
Repurposing FIASMAs against Acid Sphingomyelinase for COVID-19: A Computational Molecular Docking and Dynamic Simulation Approach
 , , , , ,
, , , , ,
Abstract
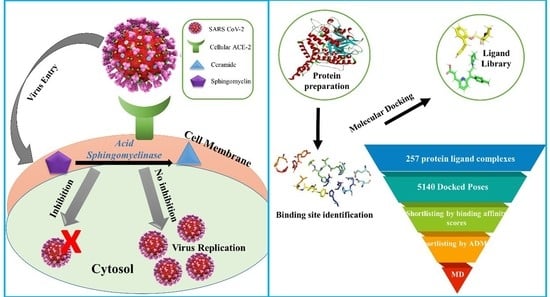
:
1. Introduction
2. Results
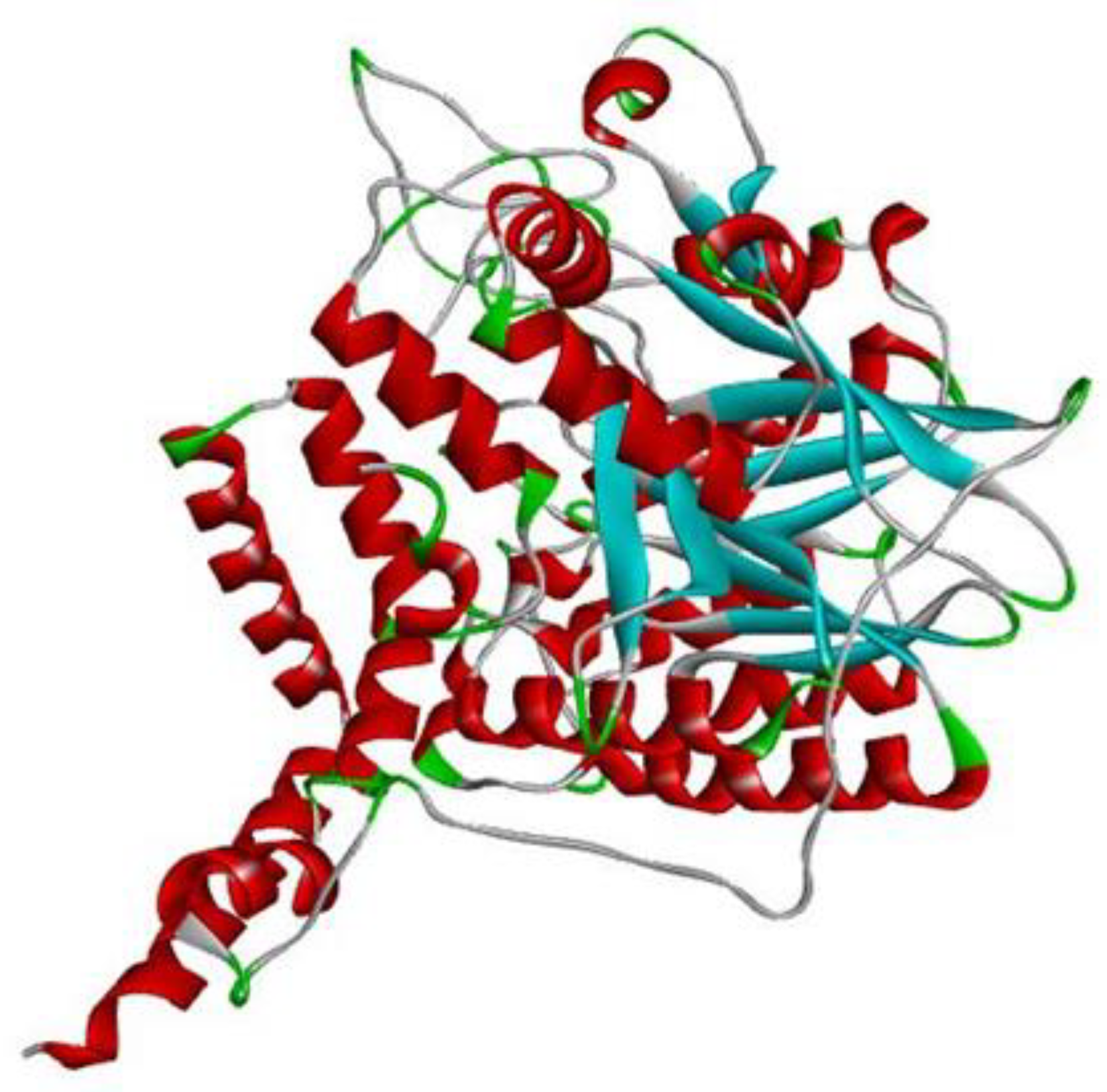
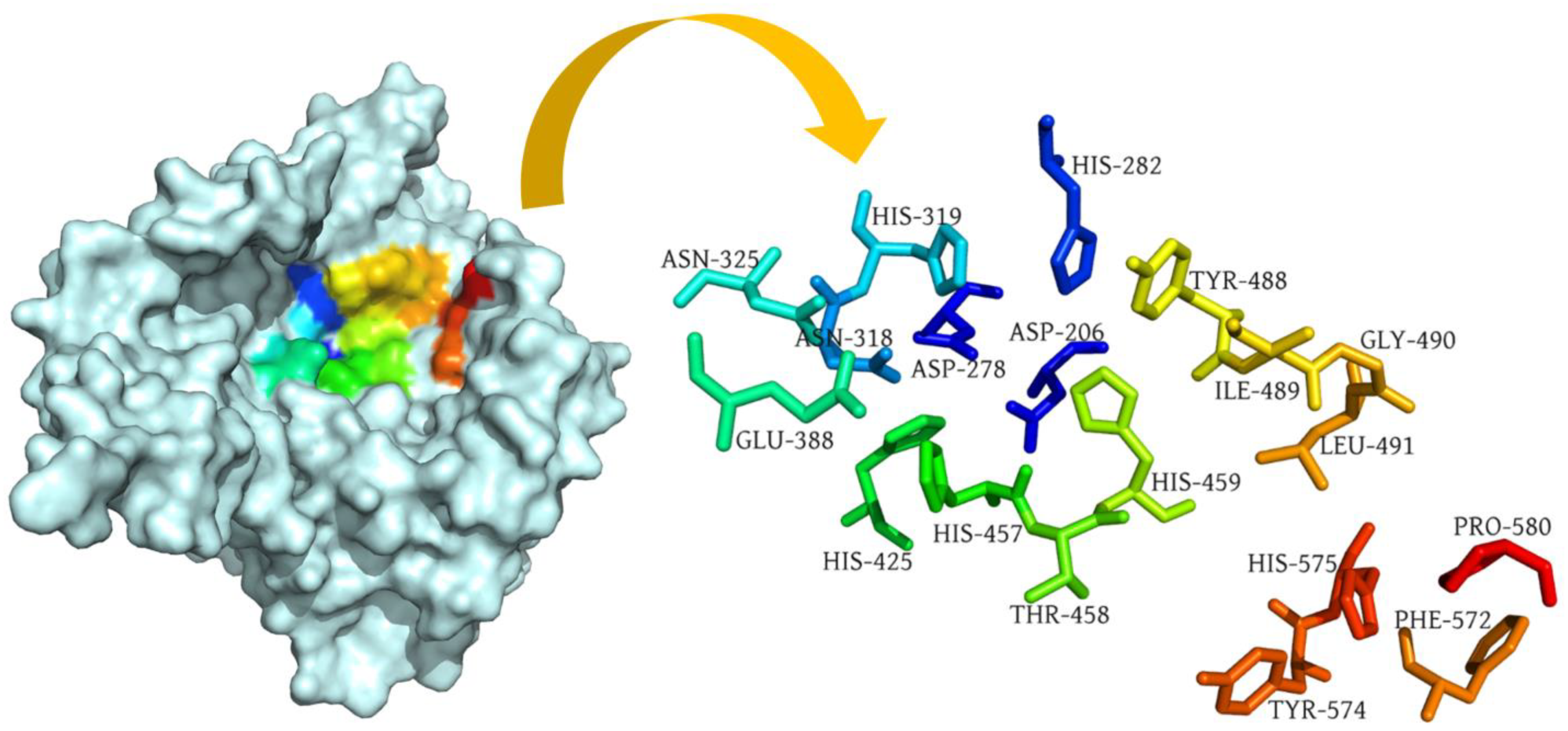
2.1. Target Protein Structure
2.2. Binding Site Prediction
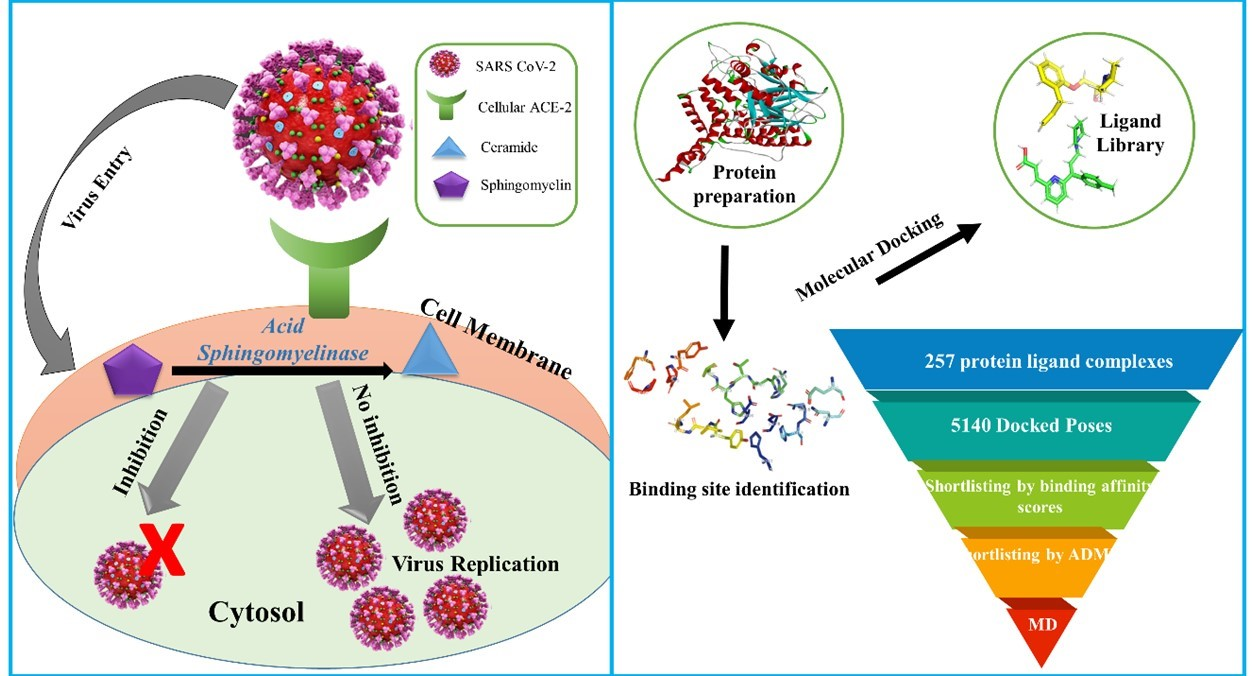
2.3. Molecular Docking and ADME Analysis
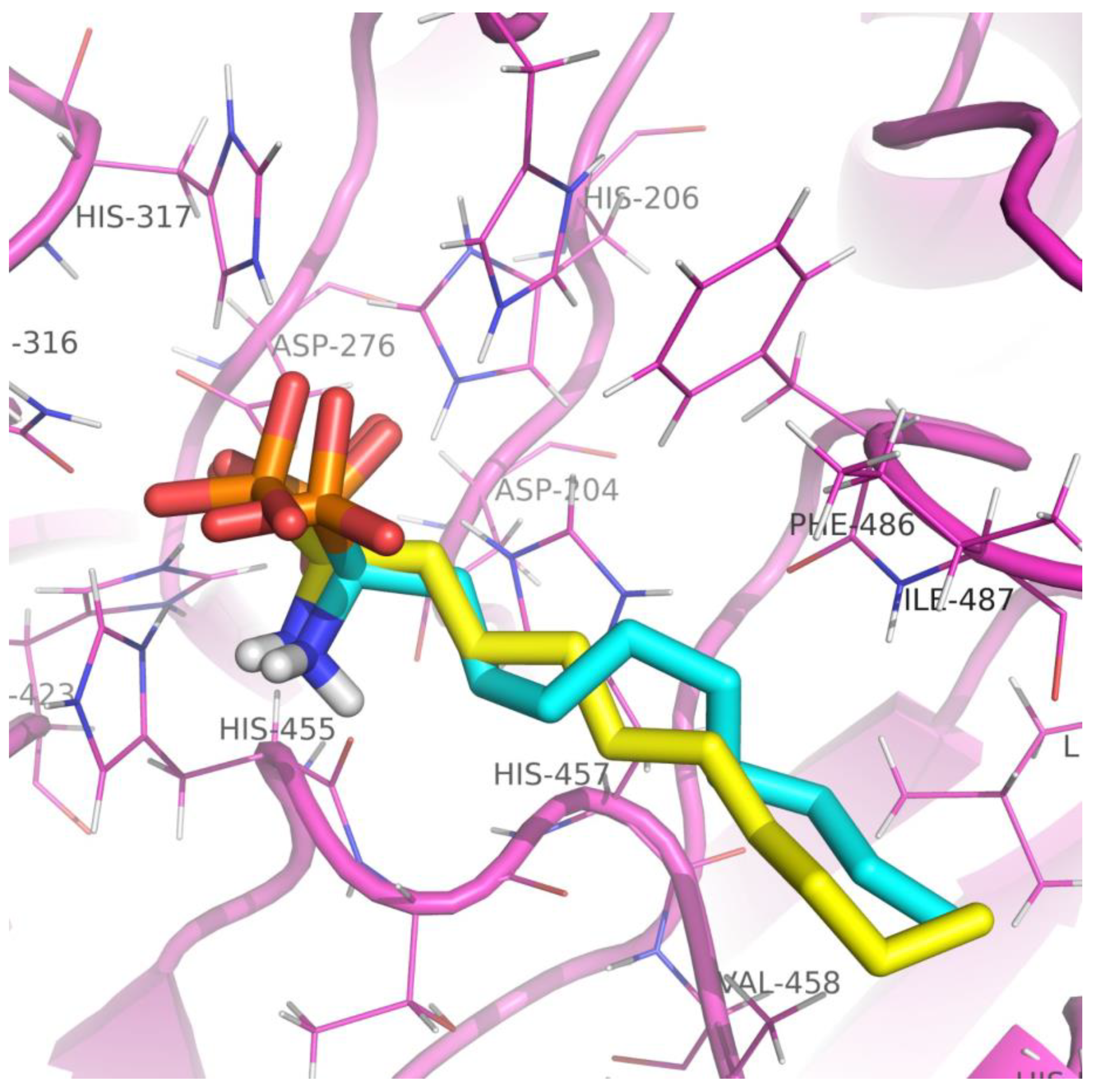
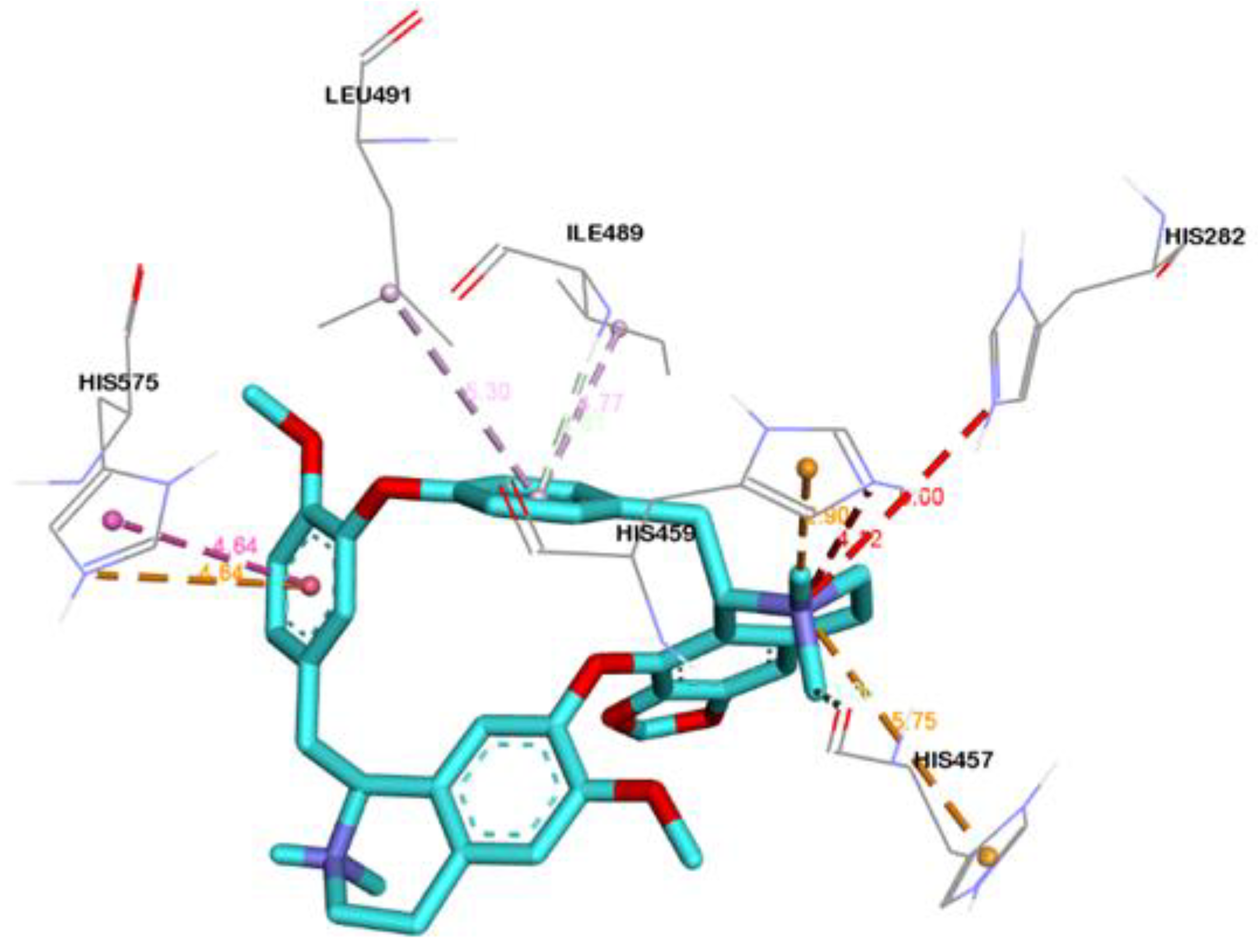
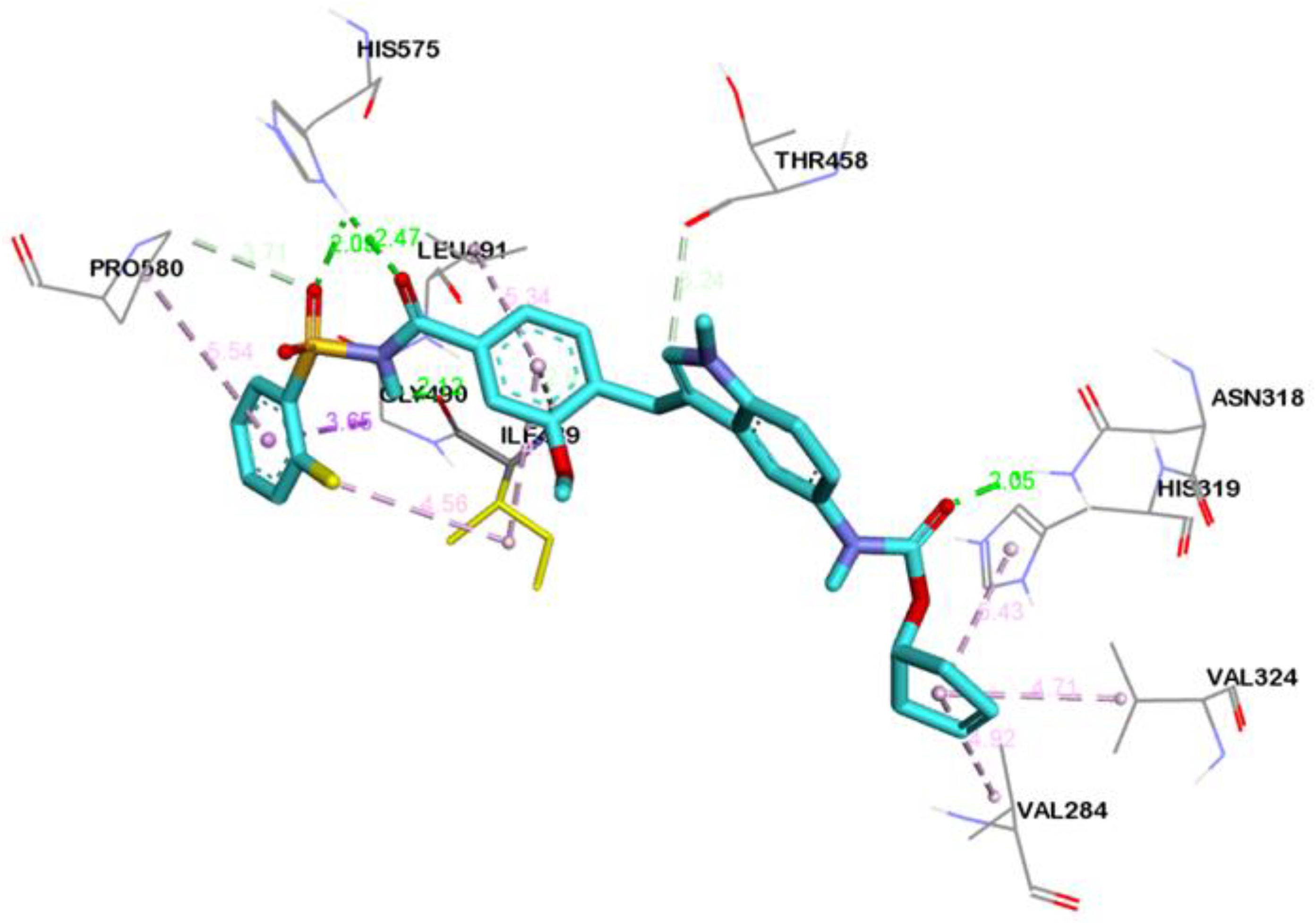
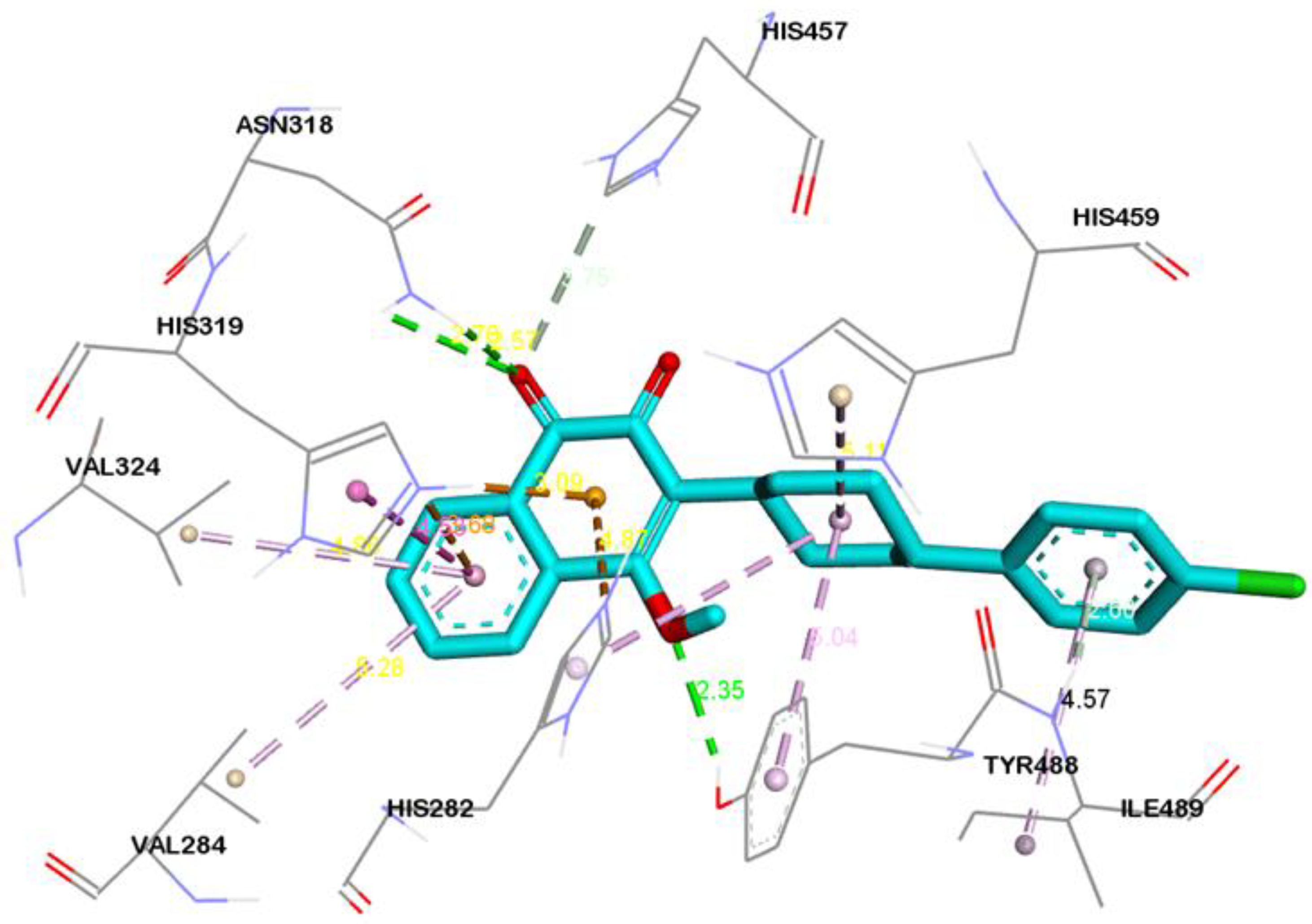
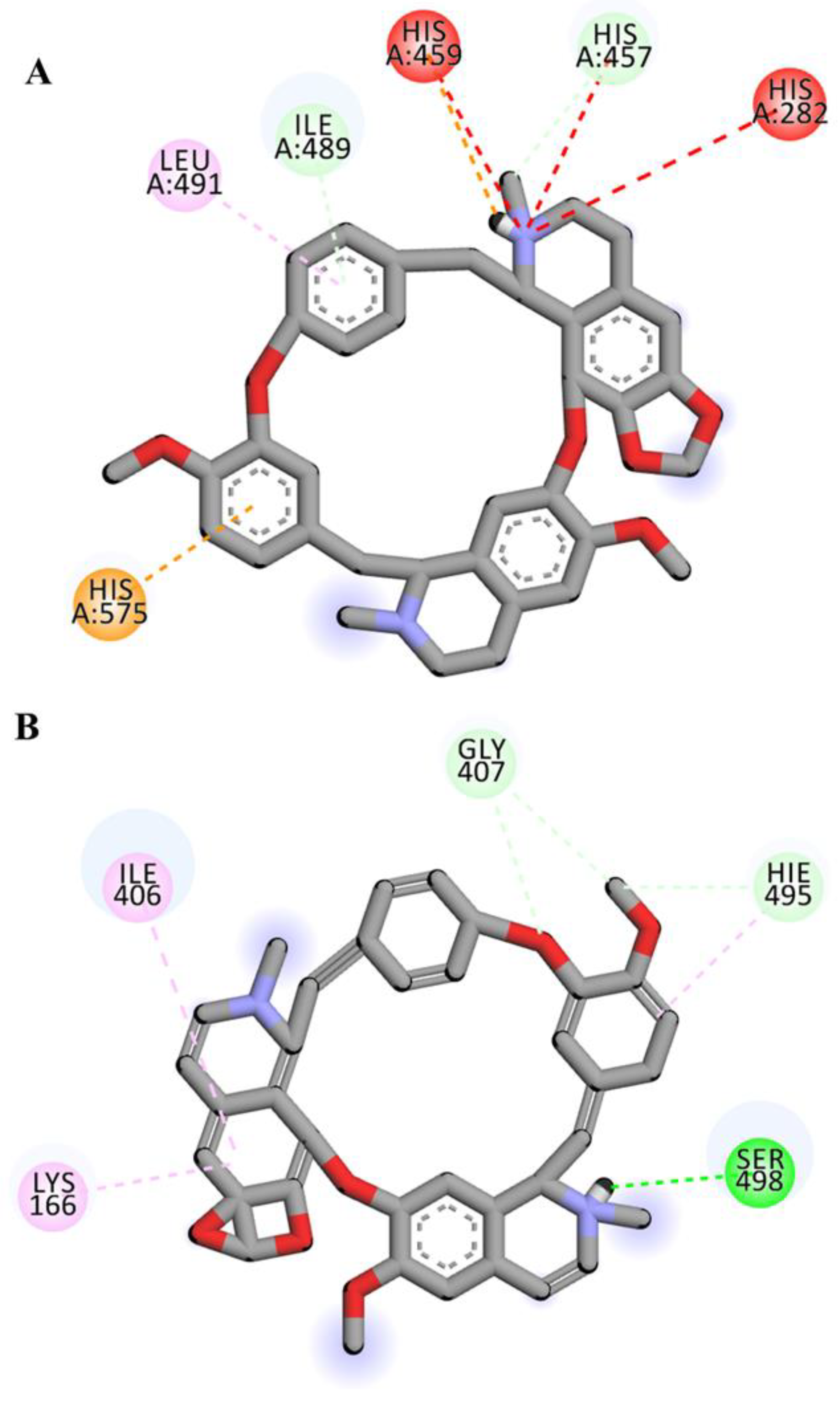
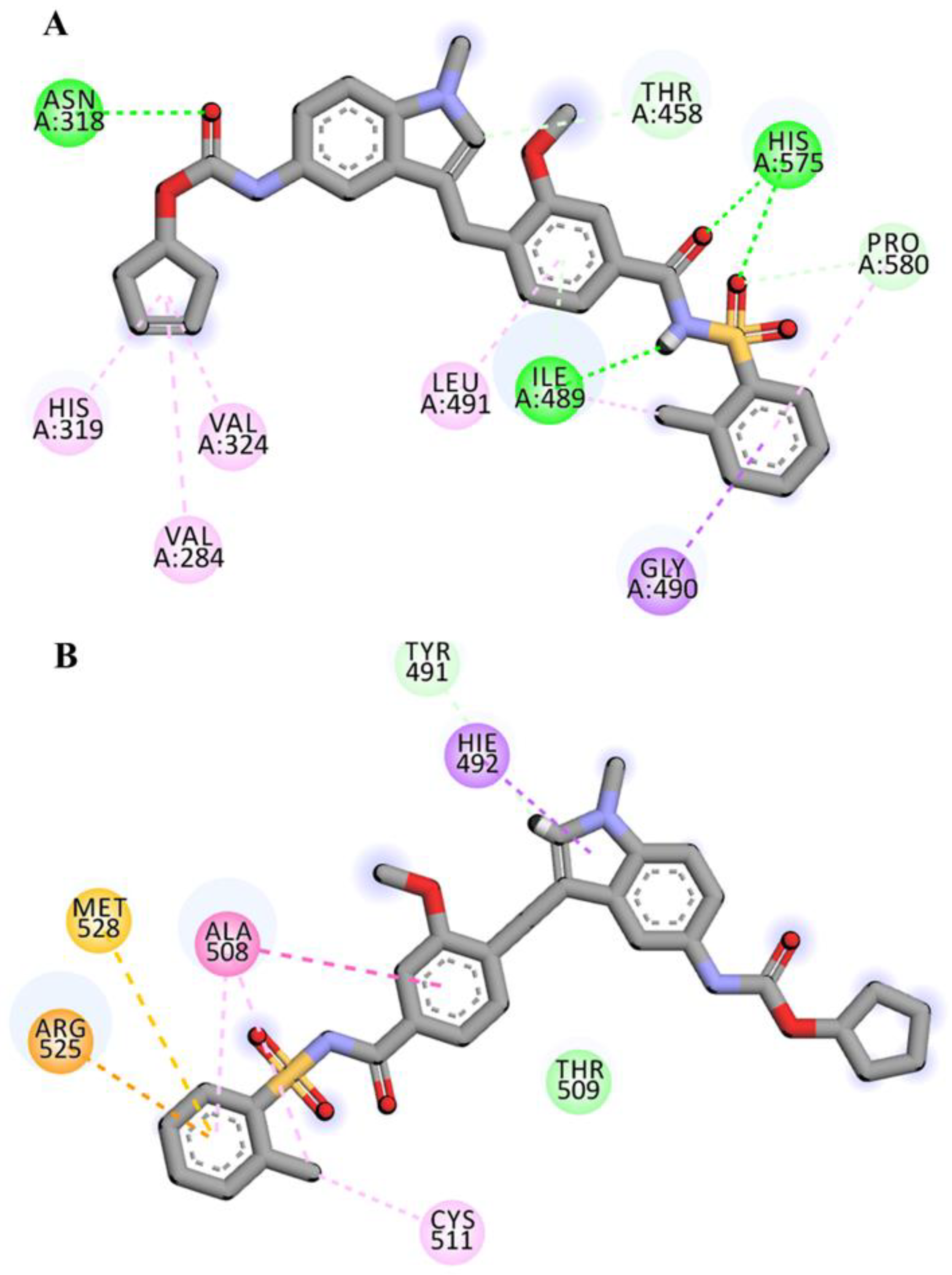
2.4. Visualization and Analysis of Docked Complexes
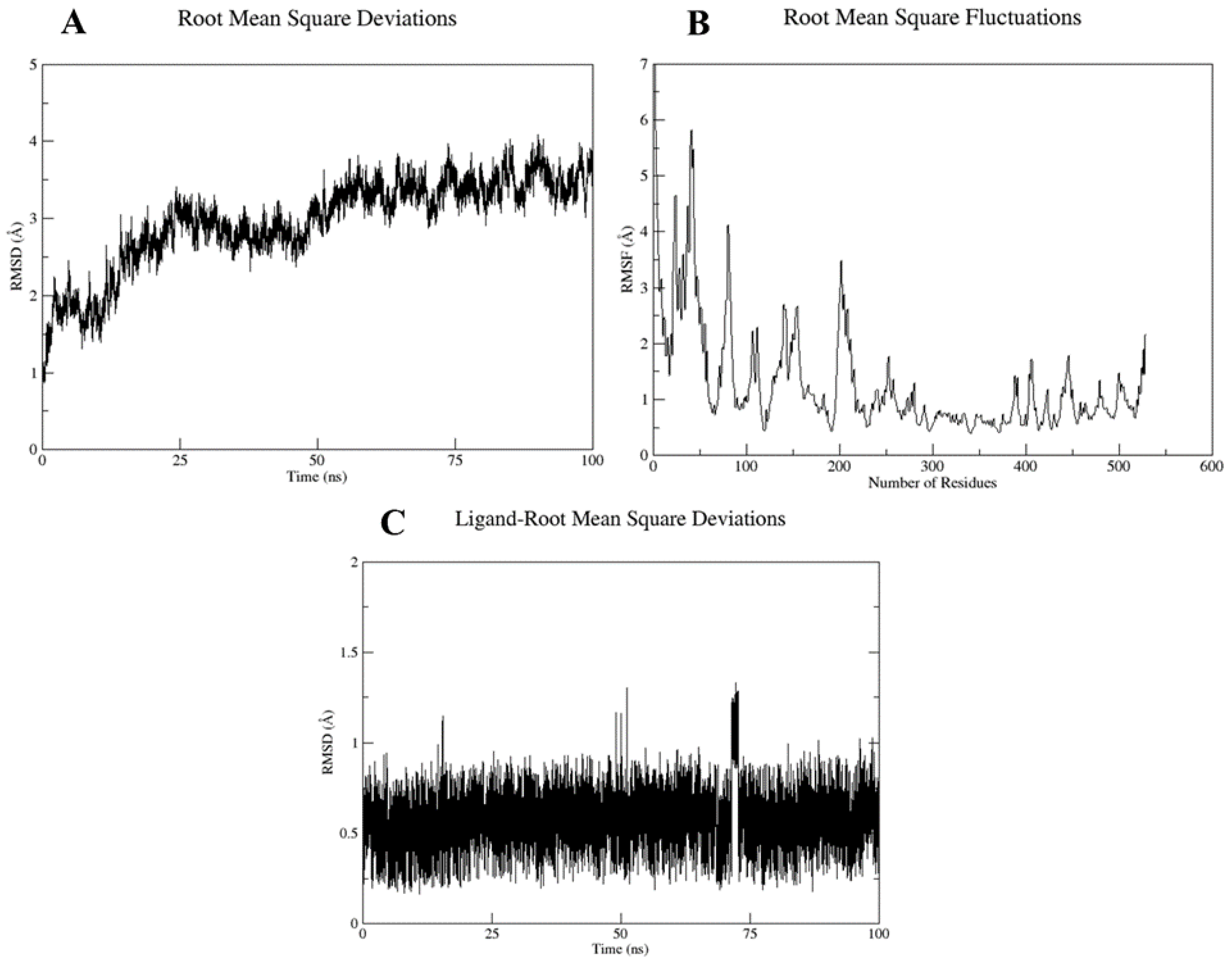
2.5. Molecular Dynamics and Simulation
3. Discussion
4. Materials and Methods
4.1. Target Protein Selection and Preparation
4.2. Binding Site Identification
4.3. Ligand Collection and Preparation
4.4. Molecular Docking
4.5. ADME Analysis
4.6. Molecular Dynamic Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, H.; Liu, S.-M.; Yu, X.-H.; Tang, S.-L.; Tang, C.-K. Coronavirus disease 2019 (COVID-19): Current status and future perspectives. Int. J. Antimicrob. Agents 2020, 55, 105951. [Google Scholar] [CrossRef] [PubMed]
- Alberca, G.G.F.; Fernandes, I.G.; Sato, M.N.; Alberca, R.W. WHAT IS COVID-? In How to Fight Harmful Microbial Bugs and Superbugs; Frontiers for Young Minds: Lausanne, Switzerland, 2021; p. 109. [Google Scholar]
- Fong, S.J.; Dey, N.; Chaki, J.; Fong, S.J.; Dey, N.; Chaki, J. An introduction to COVID-19. In Artificial Intelligence for Coronavirus Outbreak; Springer: Berlin/Heidelberg, Germany, 2021; pp. 1–22. [Google Scholar]
- Ambrosino, P.; Calcaterra, I.L.; Mosella, M.; Formisano, R.; D’anna, S.E.; Bachetti, T.; Marcuccio, G.; Galloway, B.; Mancini, F.P.; Papa, A. Endothelial dysfunction in COVID-19: A unifying mechanism and a potential therapeutic target. Biomedicines 2022, 10, 812. [Google Scholar] [CrossRef]
