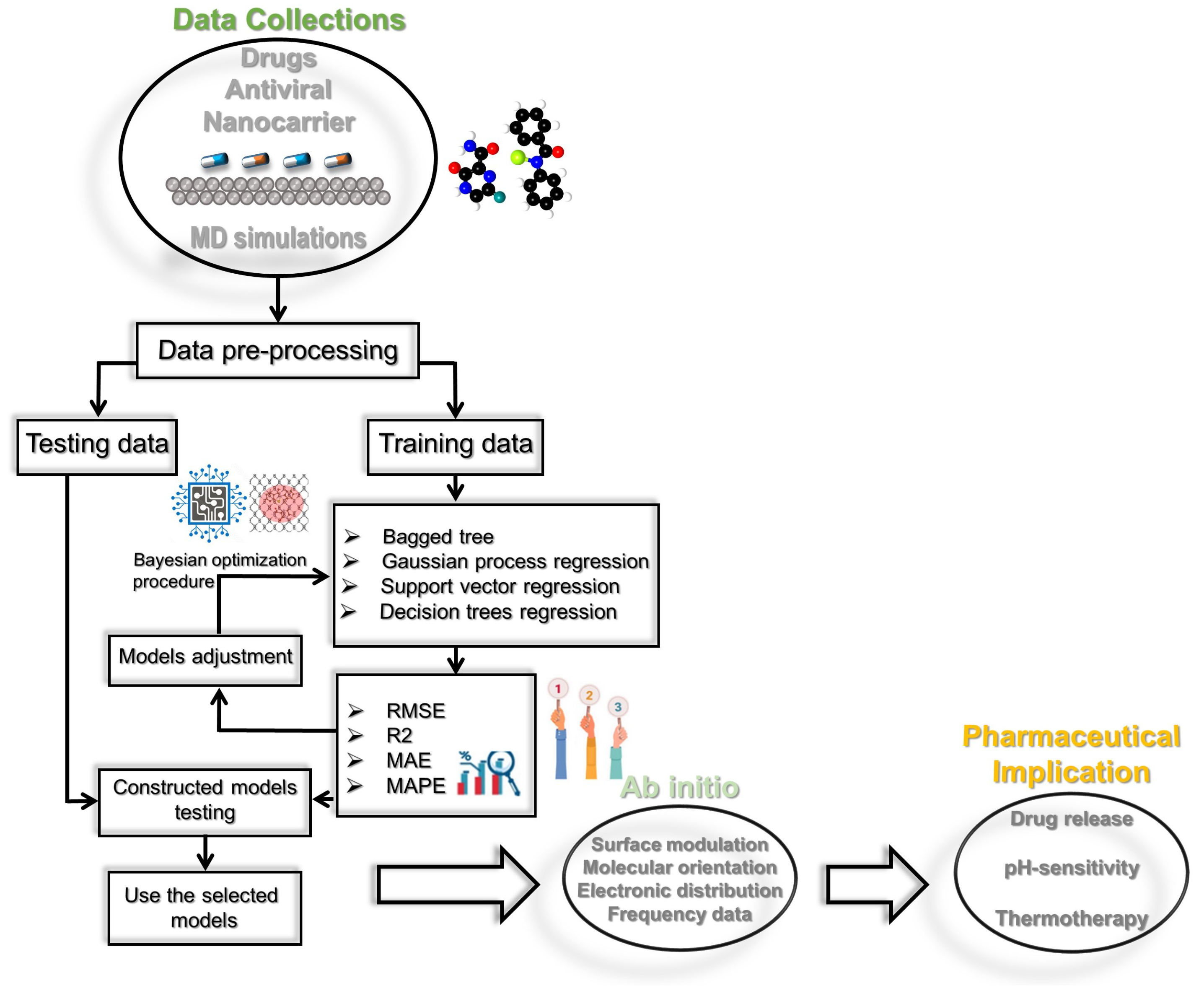
4.7. Prediction Framework
4.8. Ab Initio Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kotagiri, N.; Sudlow, G.P.; Akers, W.J.; Achilefu, S. Breaking the depth dependency of phototherapy with Cerenkov radiation and low-radiance-responsive nanophotosensitizers. Nat. Nanotechnol. 2015, 10, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.; Lee, J.C.; Robinson, J.T.; Raaz, U.; Xie, L.; Huang, N.F.; Cooke, J.P.; Dai, H. Multifunctional in vivo vascular imaging using near-infrared II fluorescence. Nat. Med. 2012, 18, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Feng, L.; Shi, X.; Liu, Z. Nano-graphene in biomedicine: Theranostic applications. Chem. Soc. Rev. 2013, 42, 530–547. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Chen, Y.; Shi, J. Insights into 2D MXenes for versatile biomedical applications: Current advances and challenges ahead. Adv. Sci. 2018, 5, 1800518. [Google Scholar] [CrossRef]
- Laref, S.; Asaduzzaman, A.; Beck, W.; Deymier, P.A.; Runge, K.; Adamowicz, L.; Muralidharan, K. Characterization of graphene–fullerene interactions: Insights from density functional theory. Chem. Phys. Lett. 2013, 582, 115–118. [Google Scholar] [CrossRef]
- Backes, C.; Abdelkader, A.M.; Alonso, C.; Andrieux-Ledier, A.; Arenal, R.; Azpeitia, J.; Balakrishnan, N.; Banszerus, L.; Barjon, J.; Bartali, R.; et al. Production and processing of graphene and related materials. 2D Mater. 2020, 7, 022001. [Google Scholar] [CrossRef]
- Gao, G.; Gao, W.; Cannuccia, E.; Taha-Tijerina, J.; Balicas, L.; Mathkar, A.; Narayanan, T.; Liu, Z.; Gupta, B.K.; Peng, J.; et al. Artificially stacked atomic layers: Toward new van der Waals solids. Nano Lett. 2012, 12, 3518–3525. [Google Scholar] [CrossRef]
- Laref, A.; Alshammari, N.; Laref, S.; Luo, S. Surface passivation effects on the electronic and optical properties of silicon quantum dots. Sol. Energy Mater. Sol. Cells 2014, 120, 622–630. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Fal’ko, V.I.; Colombo, L.; Gellert, P.; Schwab, M.; Kim, K. A roadmap for graphene. Nature 2012, 490, 192–200. [Google Scholar] [CrossRef]
- Wick, P.; Louw-Gaume, A.E.; Kucki, M.; Krug, H.F.; Kostarelos, K.; Fadeel, B.; Dawson, K.A.; Salvati, A.; Vázquez, E.; Ballerini, L.; et al. Classification framework for graphene-based materials. Angew. Chem. Int. Ed. 2014, 53, 7714–7718. [Google Scholar] [CrossRef] [PubMed]
- Laref, A.; Alsagri, M.; Alay-e Abbas, S.M.; Barakat, F.; Laref, S.; Huang, H.; Xiong, Y.; Yang, J.; Wu, X. Impact of phosphorous and sulphur substitution on Dirac cone modification and optical behaviors of monolayer graphene for nano-electronic devices. Appl. Surf. Sci. 2019, 489, 358–371. [Google Scholar] [CrossRef]
