
About the Formation of NH2OH+ from Gas Phase Reactions under Astrochemical Conditions


Abstract
:1. Introduction
2. Results
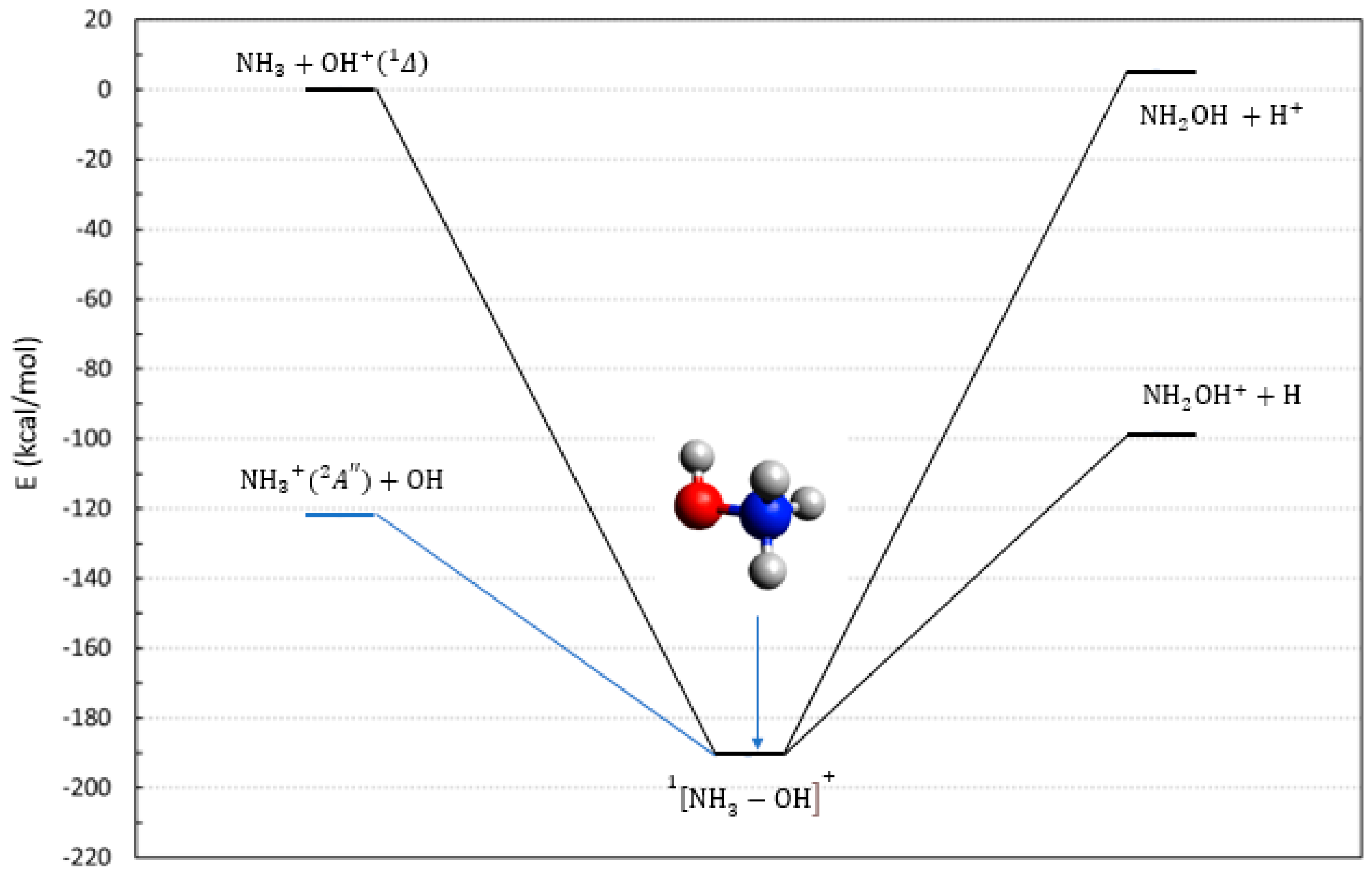
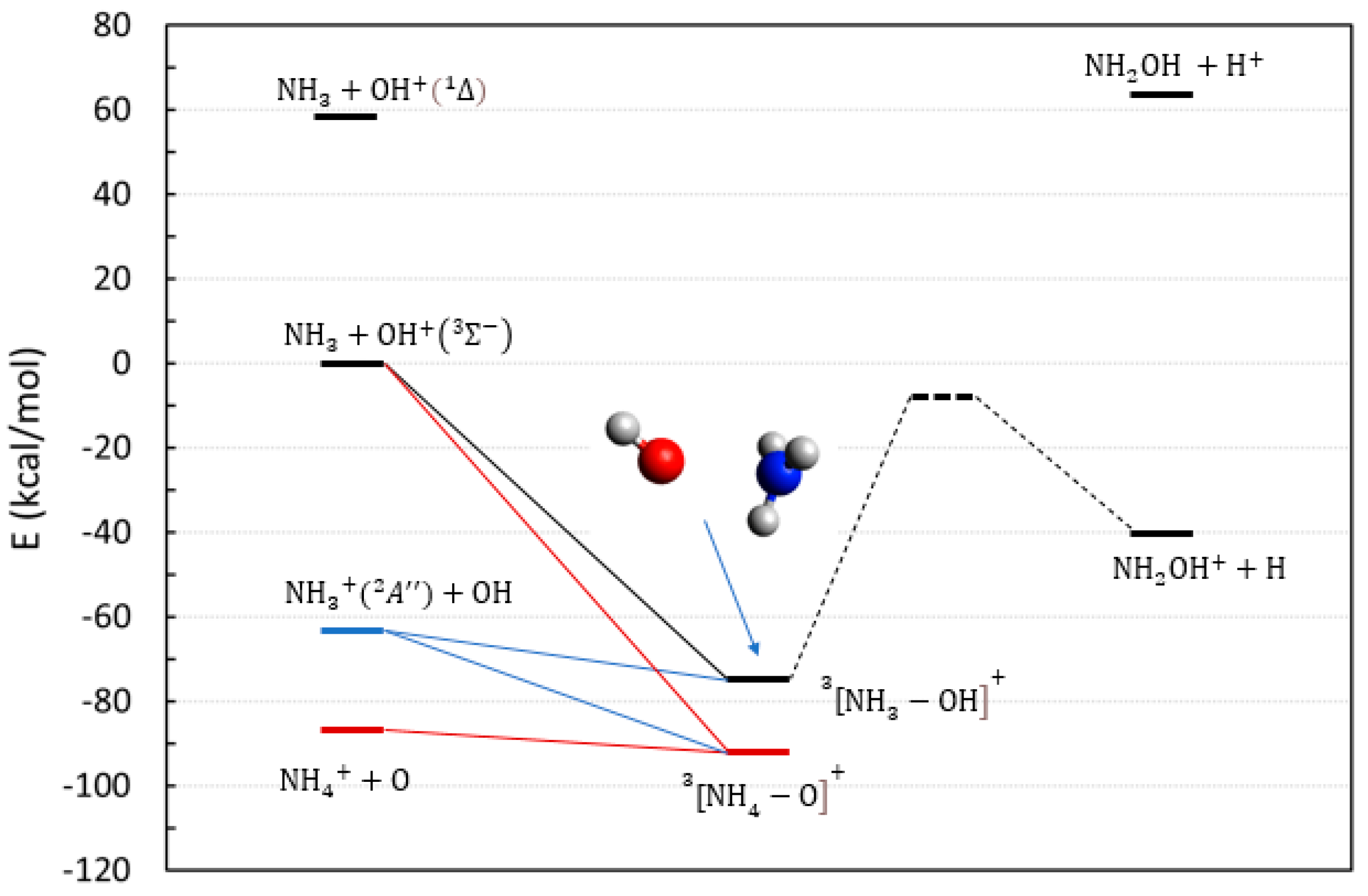
2.1. The Reactions Involving NH3, OH, and Their Cations
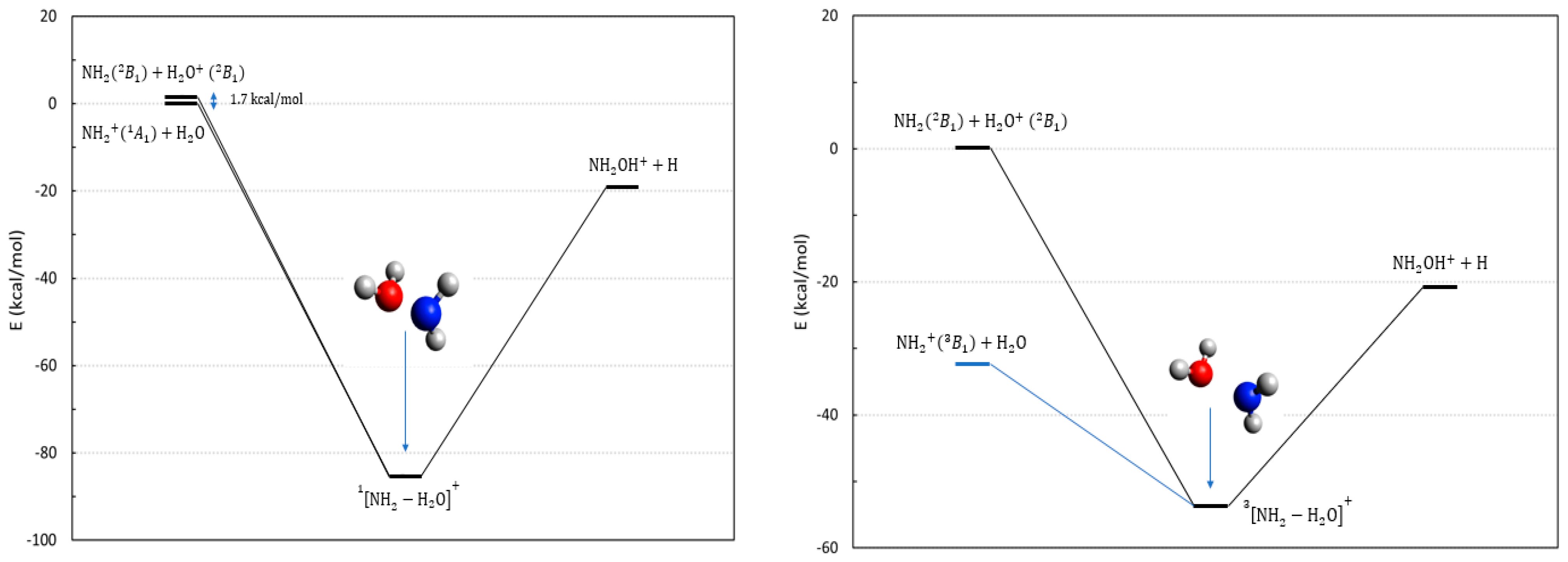
2.2. The Reactions Involving Water and Its Cation
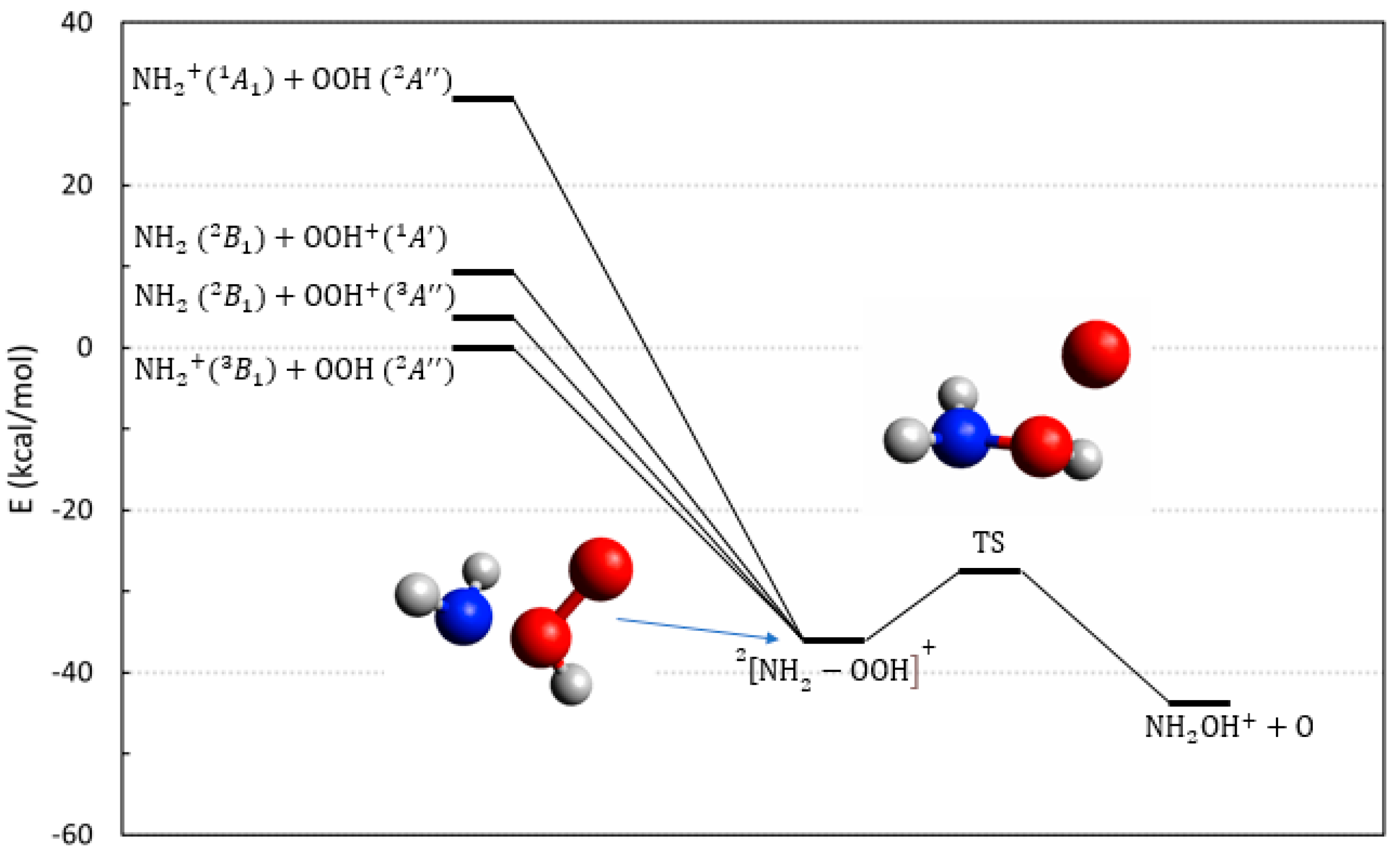
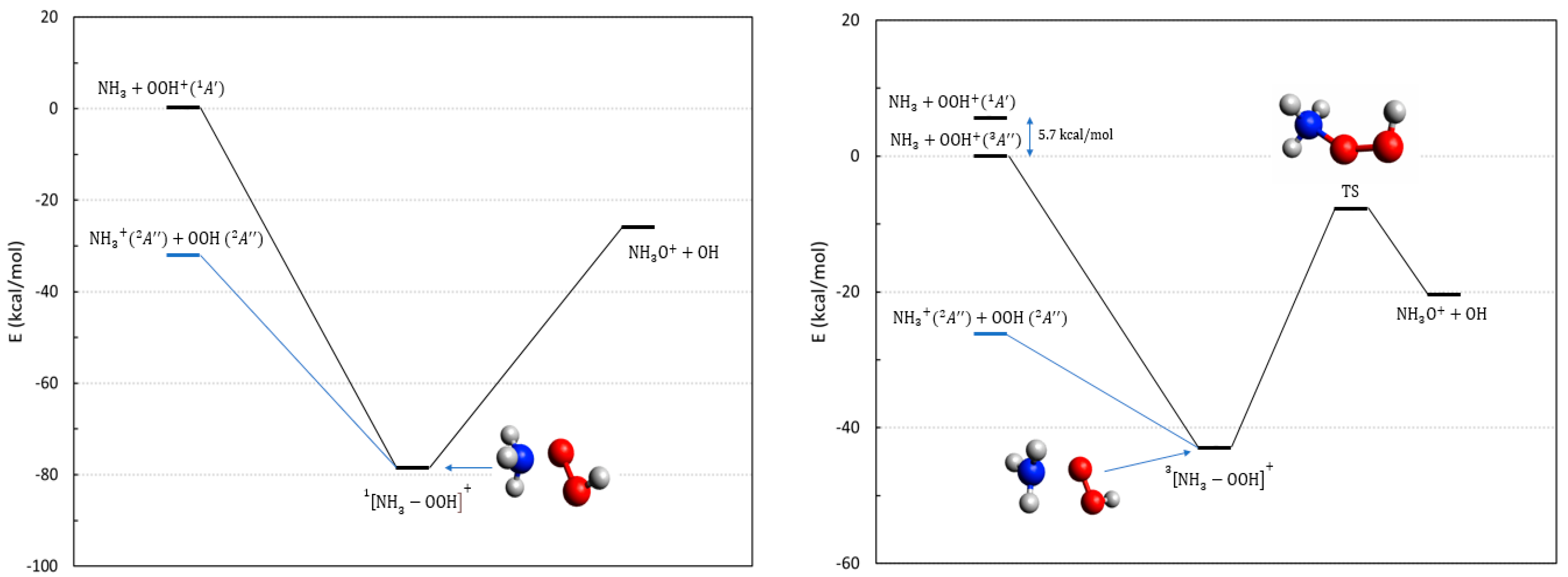
2.3. The Reactions Involving OOH, the Hydroperoxyl Radical
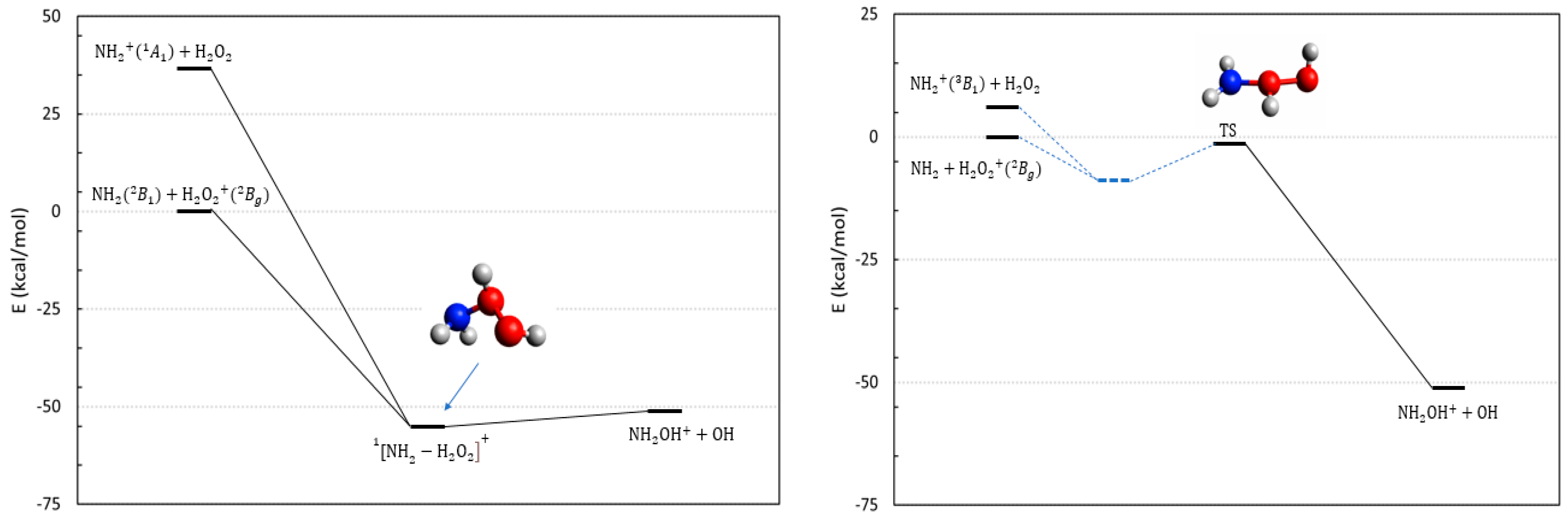
2.4. The Reactions Involving the H2O2 Peroxide
2.5. The Reactions Involving Nitrous Acid, HONO
3. Methods
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruiz-Mirazo, K.; Briones, C.; de la Escosura, A. Prebiotic Systems Chemistry: New Perspectives for the Origins of Life. Chem. Rev. 2014, 114, 285–366. [Google Scholar] [CrossRef] [PubMed]
- Powner, M.W.; Gerland, B.; Sutherland, J.D. Synthesis of Activated Pyrimidine Ribonucleotides in Prebiotically Plausible Conditions. Nature 2009, 459, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Feldmann, J.; Wiedemann, S.; Okamura, H.; Schneider, C.; Iwan, K.; Crisp, A.; Rossa, M.; Amatov, T.; Carell, T. Unified Prebiotically Plausible Synthesis of Pyrimidine and Purine RNA Ribonucleotides. Science 2019, 366, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonusas, M.; Krim, L. A Possible Answer to the Mysterious Non-Detection of Hydroxylamine in Space: The Thermal Desorption Mechanism. Mon. Not. R. Astron. Soc. 2016, 459, 1977–1984. [Google Scholar] [CrossRef]
