
Simple Synthetic Approach to N-(Pyridin-2-yl)imidates from Nitrostyrenes and 2-Aminopyridines via the N-(Pyridin-2-yl)iminonitriles as Intermediates


Abstract
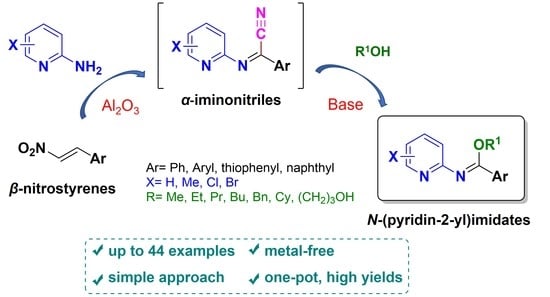
:
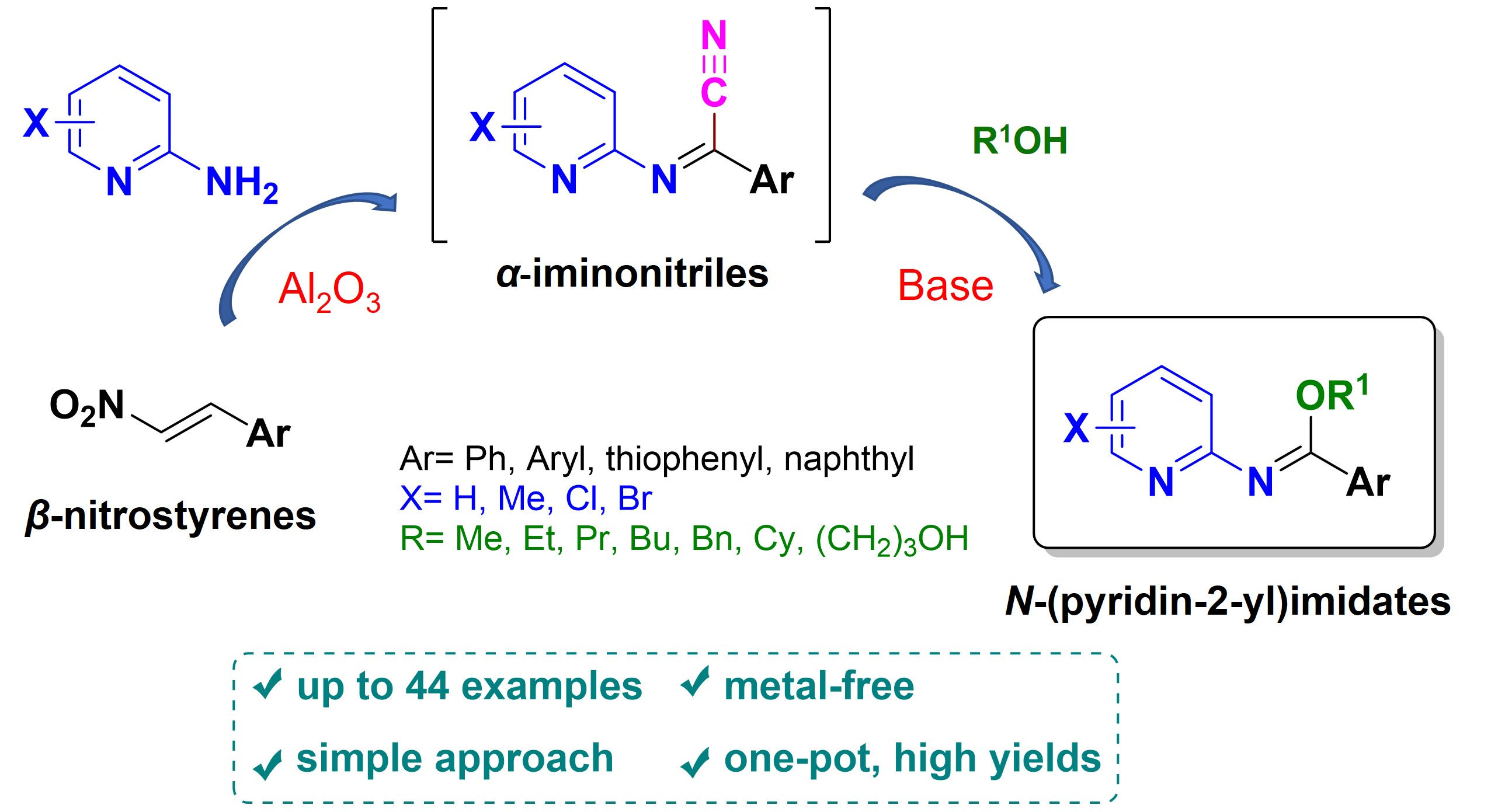
1. Introduction
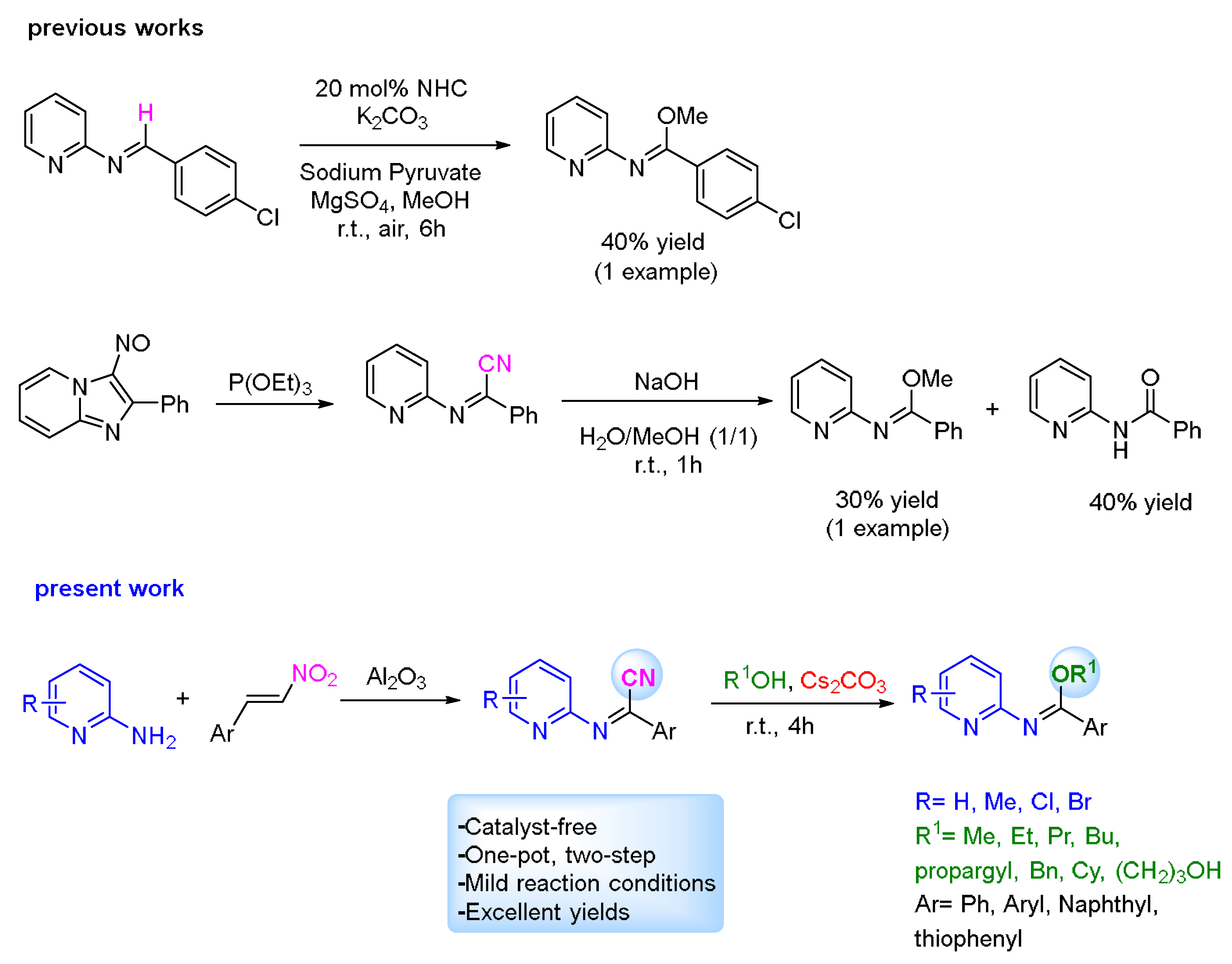
2. Results and Discussion
2.1. Evaluation of the Reaction Conditions
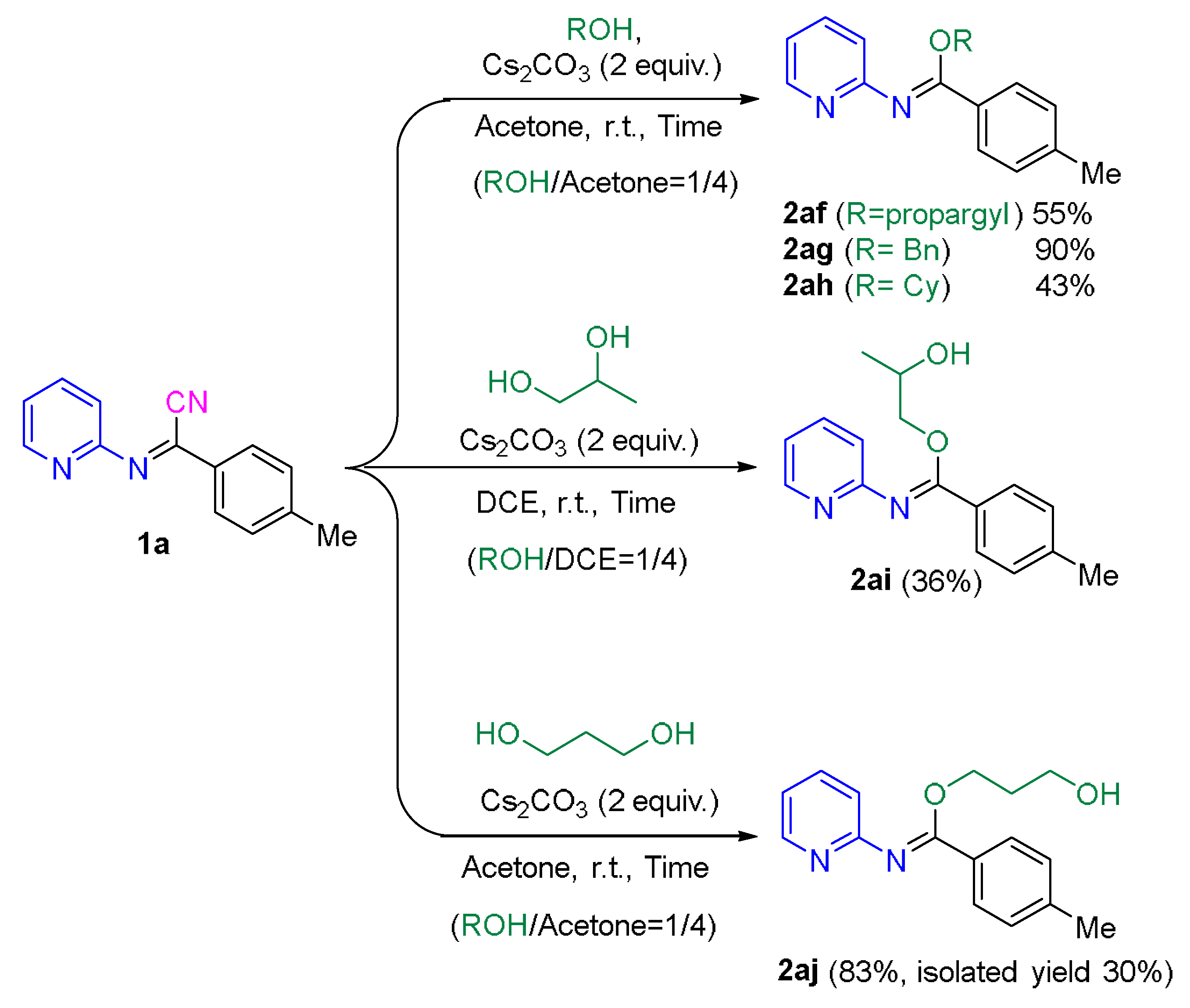
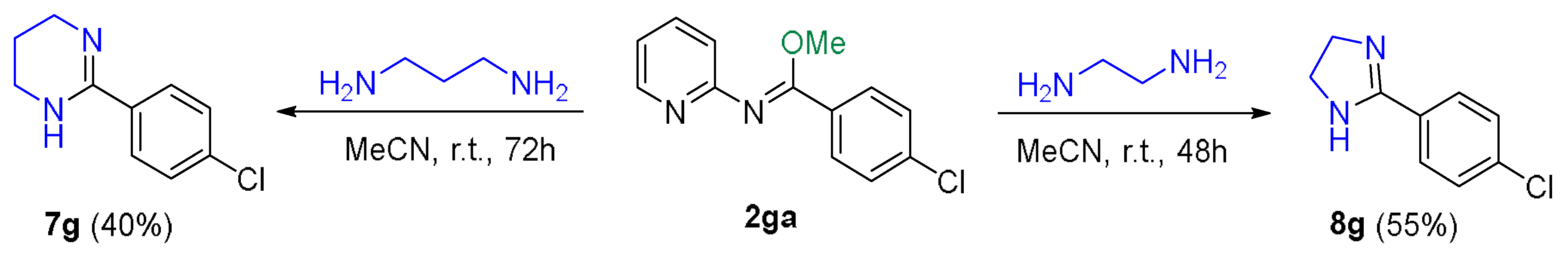
2.2. Application of the Synthetic Transformation of N-(Pyridin-2-yl)benzimidoyl Cyanides to the N-(Pyridine-2-yl)imidates
3. Materials and Methods
3.1. General and Aparatus
3.2. Synthesis of Aromatic β-Nitrostyrenes
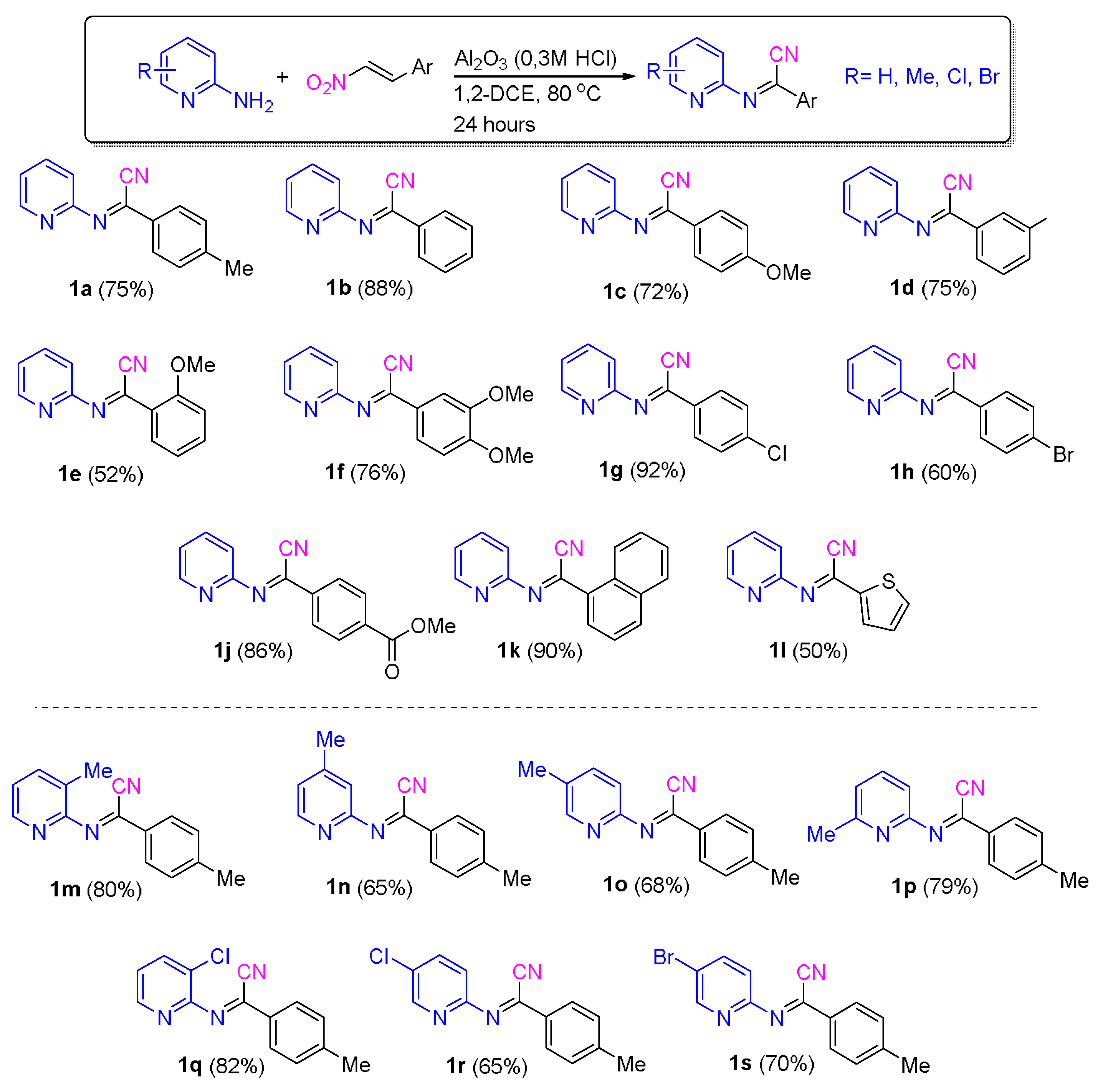
3.3. Synthesis of N-(Pyridin-2-yl)iminonitriles from Nitrostyrenes and 2-Aminopyridine
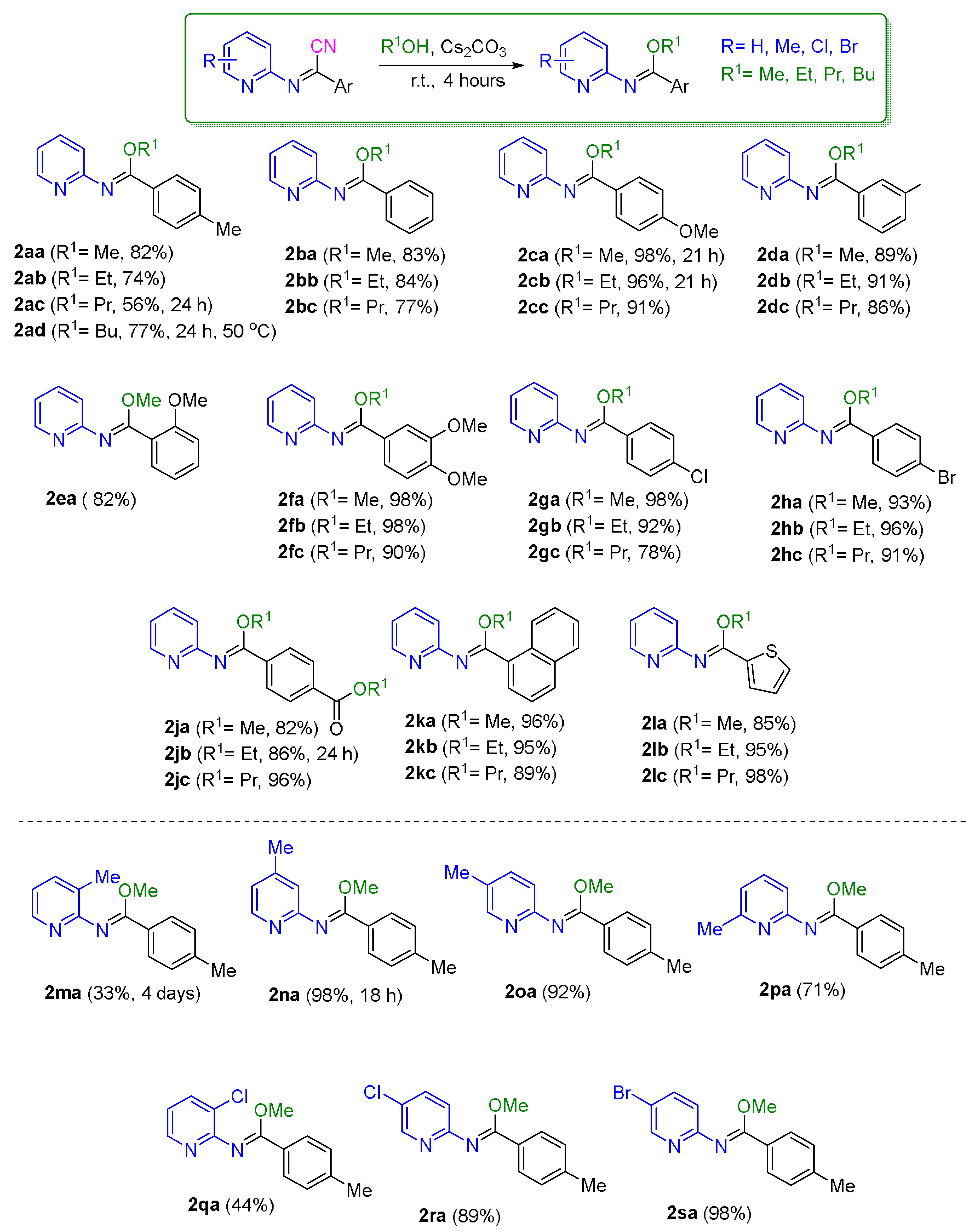
3.4. Synthesis of N-(Pyridin-2-yl)imidates from N-(Pyridin-2-yl)iminonitriles
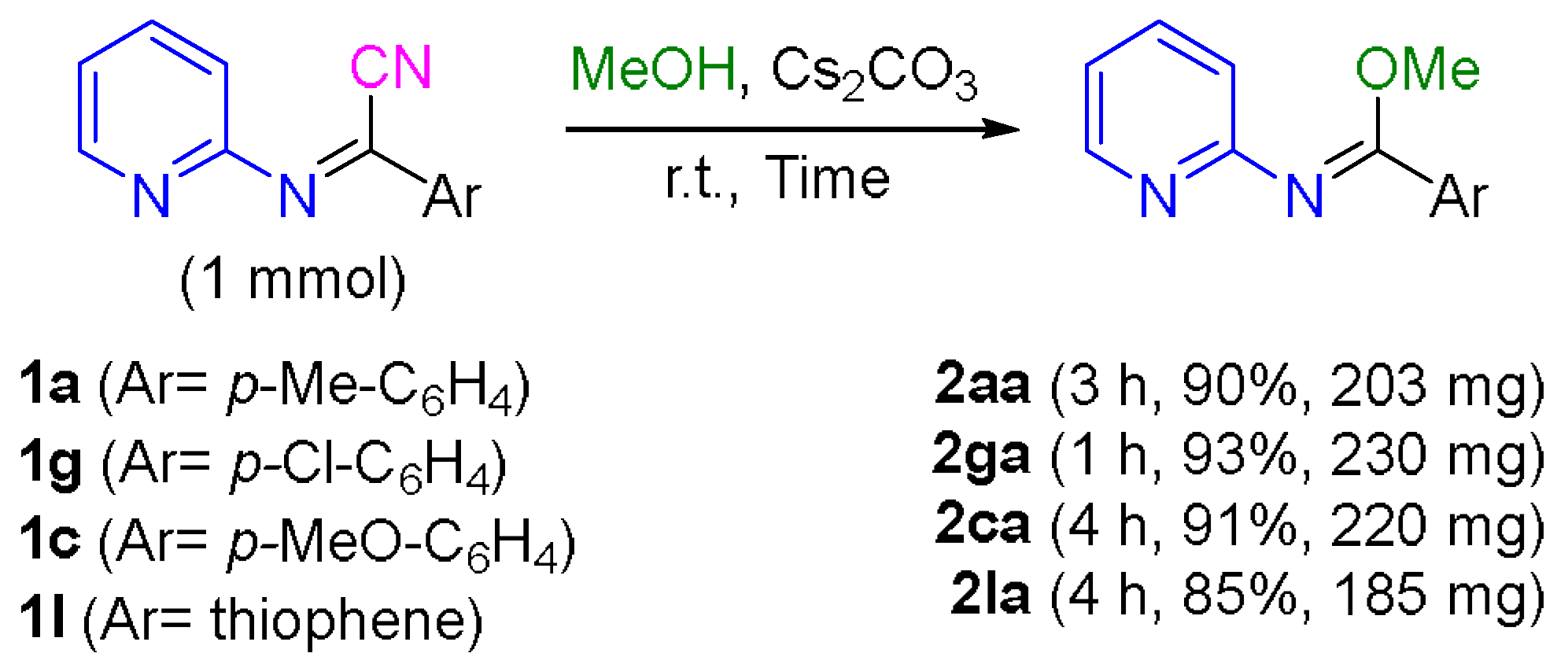
3.5. 1 mmol Scale Synthesis of N-(Pyridin-2-yl)imidates 2aa, 2ca, 2ga, and 2la
3.6. Synthesis of N,N-Heterocyclic Compounds from N-(Pyridin-2-yl)imidates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Neilson, D.G. The Chemistry of Amidines and Imidates; Patai, S., Ed.; Wiley: New York, NY, USA, 1975; pp. 385–489. [Google Scholar]
- Neilson, D.G. Chapter 9: Imidates Including Cyclic Imidates. In Amidines and Imidates; Patai, S., Rappoport, Z., Eds.; Wiley: New York, NY, USA, 1991; Volume 2, pp. 425–483. [Google Scholar]
