


Ferrocenophanium Stability and Catalysis
Abstract
:1. Introduction
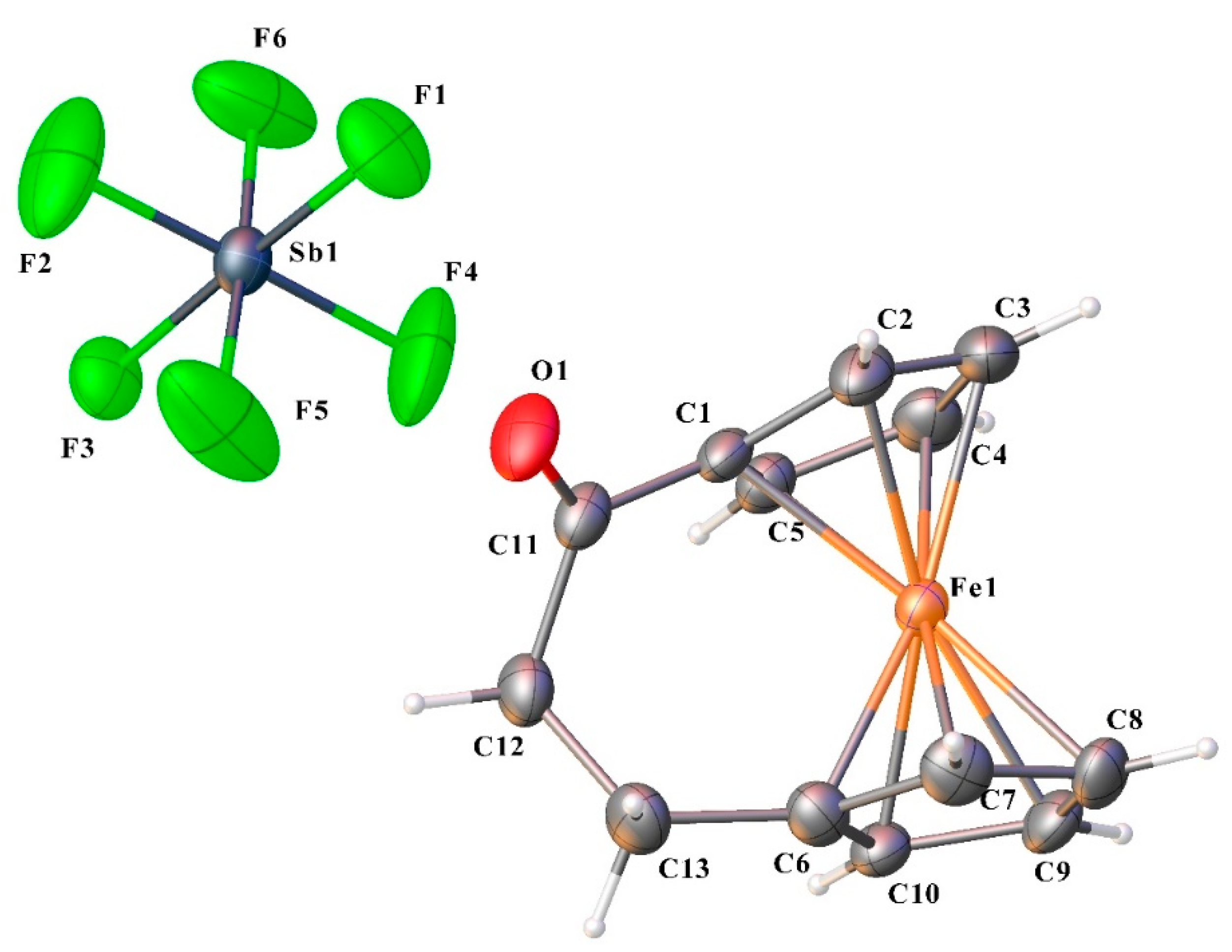
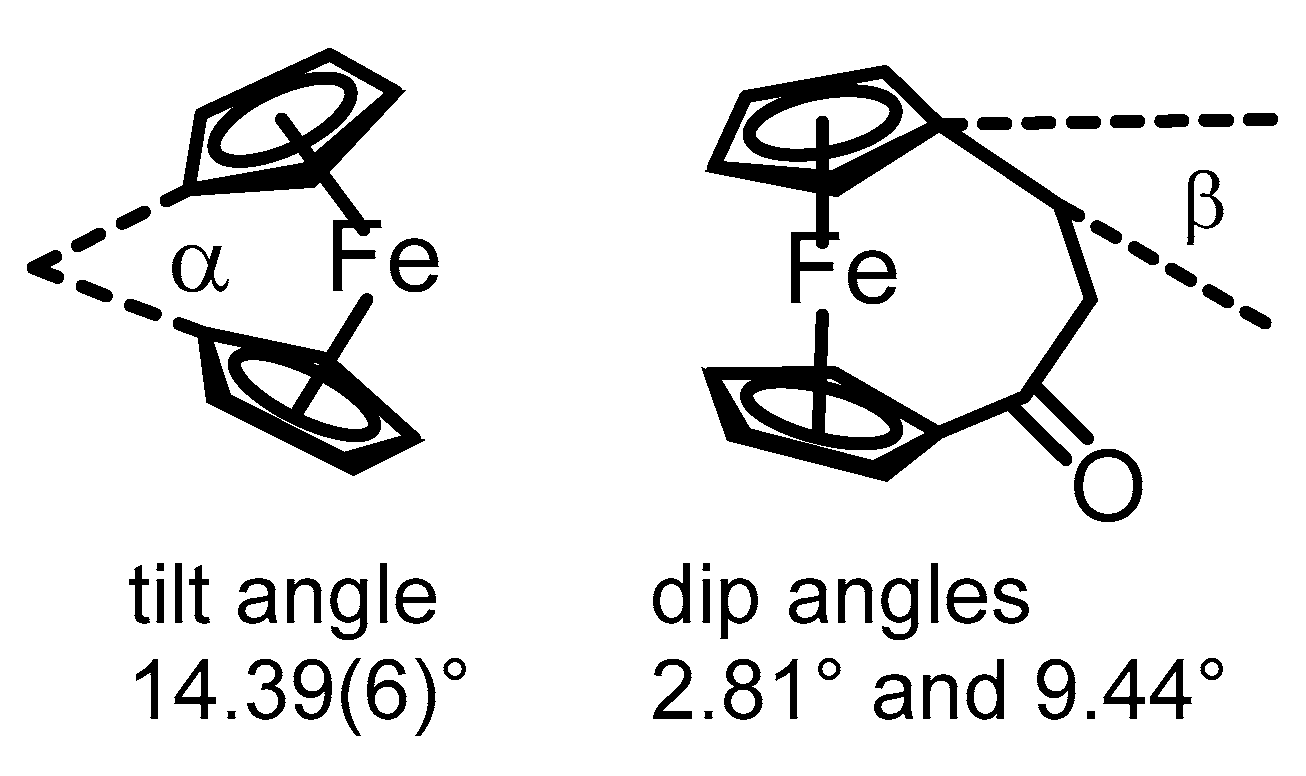
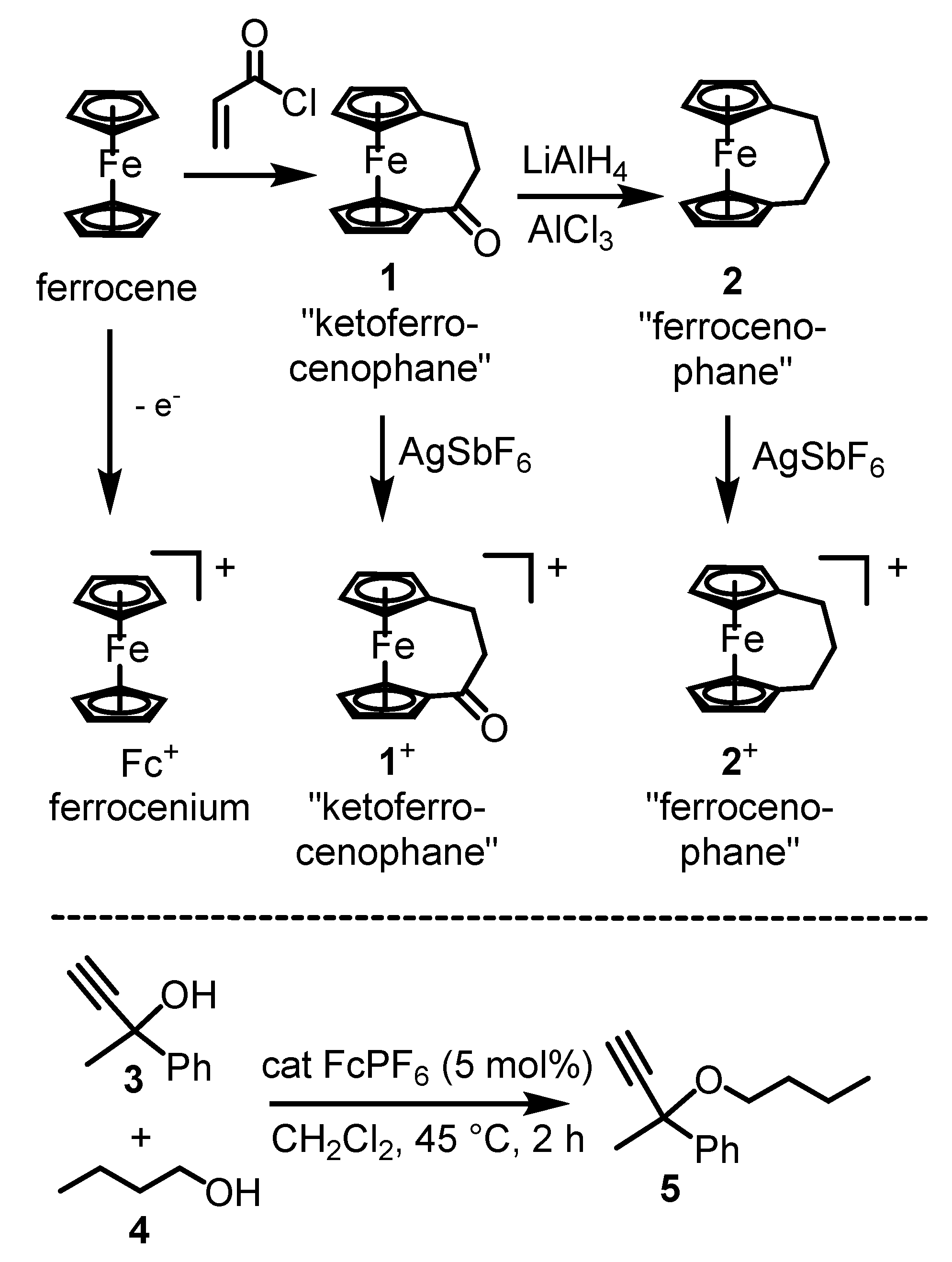
2. Results and Discussion
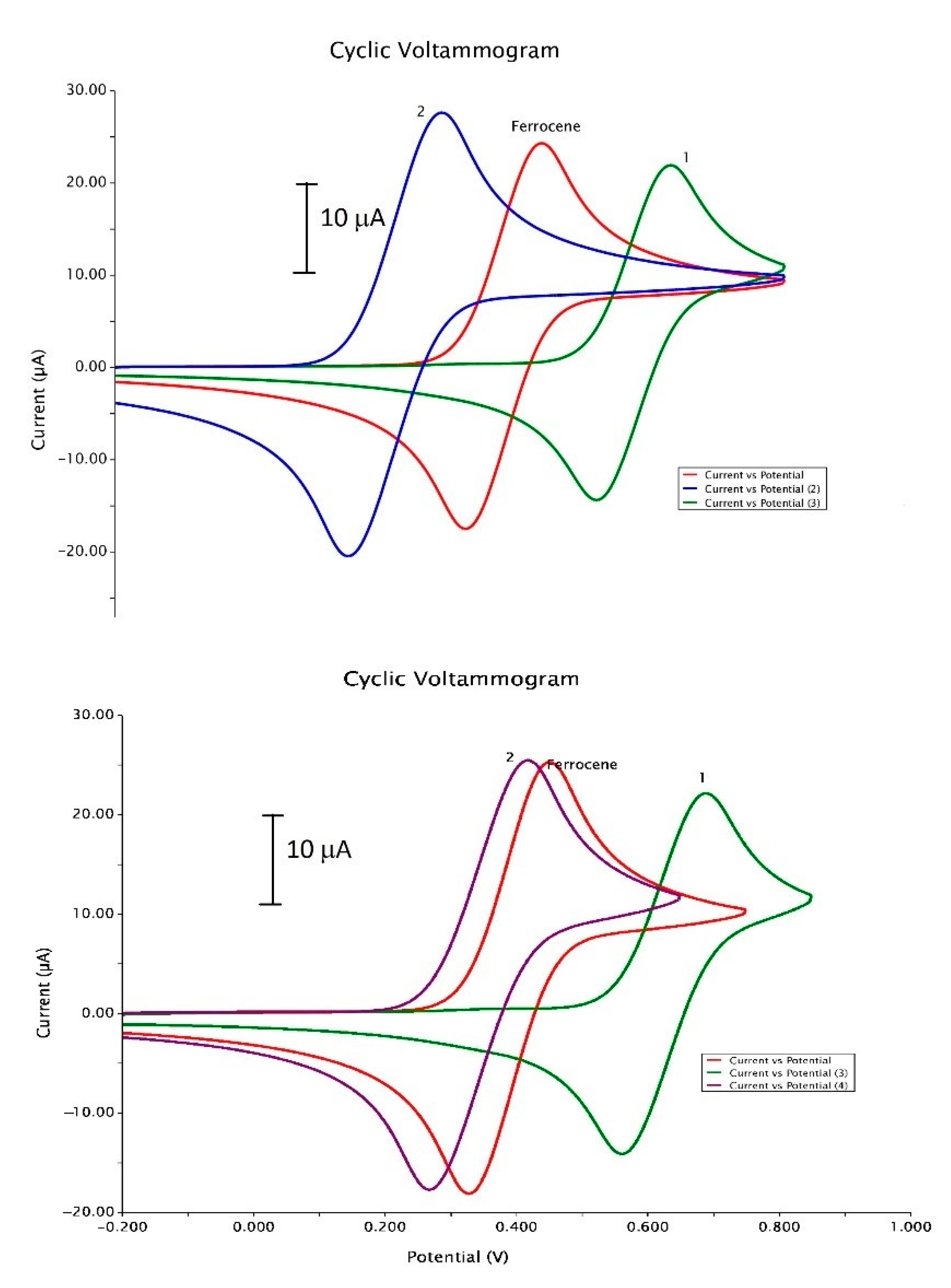
2.1. Cyclic Voltammetry Experiments
2.2. Catalytic Experiments
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A. Iron Catalyzed C–C-Bond Formation: From Canonical Cross Coupling to a Quest for New Reactivity. Bull. Chem. Soc. Jpn. 2021, 94, 666–677. [Google Scholar] [CrossRef]
- Rana, S.; Biswas, J.P.; Paul, S.; Paika, A.; Maiti, D. Organic synthesis with the most abundant transition metal–iron: From rust to multitasking catalysts. Chem. Soc. Rev. 2021, 50, 243–472. [Google Scholar] [CrossRef] [PubMed]
