
How Does Pseudo-Jahn-Teller Effect Induce the Photoprotective Potential of Curcumin?

Abstract
:
1. Introduction
2. Theory
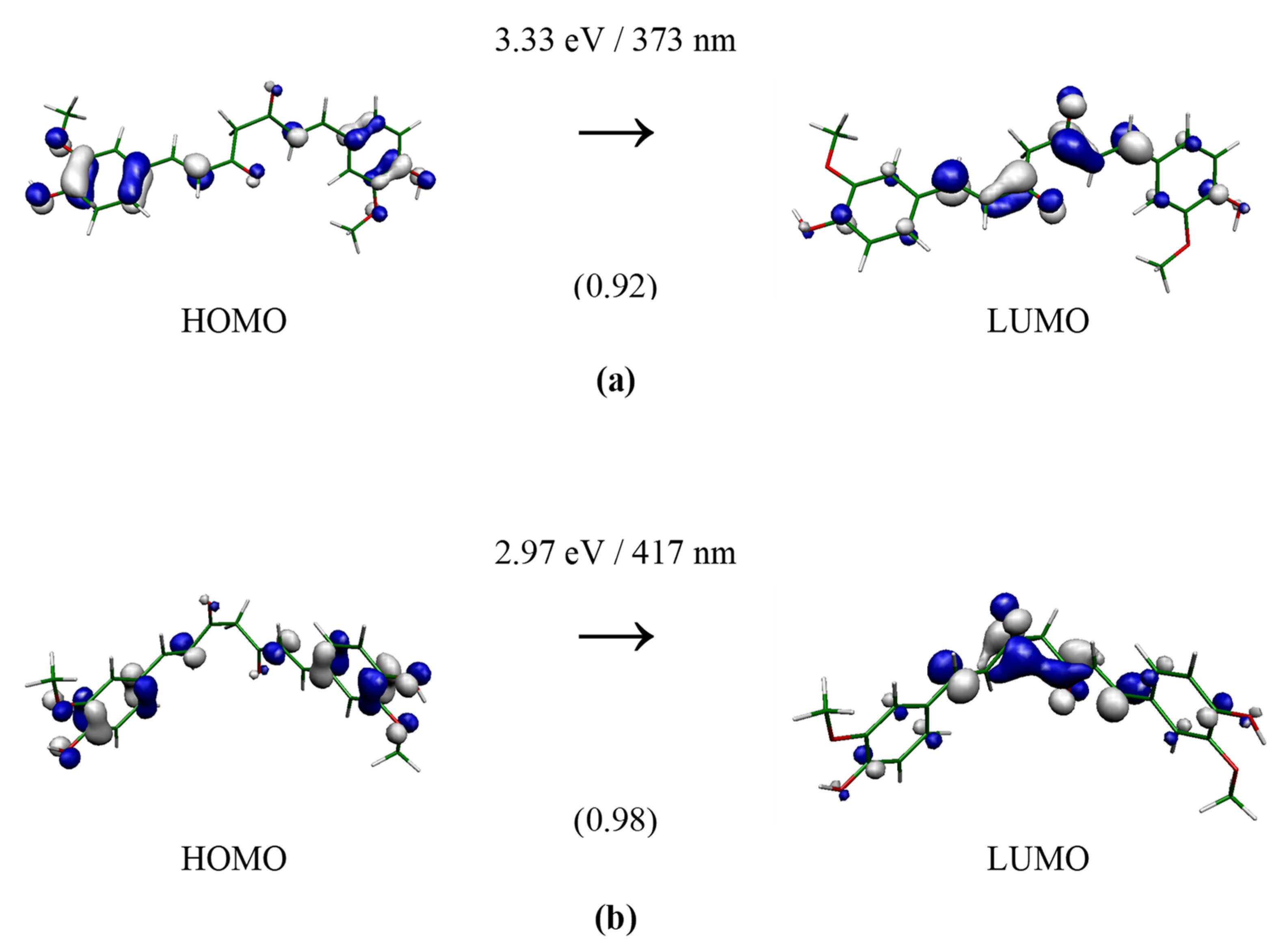
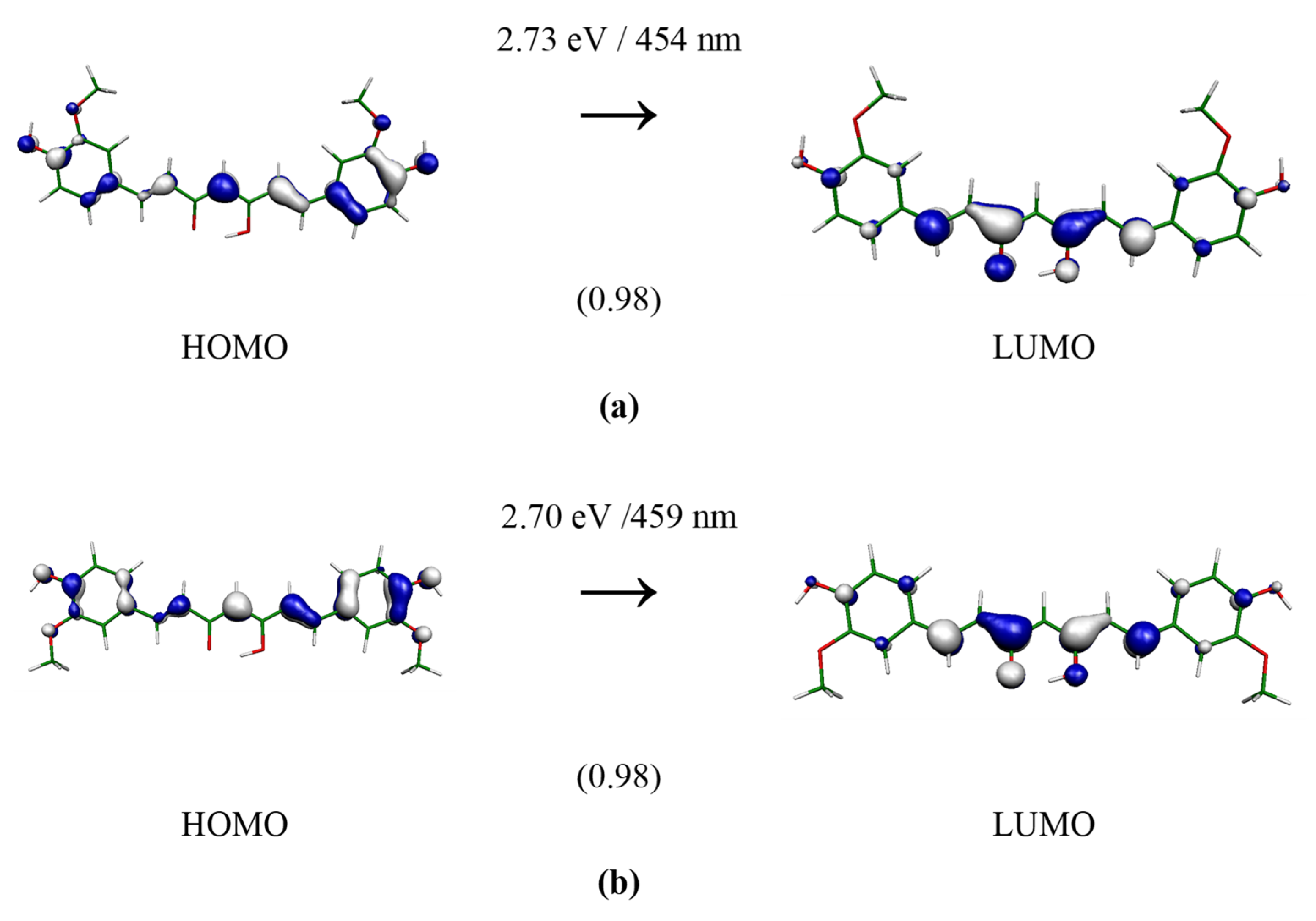
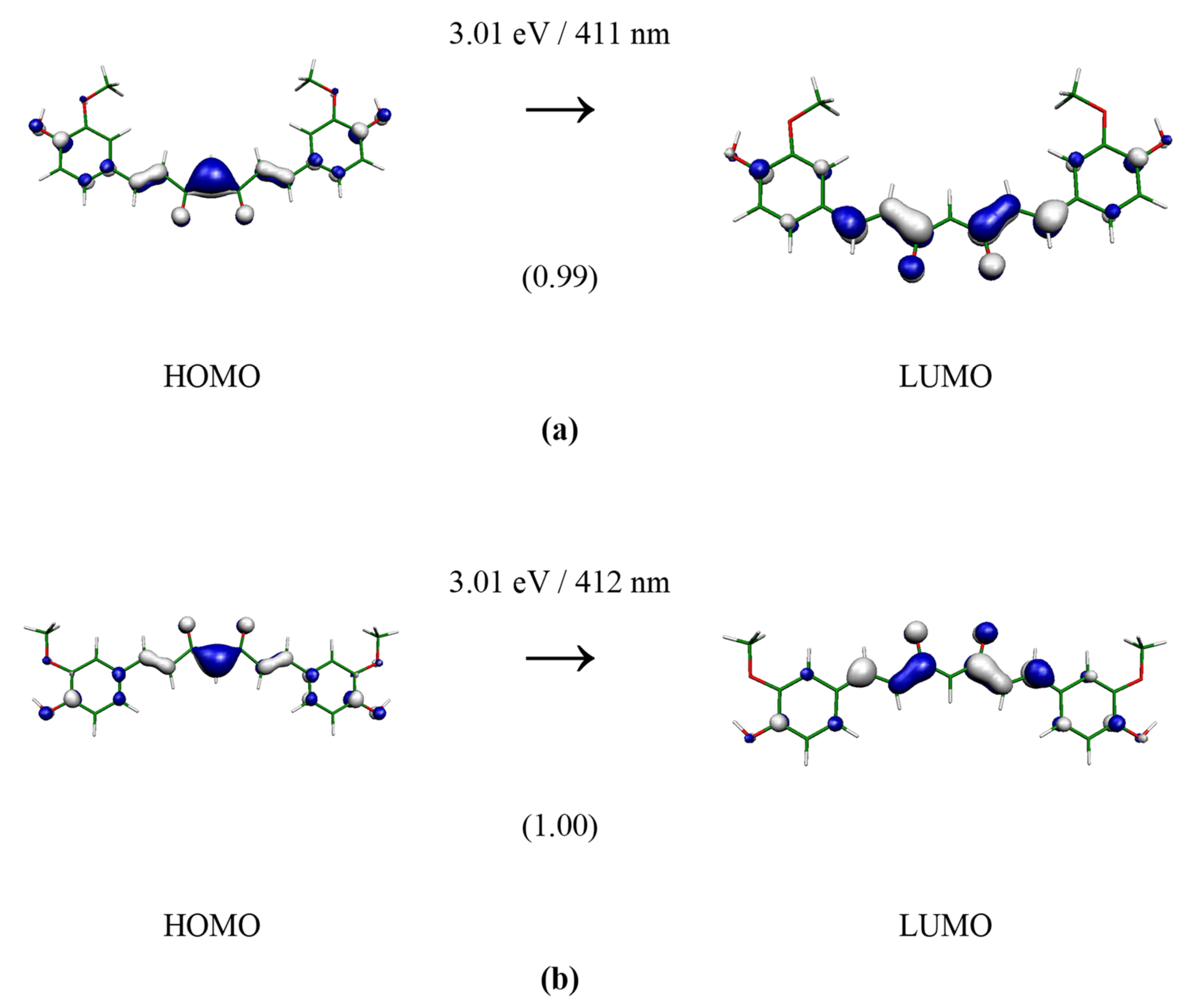
3. Results
4. Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Vendrame, R.; Braga, R.S.; Takahata, Y.; Galvao, D.S. Structure–Activity Relationship Studies of Carcinogenic Activity of Polycyclic Aromatic Hydrocarbons Using Calculated Molecular Descriptors with Principal Component Analysis and Neural Network Methods. J. Chem. Inf. Comput. Sci. 1999, 39, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Leonard, J.J.; Longo, F.R. A mechanistic study of the synthesis and spectral properties of meso-tetraphenylporphyrin. J. Am. Chem. Soc. 1972, 94, 3986–3992. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Sung, B. Pharmacological basis for the role of curcumin in chronic diseases: An age-old spice with modern targets. Trends Pharmacol. 2009, 30, 85–94. [Google Scholar] [CrossRef]
- Jovanovic, S.V.; Boone, C.W.; Steenken, S.; Trinoga, M.; Kaskey, R.B. How curcumin works preferentially with water soluble antioxidants. J. Am. Chem. Soc. 2001, 123, 3064–3068. [Google Scholar] [CrossRef]
- Sun, Y.M.; Zhang, H.Y.; Chen, D.Z.; Liu, C.B. Theoretical Elucidation on the Antioxidant Mechanism of Curcumin: A DFT Study. Org. Lett. 2002, 4, 2909–2911. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Phillips, J.; Moore-Medlin, T.; Sonavane, K.; Ekshyyan, O.; McLarty, J.; Nathan, C.A. Curcumin inhibits UV radiation-induced skin cancer in SKH-1 mice. Otolaryngol. Head Neck Surg. 2013, 148, 797–803. [Google Scholar] [CrossRef]
- Li, H.; Gao, A.; Jiang, N.; Liu, Q.; Liang, B.; Li, R.; Zhang, E.; Li, Z.; Zhu, H. Protective Effect of Curcumin Against Acute Ultraviolet B Irradiation-induced Photo-damage. Photochem. Photobiol. 2016, 92, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Das, K. Picosecond spectral relaxation of curcumin excited state in toluene–alcohol mixtures. J. Lumin. 2013, 144, 169–175. [Google Scholar] [CrossRef]
