
Green and Sustainable Ultrasound-Assisted Anodic Electrochemical Preparation of Graphene Oxide Dispersions and Their Antioxidant Properties
 , , , , and
, , , , and
Abstract
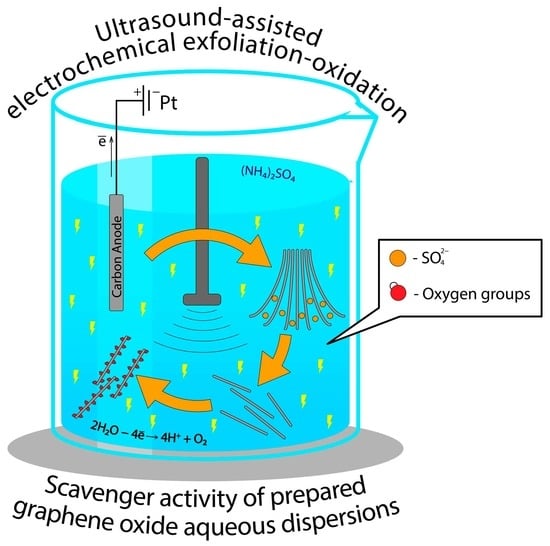
:
1. Introduction
2. Results and Discussion
- The conditions for the preparation and separation of aqueous dispersions of electrochemically generated graphene oxide (EGO) via anodic oxidation of graphite under the application of additional ultrasonic exfoliation of graphite under pulsed (modulation) conditions were determined.
- The conditions for isolation and purification of the prepared EGO with a dialysis bag (membrane) from (a) the electrolyte used in the synthesis and (b) formatted byproducts during electrolysis were determined.
- For the developed graphite anodic oxidation synthesis methods, many colloidal stability and morphology parameters are accurately reproducible. Additional structural and morphological information about the synthesized samples was obtained from the data of (a) molecular spectroscopy methods, as well as FTIR and Raman spectroscopies; (b) X-ray photoelectron spectroscopy; and (c) SEM and TEM. Dynamic light scattering (DLS) was also used to determine lateral particle sizes and zeta potentials.
- A chemiluminometry assay was used for electrochemically synthesized graphene oxide samples to evaluate their reactivity concerning reactive oxygen species (H2O2, superoxide anion radicals, and hydroxyl radicals) and to assess the induction of lipid/phospholipid peroxidation.
2.1. General Concept of EGO Synthesis
- I.
- At the cathode:
- 2H2O + 2 = H2 + 2OH− (–0.828 V) (1); the formation stage of hydrogen, which can potentially reduce EGO to a reduced sample of (rEGO) only in the case of a combined cathode–anode compartment.
- II.
- At the cathode:
- 2H2O – 4 = O2 + 4H+ (1.229 V) (2); water oxidation;
- >C=C< → >C=O, –COOH, >CH–OH, >C=O etc. (3); graphite structure oxidation [40];
- 2– 2 = (=2.010 V) (4); the formation of an additional oxidizer peroxydisulfate ion in solution.
- III.
- Reactions of hydrolysis of byproducts:
- (NH4)2S2O8 + 2H2O = 2(NH4)HSO4 + H2O2 (5); hydrolysis of electrolysis byproducts.