- Smith, A.T.; LaChance, A.M.; Zeng, S.; Liu, B.; Sun, L. Synthesis, properties, and applications of graphene oxide/reduced graphene oxide and their nanocomposites. Nano Mater. Sci. 2019, 1, 31–47. [Google Scholar] [CrossRef]
- Laref, A.; Alsagri, M.; Alay-e Abbas, S.M.; Laref, S.; Huang, H.; Xiong, Y.; Yang, J.; Khandy, S.A.; Rai, D.P.; Varshney, D.; et al. Electronic structure and optical characteristics of AA stacked bilayer graphene: A first principles calculations. Optik 2020, 206, 163755. [Google Scholar] [CrossRef]
- Huang, X.; Tang, S.; Mu, X.; Dai, Y.; Chen, G.; Zhou, Z.; Ruan, F.; Yang, Z.; Zheng, N. Freestanding palladium nanosheets with plasmonic and catalytic properties. Nat. Nanotechnol. 2011, 6, 28–32. [Google Scholar] [CrossRef]
- Anasori, B.; Lukatskaya, M.R.; Gogotsi, Y. 2D metal carbides and nitrides (MXenes) for energy storage. Nat. Rev. Mater. 2017, 2, 1–17. [Google Scholar] [CrossRef]
- Laref, S.; Wang, B.; Inal, S.; Al-Ghamdi, S.; Gao, X.; Gojobori, T. A Peculiar Binding Characterization of DNA (RNA) Nucleobases at MoOS-Based Janus Biosensor: Dissimilar Facets Role on Selectivity and Sensitivity. Biosensors 2022, 12, 442. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Martín, C.; Kurapati, R.; Bianco, A. Degradation-by-design: How chemical functionalization enhances the biodegradability and safety of 2D materials. Chem. Soc. Rev. 2020, 49, 6224–6247. [Google Scholar] [CrossRef]
- Liu, H.; Du, Y.; Deng, Y.; Peide, D.Y. Semiconducting black phosphorus: Synthesis, transport properties and electronic applications. Chem. Soc. Rev. 2015, 44, 2732–2743. [Google Scholar] [CrossRef]
- Shen, L.; Li, B.; Qiao, Y. Fe3O4 nanoparticles in targeted drug/gene delivery systems. Materials 2018, 11, 324. [Google Scholar] [CrossRef]
- Rahimi, R.; Solimannejad, M. BC3 graphene-like monolayer as a drug delivery system for nitrosourea anticancer drug: A first-principles perception. Appl. Surf. Sci. 2020, 525, 146577. [Google Scholar] [CrossRef]
- Hashemzadeh, H.; Raissi, H. Covalent organic framework as smart and high efficient carrier for anticancer drug delivery: A DFT calculations and molecular dynamics simulation study. J. Phys. D Appl. Phys. 2018, 51, 345401. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Scavone, C.; Brusco, S.; Bertini, M.; Sportiello, L.; Rafaniello, C.; Zoccoli, A.; Berrino, L.; Racagni, G.; Rossi, F.; Capuano, A. Current pharmacological treatments for COVID-19: What’s next? Br. J. Pharmacol. 2020, 177, 4813–4824. [Google Scholar] [CrossRef]