- Erez, Y.; Presiado, I.; Gepshtein, R.; Huppert, D. Temperature Dependence of the Fluorescence Properties of Curcumin. J. Phys. Chem. 2011, 115, 10962–10971. [Google Scholar] [CrossRef]
- Ghosh, R.; Mondal, J.A.; Palit, D.K. Ultrafast Dynamics of the Excited States of Curcumin in Solution. J. Phys. Chem. B 2010, 114, 12129–12143. [Google Scholar] [CrossRef] [PubMed]
- Patra, D.; Barakat, C. Synchronous fluorescence spectroscopic study of solvatochromic curcumin dye. Spectrochim. Acta A 2011, 79, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Andreev, G.N.; Schrader, B.; Schulz, H.; Fuchs, R.; Popov, S.; Handjieva, N. Non-destructive NIR-FT-Raman analyses in practice. Part I. Analyses of plants and historic textiles. Fresenius J. Anal. Chem. 2001, 371, 1009–1017. [Google Scholar] [PubMed]
- Sanphui, P.; Goud, N.R.; Khandavilli, U.B.R.; Bhanoth, S.; Nangia, A. New polymorphs of curcumin. Chem. Commun. 2011, 47, 5013–5015. [Google Scholar] [CrossRef]
- Ogilby, P.R. Singlet oxygen: There is indeed something new under the sun. Chem. Soc. Rev. 2010, 39, 3181–3209. [Google Scholar] [CrossRef]
- Bersuker, I.B. Recent Developments in the Jahn-Teller Effect theory. The Hidden Jahn-Teller Effect. In The Jahn-Teller effect. Fundamentals and Implications for Physics and Chemistry; Koppel, H., Yarkony, D.R., Barentzen, H., Eds.; Springer Series of Chemical Physics; Springer: Berlin/Heidelberg, Germany, 2009; Volume 97, pp. 3–23. [Google Scholar]
- Garcia-Fernandez, P.; Bersuker, I.B.; Boggs, J.E. Lost topological (Berry) phase factor in electronic structure calculations. Example: The ozone molecule. Phys. Rev. Lett. 2006, 96, 163005. [Google Scholar] [CrossRef]
- García-Fernández, P.; Aramburu, J.A.; Moreno, M.; Zlatar, M.; Gruden-Pavlović, M. A Practical Computational Approach to Study Molecular Instability Using the Pseudo-Jahn–Teller Effect. J. Chem. Theory Comput. 2014, 10, 1824–1833. [Google Scholar] [CrossRef]
- Liu, Y.; Bersuker, I.B.; Zou, W.; Boggs, J.E. Combined Jahn−Teller and Pseudo-Jahn−Teller Effect in the CO3 Molecule: A Seven-State Six-Mode Problem. J. Chem. Theory Comput. 2009, 5, 2679–2686. [Google Scholar] [CrossRef]