- Rivilla, V.M.; Martín-Pintado, J.; Jiménez-Serra, I.; Martín, S.; Rodríguez-Almeida, L.F.; Requena-Torres, M.A.; Rico-Villas, F.; Zeng, S.; Briones, C. Prebiotic Precursors of the Primordial RNA World in Space: Detection of NH2 OH. Astrophys. J. 2020, 899, L28. [Google Scholar] [CrossRef]
- Rivilla, V.M.; Jiménez-Serra, I.; Martín-Pintado, J.; Colzi, L.; Tercero, B.; de Vicente, P.; Zeng, S.; Martín, S.; García de la Concepción, J.; Bizzocchi, L.; et al. Molecular Precursors of the RNA-World in Space: New Nitriles in the G+0.693−0.027 Molecular Cloud. Front. Astron. Space Sci. 2022, 9, 876870. [Google Scholar] [CrossRef]
- Jiménez-Serra, I.; Martín-Pintado, J.; Rivilla, V.M.; Rodríguez-Almeida, L.; Alonso Alonso, E.R.; Zeng, S.; Cocinero, E.J.; Martín, S.; Requena-Torres, M.; Martín-Domenech, R.; et al. Toward the RNA-World in the Interstellar Medium—Detection of Urea and Search of 2-Amino-Oxazole and Simple Sugars. Astrobiology 2020, 20, 1048–1066. [Google Scholar] [CrossRef]
- Rivilla, V.M.; Jiménez-Serra, I.; Martín-Pintado, J.; Briones, C.; Rodríguez-Almeida, L.F.; Rico-Villas, F.; Tercero, B.; Zeng, S.; Colzi, L.; de Vicente, P.; et al. Discovery in Space of Ethanolamine, the Simplest Phospholipid Head Group. Proc. Natl. Acad. Sci. USA 2021, 118, e2101314118. [Google Scholar] [CrossRef]