- Sreekanth Reddy, O.; Lai, W.F. Tackling COVID-19 using remdesivir and favipiravir as therapeutic options. ChemBioChem 2021, 22, 939–948. [Google Scholar] [CrossRef]
- Guy, R.K.; DiPaola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Hu, S.; Gao, J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef]
- Delang, L.; Abdelnabi, R.; Neyts, J. Favipiravir as a potential countermeasure against neglected and emerging RNA viruses. Antivir. Res. 2018, 153, 85–94. [Google Scholar] [CrossRef]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B 2017, 93, 449–463. [Google Scholar] [CrossRef]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Lynch, E.; Kil, J. Development of ebselen, a glutathione peroxidase mimic, for the prevention and treatment of noise-induced hearing loss. Semin. Hear. 2009, 30, 047–055. [Google Scholar] [CrossRef]
- Singh, N.; Halliday, A.C.; Thomas, J.M.; Kuznetsova, O.V.; Baldwin, R.; Woon, E.C.; Aley, P.K.; Antoniadou, I.; Sharp, T.; Vasudevan, S.R.; et al. A safe lithium mimetic for bipolar disorder. Nat. Commun. 2013, 4, 1–7. [Google Scholar] [CrossRef]
- Renson, M.; Etschenberg, E.; Winkelmann, J. 2-Phenyl-1,2-benzisoselenazol-3(2H)-one Containing Pharmaceutical Preparations and Process for the Treatment of Rheumatic Diseases. U.S. Patent 4,352,799, 5 October 1982. [Google Scholar]
- Kil, J.; Lobarinas, E.; Spankovich, C.; Griffiths, S.K.; Antonelli, P.J.; Lynch, E.D.; Le Prell, C.G. Safety and efficacy of ebselen for the prevention of noise-induced hearing loss: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2017, 390, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Masaki, C.; Sharpley, A.L.; Cooper, C.M.; Godlewska, B.R.; Singh, N.; Vasudevan, S.R.; Harmer, C.J.; Churchill, G.C.; Sharp, T.; Rogers, R.D.; et al. Effects of the potential lithium-mimetic, ebselen, on impulsivity and emotional processing. Psychopharmacology 2016, 233, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gui, C.; Luo, X.; Yang, Q.; Günther, S.; Scandella, E.; Drosten, C.; Bai, D.; He, X.; Ludewig, B.; et al. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J. Virol. 2005, 79, 7095–7103. [Google Scholar] [CrossRef]
- Carmo, A.; Pereira-Vaz, J.; Mota, V.; Mendes, A.; Morais, C.; da Silva, A.C.; Camilo, E.; Pinto, C.S.; Cunha, E.; Pereira, J.; et al. Clearance and persistence of SARS-CoV-2 RNA in patients with COVID-19. J. Med. Virol. 2020, 92, 2227–2231. [Google Scholar] [CrossRef]
- Khan, M.I.; Shoaib, M.; Zubair, G.; Kumar, R.N.; Prasannakumara, B.; Mousa, A.A.A.; Malik, M.; Raja, M.A.Z. Neural artificial networking for nonlinear Darcy–Forchheimer nanofluidic slip flow. Appl. Nanosci. 2022, 1–20. [Google Scholar] [CrossRef]