- Bersuker, I.B. Modern Aspects of the Jahn-Teller Effect Theory and Applications To Molecular Problems. Chem. Rev. 2001, 101, 1067–1114. [Google Scholar] [CrossRef]
- Bersuker, I.B. The Jahn-Teller Effect; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Andjelkovic, L.; Gruden-Pavlovic, M.; Zlatar, M. Density functional theory study of the multimode Jahn-Teller problem in the open-shell corannulenes and coronenes. Chem. Phys. 2015, 460, 64–75. [Google Scholar] [CrossRef]
- Wallis, M.J.; Min, H.; Lindoy, L.F.; Li, F. Investigating the Conformations of a Family of [M2L3]4+ Helicates Using Single Crystal X-ray Diffraction. Molecules 2023, 28, 1404. [Google Scholar] [CrossRef]
- Debus, R.J. FTIR studies of metal ligands, networks of hydrogen bonds, and water molecules near the active site Mn4CaO5 cluster in Photosystem II. BBA—Bioenerg. 2015, 1847, 19–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenberg, P.; Kohn, H. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Jahn, H.A.; Teller, E. Stability of polyatomic molecules in degenerate electronic states. I. Orbital degeneracy. Proc. R. Soc. Lond. A 1937, 161, 220–235. [Google Scholar]
- Opik, U.; Pryce, M.H.L. Studies of the Jahn-Teller effect I. A survey of the static problem. Proc. R. Soc. London A 1957, 238, 425–447. [Google Scholar]
- Pelikán, P.; Breza, M. Classification of the possible symmetries of the Jahn—Teller systems. Chem. Papers 1984, 39, 255–270. [Google Scholar]
- Breza, M. Group-Theoretical Analysis of Jahn-Teller Systems. In The Jahn-Teller Effect. Fundamentals and Implications for Physics and Chemistry; Köppel, H., Yarkony, D.R., Barentzen, H., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 51–76. ISBN 978-3642034312. [Google Scholar]
- Ceulemans, A.; Beyens, D.; Vanquickenborne, L.G. Symmetry aspects of Jahn-Teller activity: Structure and reactivity. J. Am. Chem. Soc. 1984, 106, 5824–5837. [Google Scholar] [CrossRef]
- Ceulemans, A.; Vanquickenborne, L.G. The Epikernel Principle. Struct. Bonding 1989, 71, 125–159. [Google Scholar]
- Breza, M. Group-Theoretical Treatment of Pseudo-Jahn-Teller Systems. In Vibronic Interactions and the Jahn-Teller Effect: Theory and Application; Atanasov, M., Daul, C., Tregenna-Piggott, P.L.W., Eds.; Springer: Dordrecht, The Netherlands; Berlin/Heidelberg, Germany; London, UK; New York, NY, USA, 2012; pp. 59–82. [Google Scholar]