- Fiore, M.; Chieffo, C.; Lopez, A.; Fayolle, D.; Ruiz, J.; Soulère, L.; Oger, P.; Altamura, E.; Popowycz, F.; Buchet, R. Synthesis of Phospholipids Under Plausible Prebiotic Conditions and Analogies with Phospholipid Biochemistry for Origin of Life Studies. Astrobiology 2022, 22, 598–627. [Google Scholar] [CrossRef]
- Derbali, I.; Thissen, R.; Alcaraz, C.; Romanzin, C.; Zins, E.-L. Study of the Reactivity of CH3COOH +• and COOH + Ions with CH3NH2: Evidence of the Formation of New Peptide-like C(O)–N Bonds. J. Phys. Chem. A 2021, 125, 10006–10020. [Google Scholar] [CrossRef]
- Caselli, P.; Ceccarelli, C. Our Astrochemical Heritage. Astron. Astrophys. Rev. 2012, 20, 56. [Google Scholar] [CrossRef] [Green Version]
- Bergantini, A.; de Barros, A.L.F.; Toribio, N.N.; Rothard, H.; Boduch, P.; da Silveira, E.F. Infrared Spectroscopic Study on Swift-Ion Irradiation of Solid N2O–H2O Samples: Synthesis of N–O Bearing Species in Astrophysical Ices. J. Phys. Chem. A 2022, 126, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Cazaux, S.; Minissale, M.; Dulieu, F.; Hocuk, S. Dust as Interstellar Catalyst: II. How Chemical Desorption Impacts the Gas. Astron. Astrophys. 2016, 585, A55. [Google Scholar] [CrossRef]