2.2. Selection of the Ultrasonic Treatment Mode
2.3. Characterization of Prepared EGO
2.3.1. Spectroscopic and Morphological Properties
- -
- ATR-FTIR spectroscopy
- -
- UV/vis spectroscopy
- -
- Raman spectroscopy
- -
- Morphology of prepared samples by scanning electron microscopy
- -
- X-ray photoelectron spectroscopy for C:O ratio estimation
2.3.2. Quality of Prepared EGO Products (Stability and Concentration Parameters)
2.4. Chemiluminescence Assay
2.4.1. Reactivity towards ROS
2.4.2. Pro-Oxidant Activity EGO towards Lipids and Phospholipids
3. Materials and Methods
3.1. Equipment for the Experimental Setup for Ultrasonic Anodic Oxidation–Exfoliation of Graphite
3.2. Reagents
3.3. Characterization of Aqueous Graphene Oxide Dispersions
3.4. Chemiluminescent Models for Assessing the Antioxidant Potential of Aqueous Dispersions of Graphene Oxide
3.5. Elemental Analysis
3.6. Surface Analysis and Morphology
3.7. Processing of Data and Analysis Results
3.8. Procedures
3.8.1. Procedures for Optimum Ultrasonic Treatment Condition Findings
3.8.2. Procedures Describing EGO Preparation Peculiarities
3.8.3. Purification Procedures
3.8.4. Chemiluminometry Assays
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mbayachi, V.B.; Ndayiragije, E.; Sammani, T.; Taj, S.; Mbuta, E.R.; Khan, A.U. Graphene synthesis, characterization and its applications: A review. Results Chem. 2021, 3, 100163. [Google Scholar] [CrossRef]
- Ou, L.; Song, B.; Liang, H.; Liu, J.; Feng, X.; Deng, B.; Sun, T.; Shao, L. Toxicity of graphene-family nanoparticles: A general review of the origins and mechanisms. Part. Fibre Toxicol. 2016, 13, 57. [Google Scholar] [CrossRef] [Green Version]
- Smerieri, M.; Celasco, E.; Carraro, G.; Lusuan, A.; Pal, J.; Bracco, G.; Rocca, M.; Savio, L.; Vattuone, L. Enhanced Chemical Reactivity of Pristine Graphene Interacting Strongly with a Substrate: Chemisorbed Carbon Monoxide on Graphene/Nickel(1 1 1). ChemCatChem 2015, 7, 2328–2331. [Google Scholar] [CrossRef]
- Tene, T.; Tubon Usca, G.; Guevara, M.; Molina, R.; Veltri, F.; Arias, M.; Caputi, L.S.; Vacacela Gomez, C. Toward Large-Scale Production of Oxidized Graphene. Nanomaterials 2020, 10, 279. [Google Scholar] [CrossRef] [Green Version]
- Adeniyi, O.; Nwahara, N.; Mwanza, D.; Nyokong, T.; Mashazi, P. High-performance non-enzymatic glucose sensing on nanocomposite electrocatalysts of nickel phthalocyanine nanorods and nitrogen doped-reduced graphene oxide nanosheets. Appl. Surf. Sci. 2023, 609, 155234. [Google Scholar] [CrossRef]
- Cui, L.; Lin, X.; Lin, N.; Song, Y.; Zhu, Z.; Chen, X.; Yang, C.J. Graphene oxide-protected DNA probes for multiplex microRNA analysis in complex biological samples based on a cyclic enzymatic amplification method. Chem. Commun. 2012, 48, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gupta, G.H.; Singh, G.; More, N.; Keerthana, M.; Sharma, A.; Jawade, D.; Balu, A.; Kapusetti, G. Ultrahigh sensitive graphene oxide/conducting polymer composite based biosensor for cholesterol and bilirubin detection. Biosens. Bioelectron. X 2023, 13, 100290. [Google Scholar] [CrossRef]