- Liu, Y.; Bersuker, I.B.; Garcia-Fernandez, P.; Boggs, J.E. Pseudo Jahn–Teller Origin of Nonplanarity and Rectangular-Ring Structure of Tetrafluorocyclobutadiene. J. Phys. Chem. A 2012, 116, 7564–7570. [Google Scholar] [CrossRef]
- Huang, Z.; Albaqam, M.D.; Sato, T.; Iwahara, N.; Chibotaru, L.F. Jahn-Teller effect in the cubic fullerides A3C60. Phys. Rev. B 2021, 103, 134102. [Google Scholar] [CrossRef]
- Manolova, Y.; Deneva, V.; Antonov, L.; Drakalska, E.; Momekova, D.; Lambov, N. The effect of the water on the curcumin tautomerism: A quantitative approach. Spectrochim. Acta A 2014, 132, 815–820. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, D.J.; Karthick, S.N.; Hemalatha, K.V.; Raj, C.J.; Ok, S.; Choe, Y. Curcumin dye extracted from Curcuma longa L. used as sensitizers for efficient dye-sensitized solar cells. Int. J. Electrochem. Sci. 2013, 8, 8320–8328. [Google Scholar]
- Puglisi, A.; Giovannini, T.; Antonov, L.; Cappelli, C. Interplay between conformational and solvent effects in UV-visible absorption spectra: Curcumin tautomers as a case study. Phys. Chem. Chem. Phys. 2019, 21, 15504–15514. [Google Scholar] [CrossRef]
- Erez, Y.; Simkovitch, R.; Shomer, S.; Gepshtein, R.; Huppert, D. Effect of Acid on the Ultraviolet−Visible Absorption and Emission Properties of Curcumin. J. Phys. Chem. A 2014, 118, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Frisch, G.W.M.J.; Schlegel, B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functions. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Bauernschmitt, A.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J.; Mennucci, B.; Tomasi, J.; Cammi, R.; Barone, V. Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys. 2006, 124, 94107. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Ugliengo, P. MOLDRAW: A Program to Display and Manipulate Molecular and Crystal Structures, University Torino, Torino. 2012. Available online: https://www.moldraw.software.informer.com (accessed on 9 September 2019).