- Congiu, E.; Fedoseev, G.; Ioppolo, S.; Dulieu, F.; Chaabouni, H.; Baouche, S.; Lemaire, J.L.; Laffon, C.; Parent, P.; Lamberts, T.; et al. No ice hydrogenation: A solid pathway to NH2OH formation in space. Astrophys. J. 2012, 750, L12. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Vidali, G.; Lemaire, J.-L.; Garrod, R.T. Formation of hydroxylamine on dust grains via ammonia oxidation. Astrophys. J. 2015, 799, 49. [Google Scholar] [CrossRef] [Green Version]
- Tsegaw, Y.A.; Góbi, S.; Förstel, M.; Maksyutenko, P.; Sander, W.; Kaiser, R.I. Formation of Hydroxylamine in Low-Temperature Interstellar Model Ices. J. Phys. Chem. A 2017, 121, 7477–7493. [Google Scholar] [CrossRef]
- Wang, L.; Mebel, A.M.; Yang, X.; Wang, X. Ab Initio/RRKM Study of the O(1D) + NH3 Reaction: Prediction of Product Branching Ratios. J. Phys. Chem. A 2004, 108, 11644–11650. [Google Scholar] [CrossRef]
- Mousavipour, S.H.; Pirhadi, F.; HabibAgahi, A. A Theoretical Investigation on the Kinetics and Mechanism of the Reaction of Amidogen with Hydroxyl Radical. J. Phys. Chem. A 2009, 113, 12961–12971. [Google Scholar] [CrossRef]
- McGuire, B.A. 2021 Census of Interstellar, Circumstellar, Extragalactic, Protoplanetary Disk, and Exoplanetary Molecules. Astrophys. J. Suppl. Ser. 2022, 259, 30. [Google Scholar] [CrossRef]
- Ossenkopf, V.; Müller, H.S.P.; Lis, D.C.; Schilke, P.; Bell, T.A.; Bruderer, S.; Bergin, E.; Ceccarelli, C.; Comito, C.; Stutzki, J.; et al. Detection of Interstellar Oxidaniumyl: Abundant H2O + towards the Star-Forming Regions DR21, Sgr B2, and NGC6334. Astron. Astrophys. 2010, 518, L111. [Google Scholar] [CrossRef] [Green Version]