- Morales-Narváez, E.; Merkoçi, A. Graphene Oxide as an Optical Biosensing Platform. Adv. Mater. 2012, 24, 3298–3308. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, S.; Zhang, G.; Sun, X.; Lee, S.-T.; Liu, Z. Graphene in Mice: Ultrahigh In Vivo Tumor Uptake and Efficient Photothermal Therapy. Nano Lett. 2010, 10, 3318–3323. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, O.; Ghaderi, E.; Emamy, H. Nontoxic concentrations of PEGylated graphene nanoribbons for selective cancer cell imaging and photothermal therapy. J. Mater. Chem. 2012, 22, 20626–20633. [Google Scholar] [CrossRef]
- Komeily-Nia, Z.; Chen, J.-Y.; Nasri-Nasrabadi, B.; Lei, W.-W.; Yuan, B.; Zhang, J.; Qu, L.-T.; Gupta, A.; Li, J.-L. The key structural features governing the free radicals and catalytic activity of graphite/graphene oxide. Phys. Chem. Chem. Phys. 2020, 22, 3112–3121. [Google Scholar] [CrossRef]
- Song, Y.; Qu, K.; Zhao, C.; Ren, J.; Qu, X. Graphene oxide: Intrinsic peroxidase catalytic activity and its application to glucose detection. Adv. Mater. 2010, 22, 2206–2210. [Google Scholar] [CrossRef] [PubMed]
- Tayade, U.S.; Borse, A.U.; Meshram, J.S. Green reduction of graphene oxide and its applications in band gap calculation and antioxidant activity. Green. Mater. 2019, 7, 143–155. [Google Scholar] [CrossRef]
- Brodie, B.C., XIII. On the atomic weight of graphite. Philos. Trans. R. Soc. Lond. 1859, 149, 249–259. [Google Scholar]
- Hummers, W.S., Jr.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Staudenmaier, L. Verfahren zur Darstellung der Graphitsäure. Ber. Der. Dtsch. Chem. Ges. 1898, 31, 1481–1487. [Google Scholar] [CrossRef] [Green Version]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved synthesis of graphene oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.L.; Swager, T.M. Enhanced Electrochemical Expansion of Graphite for in Situ Electrochemical Functionalization. J. Am. Chem. Soc. 2012, 134, 17896–17899. [Google Scholar] [CrossRef]
- Dalal, M.H.; Lee, C.-Y.; Wallace, G.G. Simultaneous Anodic and Cathodic Exfoliation of Graphite Electrodes in an Aqueous Solution of Inorganic Salt. ChemElectroChem 2021, 8, 3168–3173. [Google Scholar] [CrossRef]
- Yu, P.; Lowe, S.E.; Simon, G.P.; Zhong, Y.L. Electrochemical exfoliation of graphite and production of functional graphene. Curr. Opin. Colloid. Interface Sci. 2015, 20, 329–338. [Google Scholar] [CrossRef]
- Raj, C.J.; Manikandan, R.; Thondaiman, P.; Sivakumar, P.; Savariraj, A.D.; Cho, W.-J.; Kim, B.C.; Jung, H. Sonoelectrochemical exfoliation of graphene in various electrolytic environments and their structural and electrochemical properties. Carbon 2021, 184, 266–276. [Google Scholar] [CrossRef]
- Parvez, K.; Wu, Z.-S.; Li, R.; Liu, X.; Graf, R.; Feng, X.; Müllen, K. Exfoliation of Graphite into Graphene in Aqueous Solutions of Inorganic Salts. J. Am. Chem. Soc. 2014, 136, 6083–6091. [Google Scholar] [CrossRef] [Green Version]
- Rao, K.S.; Senthilnathan, J.; Liu, Y.-F.; Yoshimura, M. Role of Peroxide Ions in Formation of Graphene Nanosheets by Electrochemical Exfoliation of Graphite. Sci. Rep. 2014, 4, 4237. [Google Scholar] [CrossRef] [PubMed]
- Muthoosamy, K.; Manickam, S. State of the art and recent advances in the ultrasound-assisted synthesis, exfoliation and functionalization of graphene derivatives. Ultrason. Sonochemistry 2017, 39, 478–493. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.I.; Mirabella, D.A.; Pablo Tomba, J.; Riccardi, C.C. Optimizing graphene production in ultrasonic devices. Ultrasonics 2020, 100, 105989. [Google Scholar] [CrossRef] [PubMed]