- Salari, N.; Hosseinian-Far, A.; Jalali, R.; Vaisi-Raygani, A.; Rasoulpoor, S.; Mohammadi, M.; Rasoulpoor, S.; Khaledi-Paveh, B. Prevalence of stress, anxiety, depression among the general population during the COVID-19 pandemic: A systematic review and meta-analysis. Glob. Health 2020, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Le Corre, P.; Loas, G. Repurposing functional inhibitors of acid sphingomyelinase (fiasmas): An opportunity against SARS-CoV-2 infection? J. Clin. Pharm. Ther. 2021, 46, 1213. [Google Scholar] [CrossRef]
- Negro, F. Abnormalities of lipid metabolism in hepatitis C virus infection. Gut 2010, 59, 1279–1287. [Google Scholar] [CrossRef]
- Abu-Farha, M.; Thanaraj, T.A.; Qaddoumi, M.G.; Hashem, A.; Abubaker, J.; Al-Mulla, F. The role of lipid metabolism in COVID-19 virus infection and as a drug target. Int. J. Mol. Sci. 2020, 21, 3544. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Pu, J.; Wu, Y. The role of lipid metabolism in influenza A virus infection. Pathogens 2021, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Schaulies, J.; Schneider-Schaulies, S. Viral Infections and Sphingolipids; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Yager, E.J.; Konan, K.V. Sphingolipids as potential therapeutic targets against enveloped human RNA viruses. Viruses 2019, 11, 912. [Google Scholar] [CrossRef] [Green Version]
- Schneider-Schaulies, J.; Schneider-Schaulies, S. Sphingolipids in viral infection. Biol. Chem. 2015, 396, 585–595. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Carpinteiro, A.; Edwards, M.J.; Hoffmann, M.; Kochs, G.; Gripp, B.; Weigang, S.; Adams, C.; Carpinteiro, E.; Gulbins, A.; Keitsch, S. Pharmacological inhibition of acid sphingomyelinase prevents uptake of SARS-CoV-2 by epithelial cells. Cell Rep. Med. 2020, 1, 100142. [Google Scholar] [CrossRef]
- Smith, E.L.; Schuchman, E.H. The unexpected role of acid sphingomyelinase in cell death and the pathophysiology of common diseases. FASEB J. 2008, 22, 3419. [Google Scholar] [CrossRef] [Green Version]
- Kornhuber, J.; Tripal, P.; Gulbins, E.; Muehlbacher, M. Functional inhibitors of acid sphingomyelinase (FIASMAs). In Sphingolipids: Basic Science and Drug Development; Springer: Berlin/Heidelberg, Germany, 2013; pp. 169–186. [Google Scholar]