- Joule, J.A.; Mills, K. Heterocyclic Chemistry, 5th ed.; Wiley: New York, NY, USA, 2010. [Google Scholar]
- Roger, R.; Neilson, D.G. The Chemistry of Imidates. Chem. Rev. 1961, 61, 179–211. [Google Scholar] [CrossRef]
- Thakur, R.; Jaiswal, Y.; Kumar, A. Imidates: An Emerging Synthon for N-Heterocycles. Org. Biomol. Chem. 2019, 17, 9829–9843. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.; Kumar, A. Visible-Light-Mediated β-C(Sp3)-H Amination of Glycosylimidates: En Route to Oxazoline-Fused/Spiro Nonclassical Bicyclic Sugars. Org. Lett. 2019, 21, 3108–3113. [Google Scholar] [CrossRef]
- Shi, W.Z.; Li, H.; Mu, G.C.; Lu, J.L.; Tu, Y.H.; Hu, X.G. 1,2-trans-Stereoselective Synthesis of C-Glycosides of 2-Deoxy-2-amino-sugars Involving Glycosyl Radicals. Org. Lett. 2021, 23, 2659–2663. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, B. Recent Advances in the Chemical Synthesis of C-Glycosides. Chem. Rev. 2017, 117, 12281–12356. [Google Scholar] [CrossRef]
- Pinner, A. Die Imidöather and Ihre Derivate; Oppenheim: Berlin, Germany, 1892. [Google Scholar]
- Watanabe, K.; Kogoshi, N.; Miki, H.; Torisawa, Y. Improved Pinner Reaction with CPME as a Solvent. Synth. Commun. 2009, 39, 2008–2013. [Google Scholar] [CrossRef]
- Ptiček, L.; Hok, L.; Grbčić, P.; Topić, F.; Cetina, M.; Rissanen, K.; Pavelić, S.K.; Vianello, R.; Racané, L. Amidino Substituted 2-Aminophenols: Biologically Important Building Blocks for the Amidino-Functionalization of 2-Substituted Benzoxazoles. Org. Biomol. Chem. 2021, 19, 2784–2793. [Google Scholar] [CrossRef]