- Afreen, S.; Kokubo, K.; Muthoosamy, K.; Manickam, S. Hydration or hydroxylation: Direct synthesis of fullerenol from pristine fullerene [C60] via acoustic cavitation in the presence of hydrogen peroxide. RSC Adv. 2017, 7, 31930–31939. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Manickam, S.; Lester, E.; Wu, T.; Pang, C.H. Synthesis of graphene oxide and graphene quantum dots from miscanthus via ultrasound-assisted mechano-chemical cracking method. Ultrason. Sonochemistry 2021, 73, 105519. [Google Scholar] [CrossRef]
- Stankovich, S.; Piner, R.D.; Chen, X.; Wu, N.; Nguyen, S.T.; Ruoff, R.S. Stable aqueous dispersions of graphitic nanoplatelets via the reduction of exfoliated graphite oxide in the presence of poly(sodium 4-styrenesulfonate). J. Mater. Chem. 2006, 16, 155–158. [Google Scholar] [CrossRef]
- Paredes, J.I.; Villar-Rodil, S.; Martínez-Alonso, A.; Tascón, J.M.D. Graphene Oxide Dispersions in Organic Solvents. Langmuir 2008, 24, 10560–10564. [Google Scholar] [CrossRef]
- Han, J.T.; Jang, J.I.; Kim, H.; Hwang, J.Y.; Yoo, H.K.; Woo, J.S.; Choi, S.; Kim, H.Y.; Jeong, H.J.; Jeong, S.Y.; et al. Extremely efficient liquid exfoliation and dispersion of layered materials by unusual acoustic cavitation. Sci. Rep. 2014, 4, 5133. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.; Feng, J. The effect of sonication treatment of graphene oxide on the mechanical properties of the assembled films. RSC Adv. 2016, 6, 39681–39687. [Google Scholar] [CrossRef]
- Koshani, R.; Jafari, S.M. Ultrasound-assisted preparation of different nanocarriers loaded with food bioactive ingredients. Adv. Colloid. Interface Sci. 2019, 270, 123–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, X.; Wang, Y.; Wu, L.; Hu, Y. Experimental and Modeling of Conductivity for Electrolyte Solution Systems. ACS Omega 2020, 5, 22465–22474. [Google Scholar] [CrossRef]
- Aksoy, C.; Anakli, D. Synthesis of Graphene Oxide through Ultrasonic Assisted Electrochemical Exfoliation. Open. Chem. 2019, 17, 581–586. [Google Scholar] [CrossRef]
- Myasnikov, I.Y.; Gopin, A.V.; Mikheev, I.V.; Chernysheva, M.G.; Badun, G.A. Presonication of nanodiamond hydrosols in radiolabeling by a tritium thermal activation method. Mendeleev. Commun. 2018, 28, 495–496. [Google Scholar] [CrossRef]
- Wojciech, K.; Sławomir, D. Transition metal impurities in carbon-based materials: Pitfalls, artifacts and deleterious effects. Carbon 2020, 168, 748–845. [Google Scholar] [CrossRef]
- Forbot, N.; Bolibok, P.; Wisniewski, M.; Roszek, K. Carbonaceous Nanomaterials-Mediated Defense against Oxidative Stress. Mini-Rev. Med. Chem. 2020, 20, 294–307. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell. Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Liu, W.-W.; Aziz, A. Review on the Effects of Electrochemical Exfoliation Parameters on the Yield of Graphene Oxide. ACS Omega 2022, 7, 33719–33731. [Google Scholar] [CrossRef]
- Farhat, A.; Keller, J.; Tait, S.; Radjenovic, J. Removal of persistent organic contaminants by electrochemically activated sulfate. Environ. Sci. Technol. 2015, 49, 14326–14333. [Google Scholar] [CrossRef] [PubMed]
- Le, G.T.T.; Chanlek, N.; Manyam, J.; Opaprakasit, P.; Grisdanurak, N.; Sreearunothai, P. Insight into the ultrasonication of graphene oxide with strong changes in its properties and performance for adsorption applications. Chem. Eng. J. 2019, 373, 1212–1222. [Google Scholar] [CrossRef]