- Simonis, A.; Schubert-Unkmeir, A. The role of acid sphingomyelinase and modulation of sphingolipid metabolism in bacterial infection. Biol. Chem. 2018, 399, 1135–1146. [Google Scholar] [CrossRef]
- Kornhuber, J.; Hoertel, N.; Gulbins, E. The acid sphingomyelinase/ceramide system in COVID-19. Mol. Psychiatry 2022, 27, 307–314. [Google Scholar] [CrossRef]
- Kornhuber, J.; Tripal, P.; Reichel, M.; Mühle, C.; Rhein, C.; Muehlbacher, M.; Groemer, T.W.; Gulbins, E. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): A novel pharmacological group of drugs with broad clinical applications. Cell. Physiol. Biochem. 2010, 26, 9–20. [Google Scholar] [CrossRef]
- Pauletto, P.; Bortoli, M.; Bright, F.O.; Delgado, C.P.; Nogara, P.A.; Orian, L.; da Rocha, J.B.T. In silico analysis of the antidepressant fluoxetine and similar drugs as inhibitors of the human protein acid sphingomyelinase: A related SARS-CoV-2 inhibition pathway. J. Biomol. Struct. Dyn. 2022, 29, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Chang, L.; Wang, L. Laboratory testing of SARS-CoV, MERS-CoV, and SARS-CoV-2 (2019-nCoV): Current status, challenges, and countermeasures. Rev. Med. Virol. 2020, 30, e2106. [Google Scholar] [CrossRef] [PubMed]
- Hoertel, N.; Sánchez-Rico, M.; Gulbins, E.; Kornhuber, J.; Carpinteiro, A.; Lenze, E.J.; Reiersen, A.M.; Abellán, M.; de La Muela, P.; Vernet, R. Association between FIASMAs and Reduced Risk of Intubation or Death in Individuals Hospitalized for Severe COVID-19: An observational multicenter study. Clin. Pharmacol. Ther. 2021, 110, 1498–1511. [Google Scholar] [CrossRef] [PubMed]
- Schloer, S.; Brunotte, L.; Goretzko, J.; Mecate-Zambrano, A.; Korthals, N.; Gerke, V.; Ludwig, S.; Rescher, U. Targeting the endolysosomal host-SARS-CoV-2 interface by clinically licensed functional inhibitors of acid sphingomyelinase (FIASMA) including the antidepressant fluoxetine. Emerg. Microbes Infect. 2020, 9, 2245–2255. [Google Scholar] [CrossRef]
- Vatansever, H.S.; Becer, E. Relationship between IL-6 and COVID-19: To be considered during treatment. Future Virol. 2020, 15, 817–822. [Google Scholar] [CrossRef]