- Shoaib, M.; Kausar, M.; Khan, M.I.; Zeb, M.; Gowda, R.P.; Prasannakumara, B.; Alzahrani, F.; Raja, M.A.Z. Intelligent backpropagated neural networks application on Darcy-Forchheimer ferrofluid slip flow system. Int. Commun. Heat Mass Transf. 2021, 129, 105730. [Google Scholar] [CrossRef]
- Raja, M.A.Z.; Shoaib, M.; Zubair, G.; Khan, M.I.; Gowda, R.P.; Prasannakumara, B.; Guedri, K. Intelligent neuro-computing for entropy generated Darcy–Forchheimer mixed convective fluid flow. Math. Comput. Simul. 2022, 201, 193–214. [Google Scholar] [CrossRef]
- Varun Kumar, R.; Alsulami, M.; Sarris, I.; Prasannakumara, B.; Rana, S. Backpropagated Neural Network Modeling for the Non-Fourier Thermal Analysis of a Moving Plate. Mathematics 2023, 11, 438. [Google Scholar] [CrossRef]
- Sharma, R.P.; Madhukesh, J.; Shukla, S.; Prasannakumara, B. Numerical and Levenberg–Marquardt backpropagation neural networks computation of ternary nanofluid flow across parallel plates with Nield boundary conditions. Eur. Phys. J. Plus 2023, 138, 63. [Google Scholar] [CrossRef]
- Akkermans, R.L.; Spenley, N.A.; Robertson, S.H. COMPASS III: Automated fitting workflows and extension to ionic liquids. Mol. Simul. 2021, 47, 540–551. [Google Scholar] [CrossRef]
- Laref, S.; Wang, B.; Gao, X.; Gojobori, T. Computational Studies of Auto-Active van der Waals interaction Molecules on Ultra-thin Black-Phosphorus Film. Molecules 2023, 28, 681. [Google Scholar] [CrossRef]
- Zuo, Y.; Warnock, M.; Harbaugh, A.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Knight, J.S.; Kanthi, Y.; Lawrence, D.A. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Cheng, T.; Wang, L.; Merinov, B.V.; Goddard, W.A., III. Explanation of dramatic pH-dependence of hydrogen binding on noble metal electrode: Greatly weakened water adsorption at high pH. J. Am. Chem. Soc. 2018, 140, 7787–7790. [Google Scholar] [CrossRef]
- Ou, W.; Byeon, J.H.; Thapa, R.K.; Ku, S.K.; Yong, C.S.; Kim, J.O. Plug-and-play nanorization of coarse black phosphorus for targeted chemo-photoimmunotherapy of colorectal cancer. ACS Nano 2018, 12, 10061–10074. [Google Scholar] [CrossRef]
- Xie, Y.; Zhao, K.; Sun, Y.; Chen, D. Gaussian processes for short-term traffic volume forecasting. Transp. Res. Rec. 2010, 2165, 69–78. [Google Scholar] [CrossRef]
- Harrou, F.; Saidi, A.; Sun, Y.; Khadraoui, S. Monitoring of photovoltaic systems using improved kernel-based learning schemes. IEEE J. Photovoltaics 2021, 11, 806–818. [Google Scholar] [CrossRef]