- Patil, R.; Bahadur, P.; Tiwari, S. Dispersed graphene materials of biomedical interest and their toxicological consequences. Adv. Colloid. Interface Sci. 2020, 275, 102051. [Google Scholar] [CrossRef]
- Vacacela Gomez, C.; Guevara, M.; Tene, T.; Villamagua, L.; Usca, G.T.; Maldonado, F.; Tapia, C.; Cataldo, A.; Bellucci, S.; Caputi, L.S. The liquid exfoliation of graphene in polar solvents. Appl. Surf. Sci. 2021, 546, 149046. [Google Scholar] [CrossRef]
- Fomina, P.S.; Proskurnin, M.A.; Mizaikoff, B.; Volkov, D.S. Infrared Spectroscopy in Aqueous Solutions: Capabilities and Challenges. Crit. Rev. Anal. Chem. 2022, 1–18. [Google Scholar] [CrossRef]
- Muhmood, T.; Xia, M.; Lei, W.; Wang, F.; Mahmood, A. Fe-ZrO2 imbedded graphene like carbon nitride for acarbose (ACB) photo-degradation intermediate study. Adv. Powder Technol. 2018, 29, 3233–3240. [Google Scholar] [CrossRef]
- Dimiev, A.M.; Alemany, L.B.; Tour, J.M. Graphene Oxide. Origin of Acidity, Its Instability in Water, and a New Dynamic Structural Model. ACS Nano 2013, 7, 576–588. [Google Scholar] [CrossRef]
- Toh, S.Y.; Loh, K.S.; Kamarudin, S.K.; Daud, W.R.W. Graphene production via electrochemical reduction of graphene oxide: Synthesis and characterisation. Chem. Eng. J. 2014, 251, 422–434. [Google Scholar] [CrossRef]
- Pinilla-Sánchez, A.; Chávez-Angel, E.; Murcia-López, S.; Carretero, N.M.; Palardonio, S.M.; Xiao, P.; Rueda-García, D.; Sotomayor Torres, C.M.; Gómez-Romero, P.; Martorell, J.; et al. Controlling the electrochemical hydrogen generation and storage in graphene oxide by in-situ Raman spectroscopy. Carbon 2022, 200, 227–235. [Google Scholar] [CrossRef]
- Kumar, P.V.; Bardhan, N.M.; Tongay, S.; Wu, J.; Belcher, A.M.; Grossman, J.C. Scalable enhancement of graphene oxide properties by thermally driven phase transformation. Nat. Chem. 2014, 6, 151–158. [Google Scholar] [CrossRef]
- Eda, G.; Lin, Y.-Y.; Mattevi, C.; Yamaguchi, H.; Chen, H.-A.; Chen, I.S.; Chen, C.-W.; Chhowalla, M. Blue Photoluminescence from Chemically Derived Graphene Oxide. Adv. Mater. 2010, 22, 505–509. [Google Scholar] [CrossRef]
- Li, D.; Müller, M.B.; Gilje, S.; Kaner, R.B.; Wallace, G.G. Processable aqueous dispersions of graphene nanosheets. Nat. Nanotechnol. 2008, 3, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Mellado, C.; Figueroa, T.; Baez, R.; Meléndrez, M.; Fernández, K. Effects of probe and bath ultrasonic treatments on graphene oxide structure. Mater. Today Chem. 2019, 13, 1–7. [Google Scholar] [CrossRef]
- Kacem, K.; Casanova-Chafer, J.; Ameur, S.; Nsib, M.F.; Llobet, E. Gas sensing properties of graphene oxide loaded with SrTiO3 nanoparticles. J. Alloy. Compd. 2023, 941, 169011. [Google Scholar] [CrossRef]
- Le Fevre, L.W.; Cao, J.; Kinloch, I.A.; Forsyth, A.J.; Dryfe, R.A.W. Systematic Comparison of Graphene Materials for Supercapacitor Electrodes. ChemistryOpen 2019, 8, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankovich, S.; Dikin, D.A.; Piner, R.D.; Kohlhaas, K.A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S.T.; Ruoff, R.S. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 2007, 45, 1558–1565. [Google Scholar] [CrossRef]