- Sharma, P.; Joshi, T.; Joshi, T.; Mathpal, S.; Maiti, P.; Nand, M.; Chandra, S.; Tamta, S. In silico screening of natural compounds to inhibit interaction of human ACE2 receptor and spike protein of SARS-CoV-2 for the prevention of COVID-19. J. Biomol. Struct. Dyn. 2021, 41, 646–658. [Google Scholar] [CrossRef]
- Khayrani, A.C.; Irdiani, R.; Aditama, R.; Pratami, D.K.; Lischer, K.; Ansari, M.J.; Chinnathambi, A.; Alharbi, S.A.; Almoallim, H.S.; Sahlan, M. Evaluating the potency of Sulawesi propolis compounds as ACE-2 inhibitors through molecular docking for COVID-19 drug discovery preliminary study. J. King Saud Univ.-Sci. 2021, 33, 101297. [Google Scholar] [CrossRef]
- Khaerunnisa, S.; Kurniawan, H.; Awaluddin, R.; Suhartati, S.; Soetjipto, S. Potential inhibitor of COVID-19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Preprints 2020, 2020, 2020030226. [Google Scholar]
- Khan, S.; Siddiqui, F.; Jain, S.; Sonwane, G. Discovery of potential inhibitors of SARS-CoV-2 (COVID-19) Main Protease (Mpro) from Nigella Sativa (black seed) by molecular docking study. Coronaviruses 2021, 2, 384–402. [Google Scholar] [CrossRef]
- Zhou, Y.-F.; Metcalf, M.C.; Garman, S.C.; Edmunds, T.; Qiu, H.; Wei, R.R. Human acid sphingomyelinase structures provide insight to molecular basis of Niemann–Pick disease. Nat. Commun. 2016, 7, 13082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, R.W.; Ellaway, P.H.; Prochazka, A.; Proske, U. Secondary endings of muscle spindles: Structure, reflex action, role in motor control and proprioception. Exp. Physiol. 2021, 106, 2339–2366. [Google Scholar] [CrossRef]
- Tanchuk, V.Y.; Tanin, V.O.; Vovk, A.I.; Poda, G. A new, improved hybrid scoring function for molecular docking and scoring based on AutoDock and AutoDock Vina. Chem. Biol. Drug Des. 2016, 87, 618–625. [Google Scholar] [CrossRef]
- Schissel, S.L.; Jiang, X.-c.; Tweedie-Hardman, J.; Jeong, T.-s.; Camejo, E.H.; Najib, J.; Rapp, J.H.; Williams, K.J.; Tabas, I. Secretory sphingomyelinase, a product of the acid sphingomyelinase gene, can hydrolyze atherogenic lipoproteins at neutral pH: Implications for atherosclerotic lesion development. J. Biol. Chem. 1998, 273, 2738–2746. [Google Scholar] [CrossRef] [Green Version]