- Lee, J.; Wang, W.; Harrou, F.; Sun, Y. Wind power prediction using ensemble learning-based models. IEEE Access 2020, 8, 61517–61527. [Google Scholar] [CrossRef]
- Williams, C.K.; Rasmussen, C.E. Gaussian processes for regression. In Advances in Neural Information Processing Systems 8, Proceedings of the 1995 Conference, Denver, CO, USA, 27–30 November 1995; MIT Press: Cambridge, MA, USA, 1996. [Google Scholar]
- MacKay, D.J. Gaussian Processes—A Replacement for Supervised Neural Networks? Cambridge University: Cambridge, UK, 1997. [Google Scholar]
- García-Nieto, P.J.; García-Gonzalo, E.; Puig-Bargués, J.; Duran-Ros, M.; de Cartagena, F.R.; Arbat, G. Prediction of outlet dissolved oxygen in micro-irrigation sand media filters using a Gaussian process regression. Biosyst. Eng. 2020, 195, 198–207. [Google Scholar] [CrossRef]
- Alkesaiberi, A.; Harrou, F.; Sun, Y. Efficient wind power prediction using machine learning methods: A comparative study. Energies 2022, 15, 2327. [Google Scholar] [CrossRef]
- Alali, Y.; Harrou, F.; Sun, Y. A proficient approach to forecast COVID-19 spread via optimized dynamic machine learning models. Sci. Rep. 2022, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Deng, M.; Xu, F.; Wang, H. Prediction of hourly PM2.5 using a space-time support vector regression model. Atmos. Environ. 2018, 181, 12–19. [Google Scholar] [CrossRef]
- Schulz, E.; Speekenbrink, M.; Krause, A. A tutorial on Gaussian process regression: Modelling, exploring, and exploiting functions. J. Math. Psychol. 2018, 85, 1–16. [Google Scholar] [CrossRef]
- Seeger, M. Gaussian processes for machine learning. Int. J. Neural Syst. 2004, 14, 69–106. [Google Scholar] [CrossRef]
- Nguyen, V.H.; Le, T.T.; Truong, H.S.; Le, M.V.; Ngo, V.L.; Nguyen, A.T.; Nguyen, H.Q. Applying Bayesian Optimization for Machine Learning Models in Predicting the Surface Roughness in Single-Point Diamond Turning Polycarbonate. Math. Probl. Eng. 2021, 2021, 1–16. [Google Scholar] [CrossRef]
- Murphy, K.P. Machine Learning: A Probabilistic Perspective; MIT Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Bergstra, J.; Bengio, Y. Random search for hyper-parameter optimization. J. Mach. Learn. Res. 2012, 13, 281–305. [Google Scholar]
- Protopapadakis, E.; Voulodimos, A.; Doulamis, N. An investigation on multi-objective optimization of feedforward neural network topology. In Proceedings of the 2017 8th International Conference on Information, Intelligence, Systems & Applications (IISA), Larnaca, Cyprus, 27–30 August 2017; pp. 1–6. [Google Scholar]
- Shahriari, B.; Swersky, K.; Wang, Z.; Adams, R.P.; De Freitas, N. Taking the human out of the loop: A review of Bayesian optimization. Proc. IEEE 2015, 104, 148–175. [Google Scholar] [CrossRef]