- Caron, S.; Wei, L.; Douville, J.; Ghosh, A. Preparation and Utility of Trihaloethyl Imidates: Useful Reagents for the Synthesis of Amidines. J. Org. Chem. 2010, 75, 945–947. [Google Scholar] [CrossRef]
- Marshall, E.K.; Acree, S.F. On the Reactions of Both the Ions and the Molecules of Acids Bases and Salts. J. Phys. Chem. 1915, 19, 589–608. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, F.C.; Peters, G.A. Base-Catalyzed Reaction of Nitriles with Alcohols. A Convenient Route to Imidates and Amidine Salts. J. Org. Chem. 1961, 26, 412–418. [Google Scholar] [CrossRef]
- Barcock, R.A.; Chadwick, D.J.; Storr, R.C. Synthesis of Some Novel Imidate Derivatives of Thiophene and Furan: Investigations of Their Metallation Properties and Some Synthetic Applications. Tetrahedron 1994, 50, 4149–4166. [Google Scholar] [CrossRef]
- Bartels, G.; Hinze, R.P.; Wullbrandt, D. Beiträge Zur Kenntnis Des Chromophoren Systems Der Corrine, VII. Notiz Über Die Reaktion von Diazoessigsäure-methylester Mit Lactamen; Eine Modellreaktion Zur Cyclisierung von 5,6-Dioxo-monosecocorrinen. Liebigs Ann. Chem. 1980, 1980, 168–170. [Google Scholar] [CrossRef]
- Fuchs, J.R.; Funk, R.L. Total Synthesis of (±)-Perophoramidine. J. Am. Chem. Soc. 2004, 126, 5068–5069. [Google Scholar] [CrossRef]