- Widicus Weaver, S.L.; Woon, D.E.; Ruscic, B.; McCall, B.J. IS HO+2 A DETECTABLE INTERSTELLAR MOLECULE? Astrophys. J. 2009, 697, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Wyrowski, F.; Menten, K.M.; Güsten, R.; Belloche, A. First Interstellar Detection of OH +. Astron. Astrophys. 2010, 518, A26. [Google Scholar] [CrossRef] [Green Version]
- van Lonkhuyzen, H.; de Lange, C.A.U.V. Photoelectron Spectroscopy of OH and OD Radicals. Mol. Phys. 1984, 51, 551–568. [Google Scholar] [CrossRef]
- Ball, R.; Brindley, J. The Life Story of Hydrogen Peroxide III: Chirality and Physical Effects at the Dawn of Life. Orig. Life Evol. Biospheres 2016, 46, 81–93. [Google Scholar] [CrossRef]
- Hoener, M.; Bodi, A.; Hemberger, P.; Endres, T.; Kasper, T. Threshold Photoionization Shows No Sign of Nitryl Hydride in Methane Oxidation with Nitric Oxide. Phys. Chem. Chem. Phys. 2021, 23, 1265–1272. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized Auxiliary Basis Sets and Demonstration of Efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Musiał, M. Coupled-Cluster Theory in Quantum Chemistry. Rev. Mod. Phys. 2007, 79, 291–352. [Google Scholar] [CrossRef] [Green Version]