- Pattarith, K.; Areerob, Y. Fabrication of Ag nanoparticles adhered on RGO based on both electrodes in dye-sensitized solar cells (DSSCs). Renew. Wind Water Sol. 2020, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Kim, F.; Han, T.H.; Shenoy, V.B.; Huang, J.; Hurt, R.H. Hydration-Responsive Folding and Unfolding in Graphene Oxide Liquid Crystal Phases. ACS Nano 2011, 5, 8019–8025. [Google Scholar] [CrossRef] [Green Version]
- Gengenbach, T.R.; Major, G.H.; Linford, M.R.; Easton, C.D. Practical guides for X-ray photoelectron spectroscopy (XPS): Interpreting the carbon 1s spectrum. J. Vac. Sci. Technol. A 2021, 39, 013204. [Google Scholar] [CrossRef]
- Jiříčková, A.; Jankovský, O.; Sofer, Z.; Sedmidubský, D. Synthesis and Applications of Graphene Oxide. Materials 2022, 15, 920. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.; König, E. Untersuchungen über Graphitoxyd. Z. Für. Anorg. Und. Allg. Chem. 1937, 234, 311–336. [Google Scholar] [CrossRef]
- Zhu, Y.; Stoller, M.D.; Cai, W.; Velamakanni, A.; Piner, R.D.; Chen, D.; Ruoff, R.S. Exfoliation of Graphite Oxide in Propylene Carbonate and Thermal Reduction of the Resulting Graphene Oxide Platelets. ACS Nano 2010, 4, 1227–1233. [Google Scholar] [CrossRef]
- Dubin, S.; Gilje, S.; Wang, K.; Tung, V.C.; Cha, K.; Hall, A.S.; Farrar, J.; Varshneya, R.; Yang, Y.; Kaner, R.B. A One-Step, Solvothermal Reduction Method for Producing Reduced Graphene Oxide Dispersions in Organic Solvents. ACS Nano 2010, 4, 3845–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernova, E.A.; Petukhov, D.I.; Chumakov, A.P.; Kirianova, A.V.; Sadilov, I.S.; Kapitanova, O.O.; Boytsova, O.V.; Valeev, R.G.; Roth, S.V.; Eliseev, A.A.; et al. The role of oxidation level in mass-transport properties and dehumidification performance of graphene oxide membranes. Carbon 2021, 183, 404–414. [Google Scholar] [CrossRef]
- Schramm, L.L. Emulsions, Foams, and Suspensions: Fundamentals and Applications; Wiley: Weinheim, Germany, 2005; pp. 117–154. [Google Scholar]
- Mikheev, I.V.; Byvsheva, S.M.; Sozarukova, M.M.; Kottsov, S.Y.; Proskurnina, E.V.; Proskurnin, M.A. High-Throughput Preparation of Uncontaminated Graphene-Oxide Aqueous Dispersions with Antioxidant Properties by Semi-Automated Diffusion Dialysis. Nanomaterials 2022, 12, 4159. [Google Scholar] [CrossRef]
- Kadiiska, M.B.; Maples, K.R.; Mason, R.P. A comparison of cobalt(II) and iron(II) hydroxyl and superoxide free radical formation. Arch. Biochem. Biophys. 1989, 275, 98–111. [Google Scholar] [CrossRef]
- Zhidkova, T.V.; Proskurnina, E.V.; Parfenov, E.A.; Vladimirov, Y.A. Determination of superoxide dismutase and SOD-mimetic activities by a chemical system: Co(II)/H2O2/Lucigenin. Anal. Bioanal. Chem. 2011, 401, 381–386. [Google Scholar] [CrossRef]
- Huu, T.P.; Marquetty, C.; Pasquier, C.; Hakim, J. Luminol assay for microdetermination of superoxide dismutase activity: Its application to human fetal blood. Anal. Biochem. 1984, 142, 467–472. [Google Scholar] [CrossRef]
- Bedouhene, S.; Moulti-Mati, F.; Hurtado-Nedelec, M.; Dang, P.M.; El-Benna, J. Luminol-amplified chemiluminescence detects mainly superoxide anion produced by human neutrophils. Am. J. Blood Res. 2017, 7, 41–48. [Google Scholar] [PubMed]
- Gutteridge, J.M.; Halliwell, B. The measurement and mechanism of lipid peroxidation in biological systems. Trends Biochem. Sci. 1990, 15, 129–135. [Google Scholar] [CrossRef]