- Popov, K.; Somfai, P. Synthesis of Imidates: TFA-Mediated Regioselective Amide Alkylation Using Meerwein’s Reagent. J. Org. Chem. 2016, 81, 3470–3472. [Google Scholar] [CrossRef]
- Dekorver, K.A.; North, T.D.; Hsung, R.P. An Efficient Synthesis of de Novo Imidates via Aza-Claisen Rearrangements of N-Allyl Ynamides. Synlett 2010, 16, 2397–2402. [Google Scholar] [CrossRef]
- Ghorai, S.; Lee, D. Synthesis of Imides, Imidates, Amidines, and Amides by Intercepting the Aryne-Isocyanide Adduct with Weak Nucleophiles. Org. Lett. 2019, 21, 7390–7393. [Google Scholar] [CrossRef]
- Schmitt, G.; Ebertz, W. Neue Synthese Für N-Vinylamide. Angew. Chem. 1982, 94, 650–651. [Google Scholar] [CrossRef]
- Guan, Z.; Hillrichs, K.; Ünlü, C.; Rissanen, K.; Nieger, M.; Schmidt, A. Synthesis of 2-Anilinobenzimidates, Anthranilamides, and 2,3-Dihydroquinazolin-4(1H)-Ones from N-Heterocyclic Carbenes of Indazole. Tetrahedron 2015, 71, 276–282. [Google Scholar] [CrossRef]
- Birch, D.J.; Guildford, A.J.; Tometzki, M.A.; Turner, R.W. Reaction of 3-Nitroso-2-Phenylimidazo[1,2-a]Pyridine with Triethyl Phosphite. A Revised Structure for the Product. J. Org. Chem. 1982, 47, 3547–3548. [Google Scholar] [CrossRef]
- Wang, G.; Wei, C.; Hong, X.; Fu, Z.; Huang, W. Sodium Pyruvate as a Peroxide Scavenger in Aerobic Oxidation under Carbene Catalysis. Green Chem. 2020, 22, 6819–6826. [Google Scholar] [CrossRef]
- Tzani, M.A.; Fountoulaki, S.; Lykakis, I.N. Polyoxometalate-Driven Ease Conversion of Valuable Furfural to trans-N,N-4,5-Diaminocyclopenten-2-ones. J. Org. Chem. 2022, 87, 2601–2615. [Google Scholar] [CrossRef]