- Kuzu, O.F.; Gowda, R.; Noory, M.A.; Robertson, G.P. Modulating cancer cell survival by targeting intracellular cholesterol transport. Br. J. Cancer 2017, 117, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Kornhuber, J.; Muehlbacher, M.; Trapp, S.; Pechmann, S.; Friedl, A.; Reichel, M.; Mühle, C.; Terfloth, L.; Groemer, T.W.; Spitzer, G.M. Identification of novel functional inhibitors of acid sphingomyelinase. PLoS ONE 2011, 6, e23852. [Google Scholar] [CrossRef] [Green Version]
- Riethmüller, J.; Anthonysamy, J.; Serra, E.; Schwab, M.; Döring, G.; Gulbins, E. Therapeutic efficacy and safety of amitriptyline in patients with cystic fibrosis. Cell. Physiol. Biochem. 2009, 24, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, C.-M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.-H.; Sze, K.-H.; Yang, D.; Shuai, H. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; Van Der Merwe, P.A. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. Elife 2021, 10, e70658. [Google Scholar] [CrossRef]
- Vrbanac, J.; Slauter, R. ADME in drug discovery. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development; Elsevier: Amsterdam, The Netherlands, 2017; pp. 39–67. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Bitencourt-Ferreira, G.; Veit-Acosta, M.; de Azevedo, W.F. Hydrogen bonds in protein-ligand complexes. Methods Mol. Biol. 2019, 2053, 93–107. [Google Scholar]
- Herrebout, W.; Suhm, M. Weak hydrogen bonds–strong effects? Phys. Chem. Chem. Phys. 2011, 13, 13858–13859. [Google Scholar] [CrossRef]
- Sarkhel, S.; Desiraju, G.R. N–H…O, O–H…O, and C–H…O hydrogen bonds in protein–ligand complexes: Strong and weak interactions in molecular recognition. Proteins: Struct. Funct. Bioinform. 2004, 54, 247–259. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Magnitude and mechanism of charge enhancement of CH·· O hydrogen bonds. J. Phys. Chem. A 2013, 117, 10551–10562. [Google Scholar] [CrossRef] [PubMed]
- Gamrad, W.; Dreier, A.; Goddard, R.; Pörschke, K.R. Cation–Cation Pairing by N–C–H…O Hydrogen Bonds. Angew. Chem. Int. Ed. 2015, 54, 4482–4487. [Google Scholar] [CrossRef]