- Vladimirov, Y.A.; Sharov, V.S.; Driomina, E.S.; Reznitchenko, A.V.; Gashev, S.B. Coumarin derivatives enhance the chemiluminescence accompanying lipid peroxidation. Free. Radic. Biol. Med. 1995, 18, 739–745. [Google Scholar] [CrossRef]
- Itabe, H.; Kobayashi, T.; Inoue, K. Generation of toxic phospholipid(s) during oxyhemoglobin-induced peroxidation of phosphatidylcholines. Biochim. Biophys. Acta 1988, 961, 13–21. [Google Scholar] [PubMed]
- Li, Q.T.; Yeo, M.H.; Tan, B.K. Lipid peroxidation in small and large phospholipid unilamellar vesicles induced by water-soluble free radical sources. Biochem. Biophys. Res. Commun. 2000, 273, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Panasenko, O.M.; Evgina, S.A.; Driomina, E.S.; Sharov, V.S.; Sergienko, V.I.; Vladimirov, Y.A. Hypochlorite induces lipid peroxidation in blood lipoproteins and phospholipid liposomes. Free Radic. Biol. Med. 1995, 19, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Vasiljeva, O.V.; Lyubitsky, O.B.; Klebanov, G.I.; Vladimirov, Y.A. Effect of the combined action of flavonoids, ascorbate and alpha-tocopherol on peroxidation of phospholipid liposomes induced by Fe2+ ions. Membr. Cell. Biol. 2000, 14, 47–56. [Google Scholar] [PubMed]
- Hsieh, H.-S.; Zepp, R.G. Reactivity of graphene oxide with reactive oxygen species (hydroxyl radical, singlet oxygen, and superoxide anion). Environ. Sci. Nano 2019, 6, 3734–3744. [Google Scholar] [CrossRef] [Green Version]
- Mouhat, F.; Coudert, F.-X.; Bocquet, M.-L. Structure and chemistry of graphene oxide in liquid water from first principles. Nat. Commun. 2020, 11, 1566. [Google Scholar] [CrossRef] [Green Version]
- Ederer, J.; Janoš, P.; Ecorchard, P.; Štengl, V.; Bělčická, Z.; Šťastný, M.; Pop-Georgievski, O.; Dohnal, V. Quantitative determination of acidic groups in functionalized graphene by direct titration. React. Funct. Polym. 2016, 103, 44–53. [Google Scholar] [CrossRef]
- Sozarukova, M.M.; Proskurnina, E.V.; Popov, A.L.; Kalinkin, A.L.; Ivanov, V.K. New facets of nanozyme activity of ceria: Lipo- and phospholipoperoxidase-like behaviour of CeO2 nanoparticles. RSC Adv. 2021, 11, 35351–35360. [Google Scholar] [CrossRef]
- Mikheev, I.V.; Pirogova, M.O.; Usoltseva, L.O.; Uzhel, A.S.; Bolotnik, T.A.; Kareev, I.E.; Bubnov, V.P.; Lukonina, N.S.; Volkov, D.S.; Goryunkov, A.A.; et al. Green and rapid preparation of long-term stable aqueous dispersions of fullerenes and endohedral fullerenes: The pros and cons of an ultrasonic probe. Ultrason. Sonochemistry 2021, 73, 105533. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Type of Anode (Stage 1/Stage 2) | Current, A (Stage 1/Stage 2) | Current Density (at the Working Electrode) j, mA/cm2 | Voltage, V (Stage 1/Stage 2) | Espouse Time, min | Procedure Reference Number | Separated Cathode–Anode Compartment | Comment | Brief Result |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Graphite rod ⌀ = 3.2 mm/Pt wire | 2.17/3.37 | 2.14/7.14 | 10.2/8.25 | 15 + 75 | Procedure 2 | Not used | Anode activation for 10 min using continuous ultrasound treatment | Without the formation of EGO (the anode is stratified) |
| 2 | Graphite rod ⌀ = 2 mm/Pt wire | 0.53/2.5 | 0.84/5.30 | 15.4/10.6 | 50 + 60 | Procedure 3 | With membrane | Collected the resulting graphitized foam * | EGO 2 |