- Puzzarini, C.; Barone, V. Extending the Molecular Size in Accurate Quantum-Chemical Calculations: The Equilibrium Structure and Spectroscopic Properties of Uracil. Phys. Chem. Chem. Phys. 2011, 13, 7189. [Google Scholar] [CrossRef]
- Arathala, P.; Musah, R.A. Theoretical Studies of the Gas-Phase Reactions of S-Methyl Methanesulfinothioate (Dimethyl Thiosulfinate) with OH and Cl Radicals: Reaction Mechanisms, Energetics, and Kinetics. J. Phys. Chem. A 2019, 123, 8448–8459. [Google Scholar] [CrossRef]
- Parandaman, A.; Tangtartharakul, C.B.; Kumar, M.; Francisco, J.S.; Sinha, A. A Computational Study Investigating the Energetics and Kinetics of the HNCO + (CH3)2NH Reaction Catalyzed by a Single Water Molecule. J. Phys. Chem. A 2017, 121, 8465–8473. [Google Scholar] [CrossRef]
- Stanton, J.F. Coupled-Cluster Theory, Pseudo-Jahn–Teller Effects and Conical Intersections. J. Chem. Phys. 2001, 115, 10382. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J.-P. N-Electron Valence State Perturbation Theory: A Spinless Formulation and an Efficient Implementation of the Strongly Contracted and of the Partially Contracted Variants. J. Chem. Phys. 2002, 117, 9138–9153. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.-P. Introduction of n-Electron Valence States for Multireference Perturbation Theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
- Bodo, E.; Bovolenta, G.; Simha, C.; Spezia, R. On the Formation of Propylene Oxide from Propylene in Space: Gas-Phase Reactions. Theor. Chem. Acc. 2019, 138, 97. [Google Scholar] [CrossRef]