- Snoek, J.; Larochelle, H.; Adams, R.P. Practical bayesian optimization of machine learning algorithms. In Advances in Neural Information Processing Systems 25 (NIPS 2012): 26th Annual Conference on Neural Information Processing Systems 2012; Morgan Kaufmann Publishers, Inc.: Burlington, MA, USA, 2012. [Google Scholar]
- Cui, J.; Tan, Q.; Zhang, C.; Yang, B. A novel framework of graph Bayesian optimization and its applications to real-world network analysis. Expert Syst. Appl. 2021, 170, 114524. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Chatzis, S. Gaussian process-based Bayesian optimization for data-driven unit commitment. Int. J. Electr. Power Energy Syst. 2021, 130, 106930. [Google Scholar] [CrossRef]
- Springenberg, J.T.; Klein, A.; Falkner, S.; Hutter, F. Bayesian optimization with robust Bayesian neural networks. Adv. Neural Inf. Process. Syst. 2016, 29, 4134–4142. [Google Scholar]
- Yu, P.S.; Chen, S.T.; Chang, I.F. Support vector regression for real-time flood stage forecasting. J. Hydrol. 2006, 328, 704–716. [Google Scholar] [CrossRef]
- Hong, W.C.; Dong, Y.; Chen, L.Y.; Wei, S.Y. SVR with hybrid chaotic genetic algorithms for tourism demand forecasting. Appl. Soft Comput. 2011, 11, 1881–1890. [Google Scholar] [CrossRef]
- Smola, A.J.; Schölkopf, B. A tutorial on support vector regression. Stat. Comput. 2004, 14, 199–222. [Google Scholar] [CrossRef]
- Lee, J.; Wang, W.; Harrou, F.; Sun, Y. Reliable solar irradiance prediction using ensemble learning-based models: A comparative study. Energy Convers. Manag. 2020, 208, 112582. [Google Scholar] [CrossRef]
- Zhang, Y.; Haghani, A. A gradient boosting method to improve travel time prediction. Transp. Res. Part C Emerg. Technol. 2015, 58, 308–324. [Google Scholar] [CrossRef]
- Bauer, E.; Kohavi, R. An empirical comparison of voting classification algorithms: Bagging, boosting, and variants. Mach. Learn. 1999, 36, 105–139. [Google Scholar] [CrossRef]
- Harrou, F.; Saidi, A.; Sun, Y. Wind power prediction using bootstrap aggregating trees approach to enabling sustainable wind power integration in a smart grid. Energy Convers. Manag. 2019, 201, 112077. [Google Scholar] [CrossRef]
- Ribeiro, M.H.D.M.; dos Santos Coelho, L. Ensemble approach based on bagging, boosting and stacking for short-term prediction in agribusiness time series. Appl. Soft Comput. 2020, 86, 105837. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MLFF-FP | ||||
|---|---|---|---|---|
| Methods | RMSE | MAE | MAPE | |
| BT | 0.9743 | 0.0266 | 0.0217 | 0.0049 |
| GPR | 0.9663 | 0.0304 | 0.0238 | 0.0053 |