- Tzani, M.A.; Gabriel, C.; Lykakis, I.N. Selective Synthesis of Benzimidazoles from o-Phenylenediamine and Aldehydes Promoted by Supported Gold Nanoparticles. Nanomaterials 2020, 10, 2405. [Google Scholar] [CrossRef]
- Andreou, D.; Essien, N.B.; Pubill-Ulldemolins, C.; Terzidis, M.A.; Papadopoulos, A.N.; Kostakis, G.E.; Lykakis, I.N. Skeletally Tunable Seven-Membered-Ring Fused Pyrroles. Org. Lett. 2021, 23, 6685–6690. [Google Scholar] [CrossRef] [PubMed]
- Tzani, M.A.; Kallitsakis, M.G.; Symeonidis, T.S.; Lykakis, I.N. Alumina-Supported Gold Nanoparticles as a Bifunctional Catalyst for the Synthesis of 2-Amino-3-arylimidazo[1,2-a]pyridines. ACS Omega 2018, 3, 17947–17956. [Google Scholar] [CrossRef] [Green Version]
- Andreou, D.; Kallitsakis, M.G.; Loukopoulos, E.; Gabriel, C.; Kostakis, G.E.; Lykakis, I.N. Copper-Promoted Regioselective Synthesis of Polysubstituted Pyrroles from Aldehydes, Amines, and Nitroalkenes via 1,2-Phenyl/Alkyl Migration. J. Org. Chem. 2018, 83, 2104–2113. [Google Scholar] [CrossRef]
- Kallitsakis, M.; Loukopoulos, E.; Abdul-Sada, A.; Tizzard, G.J.; Coles, S.J.; Kostakis, G.E.; Lykakis, I.N. A Copper-Benzotriazole-Based Coordination Polymer Catalyzes the Efficient One-Pot Synthesis of (N′-Substituted)-hydrazo-4-aryl-1,4-dihydropyridines from Azines. Adv. Synth. Catal. 