- Varetto, U. Molekel, Ver. 5.4.0.8; Swiss National Supercomputing Centre, Lugano, Switzerland. 2009. Available online: https://www.molekel.software.informer.com (accessed on 8 July 2017).
- Jacquemin, D.; Adamo, C. Bond Length Alternation of Conjugated Oligomers: Wave Function and DFT Benchmarks. J. Chem. Theory Comput. 2011, 7, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Perpete, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. TD-DFT Performance for the Visible Absorption Spectra of Organic Dyes: Conventional versus Long-Range Hybrids. J. Chem. Theory Comput. 2008, 4, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Nardo, L.; Paderno, R.; Andreoni, A.; Másson, M.; Haukvik, T.; Tønnesen, H.H. Role of H-bond formation in the photoreactivity of curcumin. Spectroscopy 2008, 22, 187–198. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
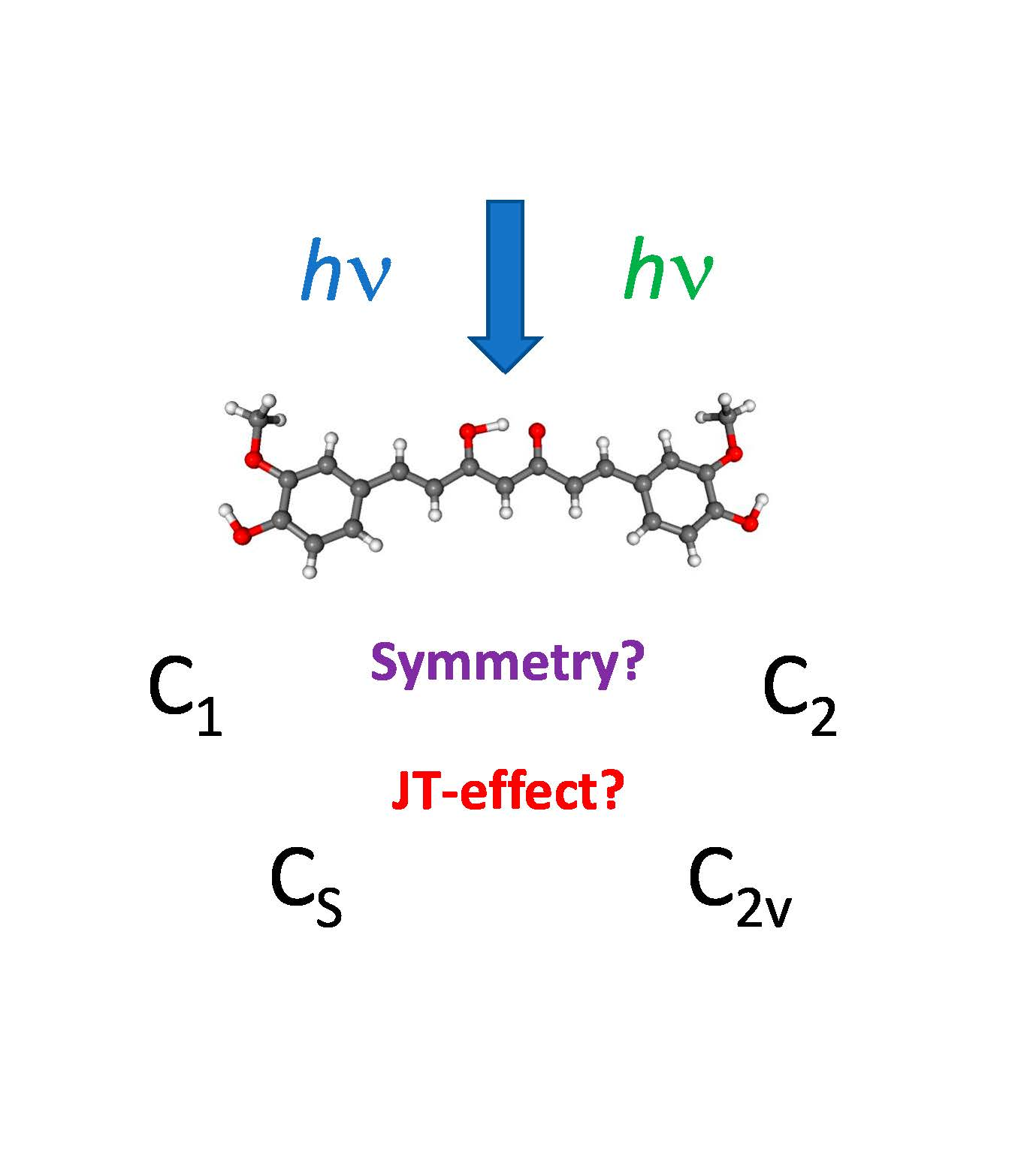
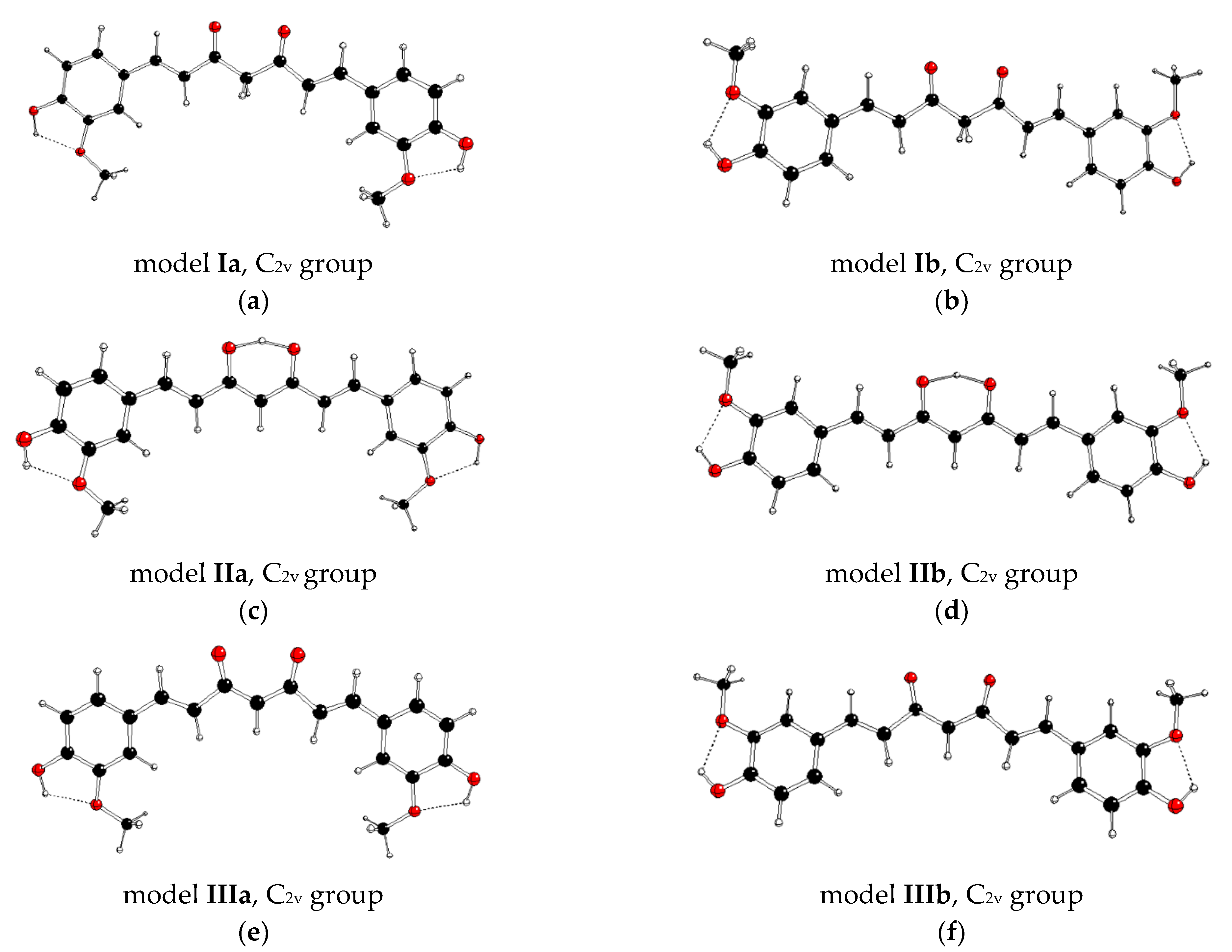
| Model | Form | C=O vs. OCH3 Conformation | Group | State | DFT Energy | Imaginary Vibration | Θ(OC-CO) (Central) | Ω (H3CO-OCH3) | d(O-H) (Central) |
|---|---|---|---|---|---|---|---|---|---|
| Ia | keto–keto | anti, anti | C2v | X1A1 | 0.00 | a2 | 0.0 | 0.0 | - |
| C2 | X1A | −19.88 | - | –111.1 | –155.0 | - | |||