- Itoh, Y.; Nakashima, Y.; Tsukamoto, S.; Kurohara, T.; Suzuki, M.; Sakae, Y.; Oda, M.; Okamoto, Y.; Suzuki, T. N+-CH··· O Hydrogen bonds in protein-ligand complexes. Sci. Rep. 2019, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.U.; Lew, W.; Williams, M.A.; Liu, H.; Zhang, L.; Swaminathan, S.; Bischofberger, N.; Chen, M.S.; Mendel, D.B.; Tai, C.Y. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: Design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997, 119, 681–690. [Google Scholar] [CrossRef]
- Lou, L.L.; Martin, J.C. Selected thoughts on hydrophobicity in drug design. Molecules 2021, 26, 875. [Google Scholar] [CrossRef]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020, 64, e00819-20. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2016, 45, gkw1000. [Google Scholar]
- Morris, G.M.; Huey, R.; Olson, A.J. Using autodock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, A.; Illes, K.; Heinz, L.X.; Superti-Furga, G.; Nagar, B. Crystal structure of mammalian acid sphingomyelinase. Nat. Commun. 2016, 7, 12196. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Dias, R.; de Azevedo, J.; Walter, F. Molecular docking algorithms. Curr. Drug Targets 2008, 9, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Shoichet, B.K.; Kuntz, I.D. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992, 13, 505–524. [Google Scholar] [CrossRef]
- Bell, E.W.; Zhang, Y. DockRMSD: An open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef] [Green Version]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Cheng, T.; He, S.; Thiessen, P.A.; Li, Q.; Gindulyte, A.; Bolton, E.E. PubChem Protein, Gene, Pathway, and Taxonomy data collections: Bridging biology and chemistry through Target-Centric Views of PubChem data. J. Mol. Biol. 2022, 434, 167514. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Shi, H.; Tian, S.; Li, Y.; Li, D.; Yu, H.; Zhen, X.; Hou, T. Absorption, distribution, metabolism, excretion, and toxicity evaluation in drug discovery. 14. Prediction of human pregnane X receptor activators by using naive Bayesian classification technique. Chem. Res. Toxicol. 