2017, 359, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Liang, P.; Zheng, J.; Zhou, Z.; Wen, X.; Liu, T.; Ye, M. Cyanide-Free Ce(III)-Catalyzed Highly Efficient Synthesis of α-Iminonitriles from 2-Aminopyridines and Nitroalkenes via Intermolecular Dehydration Reaction. ACS Omega 2018, 3, 12520–12529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasiński, R.; Kula, K.; Kącka, A.; Mirosław, B. Unexpected course of reaction between (E)-2-aryl-1-cyano-1-nitroethenes and diazafluorene: Why is there no 1,3-dipolar cycloaddition? Mon. Chem. 2017, 148, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Woliński, P.; Kącka-Zych, A.; Mirosław, B.; Wielgus, E.; Olszewska, A.; Jasiński, R. Green, one-pot synthesis of 1,2-oxazine-type herbicides via non-catalyzed Hetero Diels-Alder reactions comprising (2E)-3-aryl-2-nitroprop-2-enenitriles. J. Clean. Prod. 2022, 356, 131878. [Google Scholar] [CrossRef]
- Kallitsakis, M.G.; Tancini, P.D.; Dixit, M.; Mpourmpakis, G.; Lykakis, I.N. Mechanistic Studies on the Michael Addition of Amines and Hydrazines To Nitrostyrenes: Nitroalkane Elimination via a Retro-aza-Henry-Type Process. J. Org. Chem. 2018, 83, 1176–1184. [Google Scholar] [CrossRef]
- Siwach, A.; Verma, P.K. Synthesis and therapeutic potential of imidazole containing compounds. BMC Chem. 2021, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Szabo, B. Imidazoline antihypertensive drugs: A critical review on their mechanism of action. Pharmacol. Ther. 2002, 93, 1–35. [Google Scholar] [CrossRef]