- Coutens, A.; Ligterink, N.F.W.; Loison, J.-C.; Wakelam, V.; Calcutt, H.; Drozdovskaya, M.N.; Jørgensen, J.K.; Müller, H.S.P.; van Dishoeck, E.F.; Wampfler, S.F. The ALMA-PILS Survey: First Detection of Nitrous Acid (HONO) in the Interstellar Medium. Astron. Astrophys. 2019, 623, L13. [Google Scholar] [CrossRef]
- van Dishoeck, E.F.; Jansen, D.J.; Schilke, P.; Phillips, T.G. Detection of the Interstellar NH2 Radical. Astrophys. J. 1993, 416, L83. [Google Scholar] [CrossRef] [Green Version]
- Dunlavey, S.J.; Dyke, J.M.; Jonathan, N.; Morris, A. Vacuum ultraviolet photoelectron spectroscopy of transient species. Mol. Phys. 1980, 39, 1121–1135. [Google Scholar] [CrossRef]
- Weiss, M.J.; Lawrence, G.M. Photoelectron Spectroscopy of NH3 and ND3 Using Molecular Beams. J. Chem. Phys. 1970, 53, 214–218. [Google Scholar] [CrossRef]
- Brundle, C.R.; Turner, D.W. High resolution molecular photoelectron spectroscopy II. Water and deuterium oxide. Proc. R. Soc. London. 1968, 307, 27–36. [Google Scholar]
- Dyke, J.M.; Jonathan, N.B.H.; Morris, A.; Winter, M.J. Vacuum ultraviolet photoelectron spectroscopy of transient species. XIII: Observation of the X3A" state of HO2+. Mol. Phys. 1981, 44, 1059–1066. [Google Scholar] [CrossRef]