2015, 28, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasi, S.; Raza, S.; Azam, S.S.; Liedl, K.R.; Fuchs, J.E. Interaction mechanisms of a melatonergic inhibitor in the melatonin synthesis pathway. J. Mol. Liq. 2016, 221, 507–517. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | Binding Affinity | Molecular Weight | XLogP3-AA | H-Bond Donor | H-Bond Acceptor | Topological Polar Surface Area (Å2) | No. of Rotatable Bonds | Lipinski Violation |
|---|---|---|---|---|---|---|---|---|
| Dutasteride | −9.7 | 528.5 | 5.4 | 2 | 8 | 58.2 | 2 | No-2 |
| Cepharanthine | −9.6 | 606.7 | 6.5 | 0 | 8 | 61.9 | 2 | Yes-1 |
| Zafirlukast | −9.5 | 575.5 | 5.5 | 2 | 6 | 124 | 9 | Yes-1 |
| Carbenoxolone | −9.2 | 570.8 | 6.4 | 2 | 7 | 118 | 6 | No-2 |
| Telmisartan | −9.2 | 514.6 | 6.9 | 1 | 4 | 72.9 | 7 | No-2 |
| Atovaquone | −9.1 | 366.8 | 5.2 | 1 | 3 | 54.4 | 2 | Yes-0 |
| Doxorubicin | −8.9 | 543.5 | 1.3 | 6 | 12 | 206 | 5 | No-3 |
| Pirarubicin | −8.9 | 627.6 | 2.7 | 5 | 13 | 204 | 7 | No-2 |
| Profenamine | −8.7 | 312.5 | 4.8 | 0 | 3 | 31.8 | 5 | Yes-1 |
| Ritanserin | −8.7 | 477.6 | 5.2 | 0 | 6 | 61.2 | 5 | Yes-1 |
| Solasodine | −8.7 | 413.6 | 5.4 | 2 | 3 | 41.5 | 0 | Yes-1 |
| Tomatidine | −8.7 | 415.7 | 6.2 | 2 | 3 | 41.5 | 0 | Yes-1 |
| Astemizole | −8.6 | 458.57 | 5.97 | 4 | 1 | 42.32 | 8 | Yes-1 |
| Ligand Name | Residues Involved in Interaction | Type of Interaction | Bond Distance (Å) |
|---|---|---|---|
| Cepharantine | LIG:C---HIS457:O | Carbon Hydrogen Bond | 2.99 |
| HIS575:NE2---LIG | Pi-Cation | 4.64 | |
| LIG:H---HIS459 | Pi-Cation; Pi-Donor Hydrogen Bond | 2.89 | |
| ILE489:HN---LIG | Pi-Donor Hydrogen Bond | 2.60 | |
| HIS575---LIG | Pi-Pi T-shaped | 4.64 | |
| LIG:C---PRO580 | Alkyl | 3.65 | |
| HIS430---LIG:C | Pi-Alkyl | 5.01 | |
| HIS575---LIG:C | Pi-Alkyl | 4.64 | |
| LIG---ILE489 | Pi-Alkyl | 4.76 | |
| LIG---LEU491 | Pi-Alkyl | 5.30 | |
| Zafirlukast | ASN318:HD21---LIG:O | Hydrogen Bond | 2.05 |
| HIS575:HD1---LIG:O | Hydrogen Bond | 2.46 | |
| HIS575:HD1---LIG:O | Hydrogen Bond | 2.09 | |
| LIG:H---ILE489:O | Hydrogen Bond | 2.11 | |
| PRO580:CD---LIG:O | Carbon Hydrogen Bond | 3.70 | |
| LIG:C---THR458:O | Carbon Hydrogen Bond | 3.23 | |
| ILE489:HN---LIG | Pi-Donor Hydrogen Bond | 2.61 | |
| GLY490:CA---LIG | Pi-Sigma | 3.64 | |