| Ib | keto–keto | syn, syn | C2v | X1A1 | −0.24 | a2 | 0.0 | 0.0 | - |
| C2 | X1A | −28.15 | - | 167.7 | 179.4 | - | |||
| IIa | enol–keto | anti, anti | C2v | X1A1 | −38.12 | b1 | 0.0 | 0.0 | 1.289 (2×) |
| Cs | X1A′ | −59.73 | - | 0.0 | 0.0 | 1.023 1.522 | |||
| Cs | 11A″ | −26.81 | a″ | 0.0 | 0.0 | 0.969 1.913 | |||
| C1 | X1A | −45.50 | - | 0.7 | 38.9 | 1.029 1.499 | |||
| IIb | enol–keto | syn, syn | C2v | X1A1 | −36.78 | b1 | 0.0 | 0.0 | 1.289 (2×) |
| Cs | X1A′ | −58.42 | - | 0.0 | 0.0 | 1.023 1.521 | |||
| Cs | 11A″ | −25.47 | a″ | 0.0 | 0.0 | 0.969 1.915 | |||
| C1 | X1A | −48.18 | - | 0.3 | 6.5 | 1.030 1.493 | |||
| IIIa | enolate | anti, anti | C2v | X1A1 | 1438.35 | - | 0.0 | 0.0 | - |
| IIIb | enolate | syn, syn | C2v | X1A1 | 1441.61 | - | 0.0 | 0.0 | - |
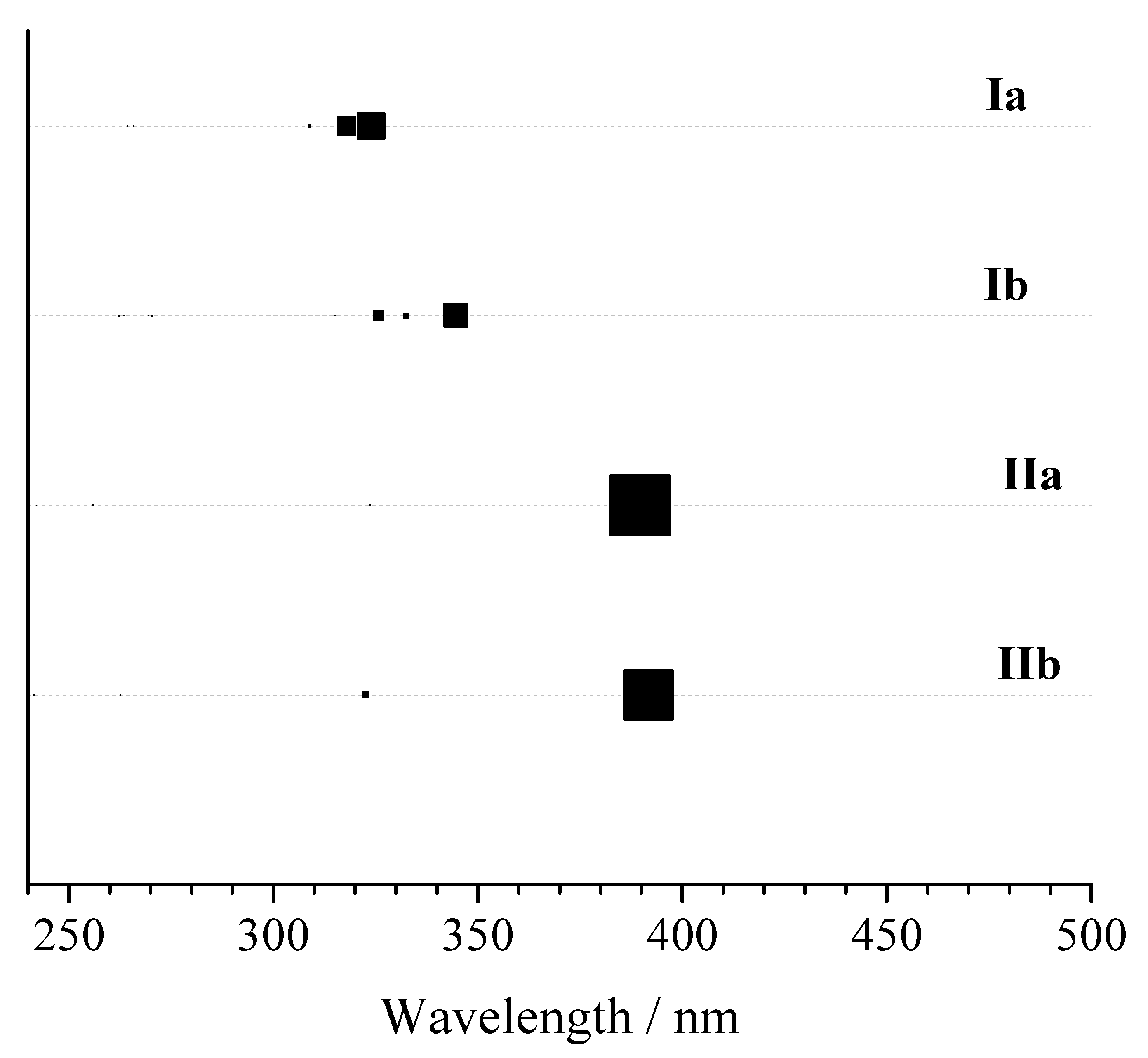
| Model | Group/GS | Excited el. State | Eexc | f | Group/GS | Excited el. State | Eexc | f |
|---|---|---|---|---|---|---|---|---|
| Ia | C2v/X1A1 | 11B1 | 3.443 | 0.00 | C2/X1A | 11B | 3.616 | 0.00 |
| 11A2 | 3.621 | 0.00 | 11A | 3.688 | 0.01 | |||
| 11B2 | 4.191 | 1.38 | 21B | 4.154 | 1.39 | |||
| 11A1 | 4.378 | 0.13 | 21A | 4.340 | 0.10 | |||
| 21B2 | 4.900 | 0.08 | 31B | 4.714 | 0.00 | |||
| Ib | C2v/X1A1 | 11B1 | 3.441 | 0.00 | C2/X1A | 11B | 3.484 | 0.02 |
| 11A2 | 3.621 | 0.00 | 11A | 3.699 | 0.00 | |||