| SVR | 0.9767 | 0.0253 | 0.0208 | 0.0047 |
| RT | 0.9674 | 0.0299 | 0.0238 | 0.0053 |
| MLFF-EB | ||||
| Methods | RMSE | MAE | MAPE | |
| BT | 0.9513 | 0.1001 | 0.0655 | 0.0115 |
| GPR | 0.9614 | 0.0891 | 0.0606 | 0.0106 |
| SVR | 0.9447 | 0.1067 | 0.0809 | 0.0142 |
| RT | 0.9501 | 0.1014 | 0.0654 | 0.0114 |
| MLFF-FP_EB | ||||
| Methods | RMSE | MAE | MAPE | |
| BT | 0.9489 | 0.1459 | 0.1011 | 0.0179 |
| GPR | 0.9669 | 0.1173 | 0.0718 | 0.0127 |
| SVR | 0.9665 | 0.1182 | 0.0710 | 0.0125 |
| RT | 0.9259 | 0.1756 | 0.1109 | 0.0196 |
| RMSE | MAE | MAPE | ||
|---|---|---|---|---|
| BT | 0.9582 | 0.0909 | 0.0628 | 0.0114 |
| GPR | 0.9649 | 0.0789 | 0.0521 | 0.0095 |
| SVR | 0.9626 | 0.0834 | 0.0576 | 0.0105 |
| RT | 0.9478 | 0.1023 | 0.0667 | 0.0121 |
| MLFF-FP | ||||
|---|---|---|---|---|
| RMSE | MAE | MAPE | ||
| BT | 0.9772 | 0.0279 | 0.0218 | 0.2466 |
| GPR | 0.9792 | 0.0266 | 0.0208 | 0.2348 |
| SVR | 0.9772 | 0.0279 | 0.0211 | 0.2382 |
| RT | 0.9775 | 0.0277 | 0.0221 | 0.2502 |
| MLFF-EB | ||||
| RMSE | MAE | MAPE | ||
| BT | 0.9685 | 0.0758 | 0.0493 | 0.4496 |
| GPR | 0.9735 | 0.0695 | 0.0468 | 0.4277 |
| SVR | 0.9705 | 0.0734 | 0.0492 | 0.4503 |
| RT | 0.9729 | 0.0703 | 0.0486 | 0.4448 |
| MLFF-FP_EB | ||||
| RMSE | MAE | MAPE | ||
| BT | 0.9648 | 0.1253 | 0.0843 | 2.5297 |
| GPR | 0.9725 | 0.1107 | 0.0732 | 2.2111 |
| SVR | 0.9710 | 0.1138 | 0.0771 | 2.3273 |
| RT | 0.9689 | 0.1178 | 0.0795 | 2.3512 |
| RMSE | MAE | MAPE | ||
|---|---|---|---|---|
| BT | 0.9701 | 0.0763 | 0.0518 | 1.0753 |
| GPR | 0.9751 | 0.0689 | 0.0469 | 0.9579 |
| SVR | 0.9729 | 0.0717 | 0.0491 | 1.0053 |
| RT | 0.9731 | 0.0719 | 0.0501 | 1.0154 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laref, S.; Harrou, F.; Wang, B.; Sun, Y.; Laref, A.; Laleg-Kirati, T.-M.; Gojobori, T.; Gao, X. Synergy of Small Antiviral Molecules on a Black-Phosphorus Nanocarrier: Machine Learning and Quantum Chemical Simulation Insights. Molecules 2023, 28, 3521. https://doi.org/10.3390/molecules28083521
Laref S, Harrou F, Wang B, Sun Y, Laref A, Laleg-Kirati T-M, Gojobori T, Gao X. Synergy of Small Antiviral Molecules on a Black-Phosphorus Nanocarrier: Machine Learning and Quantum Chemical Simulation Insights. Molecules. 2023; 28(8):3521. https://doi.org/10.3390/molecules28083521
Chicago/Turabian StyleLaref, Slimane, Fouzi Harrou, Bin Wang, Ying Sun, Amel Laref, Taous-Meriem Laleg-Kirati, Takashi Gojobori, and Xin Gao. 2023. "Synergy of Small Antiviral Molecules on a Black-Phosphorus Nanocarrier: Machine Learning and Quantum Chemical Simulation Insights" Molecules 28, no. 8: 3521. https://doi.org/10.3390/molecules28083521