| 3 | Graphite rod ⌀ = 2 mm/Pt wire | 0.53/2.5 | 0.84/5.30 | 15.4/10.6 | 50 + 60 | Procedure 3 | With membrane 10 × 10 cm 3.5 kDa | Collected the solution * | Sample with a negligible ratio of C/O |
| 4 | Graphite rod ⌀ = 3.2 mm/n/a | 0.15/n/a | 0.15/n/a | 30/n/a | 15 h 15 min | Procedure 4 | Salt bridge with 3.5 M KCl | Without ultrasonic treatment | Sample with a negligible ratio of C/O |
| 5 | Graphite rod ⌀ = 3.2 mm/n/a | 0.1/n/a | 0.10/n/a | 30/n/a | 5 h | Procedure 5 | Salt bridge with 3.5 M KCl | n/a * | EGO 5 |
| 6 | Graphite rod ⌀ = 3.2 mm/n/a | 3/n/a | 2.96/n/a | 12.3/n/a | 1.5 h | Procedure 6 | Not used | n/a * | EGO 6 |
| Type of Amplitude Modulation of Ultrasound | Ultrasound Processing Time (Working Time) (s) | Rest Time (s) | c(Ti) *, ppm | c(Al) *, ppm | Number of Modulations | Total Time (min) |
|---|---|---|---|---|---|---|
| Continuous mode | 30 min | 0 | 36 | 1.85 | 0 | 30 |
| 1/5 | 2 | 9.9 | 280 | 525 | 900 | 180 |
| 1/3 | 2 | 6 | 135 | 245 | 900 | 120 |
| 1/2 | 2 | 4 | 70 | 120 | 900 | 90 |
| 2/3 | 2 | 3 | 55 | 100 | 900 | 75 |
| 1 | 2 | 2 | 30 | 50 | 900 | 60 |
| 2 | 4 | 2 | 15 | 20 | 450 | 45 |
| 3 | 6 | 3 | 12 | 15 | 300 | 45 |
| 4 | 8 | 2 | 9.3 | 13 | 225 | 38 |
| 5 | 9 | 1.8 | 8.4 | 11 | 200 | 36 |
| 10 | 9 | 0.9 | 2.2 | 3.2 | 200 | 33 |
| Spectra | Binding Energies, eV | Fraction of Atoms, % | Bond Type | ||
|---|---|---|---|---|---|
| EGO 5 | EGO 2 | EGO 6 | |||
| C 1s | 284.1–284.2 | 47.0 | 37.2 | 0.0 | C−C/C−H (sp2) |
| 284.9–285.0 | 17.6 | 39.4 | 74.9 | C−C/C−H (sp3) | |
| 286.5–286.7 | 24.8 | 14.6 | 14.1 | C−O | |
| 287.8–287.9 | 5.3 | 4.0 | 4.6 | O−C−O, C=O | |
| 288.8–289.0 | 5.3 | 4.8 | 6.4 | O=C−O | |
| C, at. % | 74.5 | 64.0 | 70.0 | ||
| O, at. % | 24.6 | 28.9 | 22.3 | ||
| C:O ratio | 4.04 | 2.95 | 4.19 | ||
| Sample Badge | Procedure | GO Flake Size, nm | PDI | Zeta Potential, mV | cTOC, ppm | Yield, % | |
|---|---|---|---|---|---|---|---|
| EGO 2 | 3 | 270 ± 30 | 80 ± 10 | 0.567 | −18.7 ± 1.1 | 80 ± 10 | 9.2 |
| EGO 5 | 5 | 360 ± 35 | 140 ± 15 | 0.373 | −23.4 ± 1.1 | 76 ± 10 | 8.1 |
| EGO 6 | 5 | 190 ± 20 | 140 ± 15 | 0.580 | −22.5 ± 1.1 | 84 ± 10 | 9.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ratova, D.-M.V.; Mikheev, I.V.; Chermashentsev, G.R.; Maslakov, K.I.; Kottsov, S.Y.; Stolbov, D.N.; Maksimov, S.V.; Sozarukova, M.M.; Proskurnina, E.V.; Proskurnin, M.A. Green and Sustainable Ultrasound-Assisted Anodic Electrochemical Preparation of Graphene Oxide Dispersions and Their Antioxidant Properties. Molecules 2023, 28, 3238. https://doi.org/10.3390/molecules28073238
Ratova D-MV, Mikheev IV, Chermashentsev GR, Maslakov KI, Kottsov SY, Stolbov DN, Maksimov SV, Sozarukova MM, Proskurnina EV, Proskurnin MA. Green and Sustainable Ultrasound-Assisted Anodic Electrochemical Preparation of Graphene Oxide Dispersions and Their Antioxidant Properties. Molecules. 2023; 28(7):3238. https://doi.org/10.3390/molecules28073238
Chicago/Turabian StyleRatova, Daria-Maria V., Ivan V. Mikheev, Grigoryi R. Chermashentsev, Konstantin I. Maslakov, Sergei Yu. Kottsov, Dmitrii N. Stolbov, Sergey V. Maksimov, Madina M. Sozarukova, Elena V. Proskurnina, and Mikhail A. Proskurnin. 2023. "Green and Sustainable Ultrasound-Assisted Anodic Electrochemical Preparation of Graphene Oxide Dispersions and Their Antioxidant Properties" Molecules 28, no. 7: 3238. https://doi.org/10.3390/molecules28073238