| 11B2 | 4.171 | 1.34 | 21B | 3.960 | 0.91 | |||
| 11A1 | 4.353 | 0.13 | 21A | 4.190 | 0.32 | |||
| 21B2 | 4.867 | 0.02 | 31B | 4.813 | 0.01 | |||
| IIa | C2v/X1A1 | 11B1 | 3.388 | 1.75 | Cs(σv)/X1A′ | 11A′ | 3.475 | 1.81 |
| 11B2 | 4.048 | 0.00 | 11A″ | 3.850 | 0.00 | |||
| 11A1 | 4.164 | 0.07 | 21A′ | 4.159 | 0.08 | |||
| 21B1 | 4.472 | 0.07 | 31A′ | 4.610 | 0.02 | |||
| 21A1 | 4.770 | 0.00 | 41A′ | 4.792 | 0.00 | |||
| IIa | Cs/11A″ | 11A′ | 3.371 | 1.77 | C1/X1A | 11A | 3.446 | 1.40 |
| 11A″ | 3.217 | 0.00 | 21A | 3.884 | 0.00 | |||
| 21A′ | 3.957 | 0.13 | 31A | 4.177 | 0.19 | |||
| 31A′ | 4.551 | 0.02 | 41A | 4.583 | 0.05 | |||
| 41A′ | 4.687 | 0.01 | 51A | 4.797 | 0.00 | |||
| IIb | C2v/X1A1 | 11B1 | 3.372 | 1.78 | Cs(σv)/X1A′ | 11A′ | 3.459 | 1.83 |
| 11B2 | 4.044 | 0.00 | 11A″ | 3.848 | 0.00 | |||
| 11A1 | 4.120 | 0.07 | 21A′ | 4.118 | 0.07 | |||
| 21B1 | 4.441 | 0.04 | 31A′ | 4.593 | 0.01 | |||
| 21A1 | 4.790 | 0.00 | 41A′ | 4.800 | 0.01 | |||
| IIb | Cs/11A″ | 11A′ | 3.352 | 1.80 | C1/X1A | 11A | 3.424 | 1.48 |
| 11A″ | 3.216 | 0.00 | 21A | 3.892 | 0.00 | |||
| 21A′ | 3.916 | 0.11 | 31A | 4.149 | 0.17 | |||
| 31A′ | 4.536 | 0.02 | 41A | 4.573 | 0.05 | |||
| 41A′ | 4.695 | 0.02 | 51A | 4.810 | 0.00 | |||
| IIIa | C2v/X1A1 | 11B2 | 2.952 | 0.00 | ||||
| 11B1 | 3.045 | 0.72 | ||||||