- Messer, W.S.; Abuh, Y.F.; Liu, Y.; Periyasamy, S.; Ngur, D.O.; Edgar, M.A.; El-Assadi, A.A.; Sbeih, S.; Dunbar, P.G.; Roknich, S.; et al. Synthesis and Biological Characterization of 1,4,5,6-Tetrahydropyrimidine and 2-Amino-3,4,5,6-Tetrahydropyridine Derivatives as Selective M1 Agonists. J. Med. Chem. 1997, 40, 1230–1246. [Google Scholar] [CrossRef] [PubMed]
- Kopp, S.R.; Kotze, A.C.; McCarthy, J.S.; Traub, R.J.; Coleman, G.T. Pyrantel in Small Animal Medicine: 30 Years On. Vet. J. 2008, 178, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Zhang, S.; Zhao, Y.; Wang, D.Z. New Approach to Oximes through Reduction of Nitro Compounds Enabled by Visible Light Photoredox Catalysis. Org. Lett. 2013, 15, 2660–2663. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Products (%) [b] | ||||||||
| Entry | Base (eq.) [a] | Time (h) | 1a | 2aa | 3a | 4 | 5a | 6a |
| 1 | Et3N (1.5) | 4 | 50 | 50 | - | - | - | - |
| 2 | Imidazole (1.5) | 4 | 87 | 13 | - | - | - | - |
| 3 | tBuOK (1.5) | 4 | - | 70 | 10 | 10 | 3 | 7 |
| 4 | tBuOK (2) | 4 | - | 90 | 4 | 3 | 2 | 1 |
| 5 | AcONa (1.5) | 4 | 62 | 38 | - | - | - | - |
| 6 | AcONa (2) | 4 | 58 | 42 | - | - | - | - |
| 7 | DBU (1.5) | 4 | - | 100 | - | - | - | - |
| 8 | DBU (2) | 4 | - | 100 | - | - | - | - |
| 9 | NaHCO3 (1.5) | 4 | 36 | 56 | 8 | - | - | - |
| 10 | Cs2CO3 (1.5) | 4 | - | 100 | - | - | - | - |
| 11 [c] | Cs2CO3 (1) | 4 | - | 92 | 2 | 3 | 2 | 1 |
| 12 [d] | Cs2CO3 (1) | 4 | - | 91 | 3 | 3 | 1 | 2 |
| 13 | K2CO3 (1.5) | 4 | - | 91 | 3 | 3 | 1 | 2 |