| VAL284---LIG | Alkyl | 4.91 | |
| VAL324---LIG | Alkyl | 4.70 | |
| LIG:C---ILE489 | Alkyl | 4.55 | |
| LIG:C---ILE489 | Alkyl | 4.81 | |
| HIS319---LIG | Pi-Alkyl | 5.42 | |
| LIG--- ILE489 | Pi-Alkyl | 4.70 | |
| LIG---LEU491 | Pi-Alkyl | 5.34 | |
| Atovaquone | ASN318:HD21---LIG:O | Hydrogen Bond | 2.57 |
| ASN318:HD22---LIG:O | Hydrogen Bond | 2.76 | |
| TYR488:HH---LIG:O | Hydrogen Bond | 2.35 | |
| HIS457:CE1---LIG:O | Carbon Hydrogen Bond | 3.75 | |
| HIS282:NE2---LIG | Pi-Cation | 4.87 | |
| HIS319:NE2---LIG | Pi-Cation | 3.68 | |
| HIS319:HE2---LIG | Pi-Cation; Pi-Donor Hydrogen Bond | 3.08 | |
| ILE489:HN---LIG | Pi-Donor Hydrogen Bond | 2.60 | |
| HIS319---LIG | Pi-Pi T-shaped | 4.52 | |
| LIG:CL---VAL460 | Alkyl | 5.40 | |
| LIG:CL---LEU491 | Alkyl | 4.24 | |
| HIS459---LIG | Pi-Alkyl | 5.11 | |
| TYR488---LIG | Pi-Alkyl | 5.03 | |
| HIS575---LIG:CL | Pi-Alkyl | 4.93 | |
| LIG---VAL284 | Pi-Alkyl | 5.27 | |
| LIG---VAL324 | Pi-Alkyl | 4.85 | |
| LIG---ILE489 | Pi-Alkyl | 4.56 |
| Parameters | Complex 1 | Complex 2 |
|---|---|---|
| RMSD of complex | 2.99 Å | 3.17 Å |
| Ligand RMSD | 0.57 Å | 2.62 Å |
| RMSF | 1.27 Å | 1.33 Å |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naz, A.; Asif, S.; Alwutayd, K.M.; Sarfaraz, S.; Abbasi, S.W.; Abbasi, A.; Alenazi, A.M.; Hasan, M.E. Repurposing FIASMAs against Acid Sphingomyelinase for COVID-19: A Computational Molecular Docking and Dynamic Simulation Approach. Molecules 2023, 28, 2989. https://doi.org/10.3390/molecules28072989
Naz A, Asif S, Alwutayd KM, Sarfaraz S, Abbasi SW, Abbasi A, Alenazi AM, Hasan ME. Repurposing FIASMAs against Acid Sphingomyelinase for COVID-19: A Computational Molecular Docking and Dynamic Simulation Approach. Molecules. 2023; 28(7):2989. https://doi.org/10.3390/molecules28072989
Chicago/Turabian StyleNaz, Aliza, Sumbul Asif, Khairiah Mubarak Alwutayd, Sara Sarfaraz, Sumra Wajid Abbasi, Asim Abbasi, Abdulkareem M. Alenazi, and Mohamed E. Hasan. 2023. "Repurposing FIASMAs against Acid Sphingomyelinase for COVID-19: A Computational Molecular Docking and Dynamic Simulation Approach" Molecules 28, no. 7: 2989. https://doi.org/10.3390/molecules28072989