| 11A2 | 3.612 | 0.00 | ||||||
| 11A1 | 3.637 | 0.01 | ||||||
| 21B1 | 4.404 | 0.00 | ||||||
| IIIb | C2v/X1A1 | 11B2 | 2.955 | 0.00 | ||||
| 11B1 | 3.056 | 0.79 | ||||||
| 11A2 | 3.621 | 0.00 | ||||||
| 11A1 | 3.680 | 0.00 | ||||||
| 21B1 | 4.373 | 0.02 |
| Model | Parent Group | Subgroup | EJT | ||
|---|---|---|---|---|---|
| Symmetry | Electron State | Symmetry | Electron State | /eV | |
| Ia | C2v | X1A1 | C2 | X1A | 0.206 |
| Ib | C2v | X1A1 | C2 | X1A | 0.289 |
| IIa | C2v | X1A1 | Cs(σv) | X1A′ | 0.224 |
| Cs | 11A″ | C1 | X1A | 0.194 | |
| IIb | C2v | X1A1 | Cs(σv) | X1A′ | 0.224 |
| Cs | 11A″ | C1 | X1A | 0.235 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Štellerová, D.; Lukeš, V.; Breza, M. How Does Pseudo-Jahn-Teller Effect Induce the Photoprotective Potential of Curcumin? Molecules 2023, 28, 2946. https://doi.org/10.3390/molecules28072946
Štellerová D, Lukeš V, Breza M. How Does Pseudo-Jahn-Teller Effect Induce the Photoprotective Potential of Curcumin? Molecules. 2023; 28(7):2946. https://doi.org/10.3390/molecules28072946
Chicago/Turabian StyleŠtellerová, Dagmar, Vladimír Lukeš, and Martin Breza. 2023. "How Does Pseudo-Jahn-Teller Effect Induce the Photoprotective Potential of Curcumin?" Molecules 28, no. 7: 2946. https://doi.org/10.3390/molecules28072946