| 14 | NaOH (1) | 4 | - | 94 | 6 | - | - | - |
| 15 | - | 4 | 82 | 18 | - | - | - | - |
| 16 [e] | - | 24 | 51 | 49 | - | - | - | - |
| 17 [f] | - | 24 | - | 10 | - | 45 | 10 | 35 |
 | |||||||
|---|---|---|---|---|---|---|---|
| Products (%) [b] | |||||||
| Entry | Solvent [a] | MeOH/Solvent | 2aa | 3a | 4 | 5a | 6a |
| 1 | THF | 1/1 | 98 | 2 | - | - | - |
| 2 | THF | 1/4 | 98 | 2 | - | - | - |
| 3 | DCE | 1/4 | 95 | 5 | - | - | - |
| 4 | Acetone | 1/4 | 97 | 3 | - | - | - |
| 5 | CH3CN | 1/4 | 99 | 1 | - | - | - |
| 6 | DMSO | 1/1 | 99 | 1 | - | - | - |
| 7 | EtOAc | 1/1 | 95 | 5 | - | - | - |
| 8 [c,d] | H2O | 1/1 | 44 | 43 | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaidali, A.G.; Lykakis, I.N. Simple Synthetic Approach to N-(Pyridin-2-yl)imidates from Nitrostyrenes and 2-Aminopyridines via the N-(Pyridin-2-yl)iminonitriles as Intermediates. Molecules 2023, 28, 3321. https://doi.org/10.3390/molecules28083321
Chaidali AG, Lykakis IN. Simple Synthetic Approach to N-(Pyridin-2-yl)imidates from Nitrostyrenes and 2-Aminopyridines via the N-(Pyridin-2-yl)iminonitriles as Intermediates. Molecules. 2023; 28(8):3321. https://doi.org/10.3390/molecules28083321
Chicago/Turabian StyleChaidali, Andriani G., and Ioannis N. Lykakis. 2023. "Simple Synthetic Approach to N-(Pyridin-2-yl)imidates from Nitrostyrenes and 2-Aminopyridines via the N-(Pyridin-2-yl)iminonitriles as Intermediates" Molecules 28, no. 8: 3321. https://doi.org/10.3390/molecules28083321