- Ashmore, F.S.; Burgess, A.R. Study of some medium size alcohols and hydroperoxides by photoelectron spectroscopy. J. Chem. Soc. Faraday Trans. 1977, 73, 1247–1261. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | 2s + 1 | Def2-TZVPP | Def2-QZVPP |
|---|---|---|---|
| 1 | 1 | −98.9 | −99.8 |
| 2 | 1 | +5.0 | +2.2 |
| 3 | 3 | −40.3 | −41.8 |
| 4 | 3 | +22.9 | +22.5 |
| 5 | 1/3 | −20.8 | −21.8 |
| 6 | 1 | −19.1 | −21.7 |
| 7 | 2/4 | −47.4 | - |
| 8 | 2 | −53.1 | −52.5 |
| 9 | 2/4 | −43.7 | - |
| 10 | 2 | −74.3 | −73.6 |
| 11 | 3 | −41.8 | - |
| 12 | 1 | −47.5 | −47.5 |
| 13 | 1/3 | −15.5 | −25.0 |
| 14 | 1/3 | −51.2 | −48.5 |
| 15 | 3 | −57.2 | - |
| 16 | 1 | −87.8 | −88.7 |
| 17 | 1/3 | - | −59.5 |
| 18 | 1 | - | −59.9 |
| 19 | 3 | - | −58.0 |
| 20 | 3 | +0.3 a | +0.8 |
| 21 | 2 | −13.3 a | −13.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dilena, G.; Pistillo, S.; Bodo, E. About the Formation of NH2OH+ from Gas Phase Reactions under Astrochemical Conditions. Molecules 2023, 28, 2932. https://doi.org/10.3390/molecules28072932
Dilena G, Pistillo S, Bodo E. About the Formation of NH2OH+ from Gas Phase Reactions under Astrochemical Conditions. Molecules. 2023; 28(7):2932. https://doi.org/10.3390/molecules28072932
Chicago/Turabian StyleDilena, Gabriele, Simone Pistillo, and Enrico Bodo. 2023. "About the Formation of NH2OH+ from Gas Phase Reactions under Astrochemical Conditions" Molecules 28, no. 7: 2932. https://doi.org/10.3390/molecules28072932