- Bisz, E. Iron-Catalyzed Cross-Coupling Reactions of Alkyl Grignards with Aryl Chlorobenzenesulfonates. Molecules 2021, 26, 5895. [Google Scholar] [CrossRef]
- Wei, D.; Netkaew, C.; Darcel, C. Multi-Step Reactions Involving Iron-Catalysed Reduction and Hydrogen Borrowing Reactions. Eur. J. Inorg. Chem. 2019, 2471–2487. [Google Scholar] [CrossRef]
- Wei, D.; Darcel, C. Iron Catalysis in Reduction and Hydrometalation Reactions. Chem. Rev. 2019, 119, 2550–2610. [Google Scholar] [CrossRef]
- Piontek, A.; Bisz, E.; Szostak, M. Iron-Catalyzed Cross-Couplings in the Synthesis of Pharmaceuticals: In Pursuit of Sustainability. Angew. Chem. Int. Ed. 2018, 57, 11116–11128. [Google Scholar] [CrossRef]
- Fürstner, A. Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes to Make This Base Metal a Multitasking Champion. ACS Cent. Sci. 2016, 2, 778–789. [Google Scholar] [CrossRef]
- Bauer, I.; Knölker, H.J. Iron catalysis in organic synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef]
- Bauer, E.B. (Ed.) Iron Catalysis II.; Springer: New York, NY, USA, 2015. [Google Scholar]
- Mihovilovic, D.; Schnürch, M. Ligand-Assisted Iron Catalysis in the Direct Functionalization of C–H Bonds. ChemCatChem 2014, 6, 2194–2196. [Google Scholar] [CrossRef]
- Gopalaiah, K. Chiral iron catalysts for asymmetric synthesis. Chem. Rev. 2013, 113, 3248–3296. [Google Scholar] [CrossRef]
- MacLeod, K.C.; Holland, P.L. Recent developments in the homogeneous reduction of dinitrogen by molybdenum and iron. Nat. Chem. 2013, 5, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Welcher, A.; Jacobi von Wangelin, A. Iron(0) Nanoparticle Catalysts in Organic Synthesis. Curr. Org. Chem. 2013, 17, 326–355. [Google Scholar] [CrossRef]
- Sun, X.; Li, J.; Huang, X.; Sun, C. Recent Advances in Iron-Catalyzed C-H Bond Activation Reactions. Curr. Inorg. Chem. 2012, 2, 64–85. [Google Scholar] [CrossRef]
- Sun, C.-L.; Li, B.-J.; Shi, Z.-J. Direct C− H transformation via iron catalysis. Chem. Rev. 2011, 111, 1293–1314. [Google Scholar] [CrossRef]
- Sarhan, A.A.O.; Bolm, C. Iron (III) chloride in oxidative C–C coupling reactions. Chem. Soc. Rev. 2009, 38, 2730–2744. [Google Scholar] [CrossRef]
- Lee, H.; He, T.; Cook, S.P. Iron-Catalyzed, Directed Benzylic Borylation. Org. Lett. 2023, 25, 1–4. [Google Scholar] [CrossRef]
- Xu, H.; Li, M.-J.; Chen, H.; Huang, F.-H.; Zhu, Q.-Y.; Wang, G.-W.; Zhang, Z. I2/FeCl3-Catalyzed Domino Reaction of Aurones with Enamino Esters for the Synthesis of Highly Functionalized Pyrroles. Org. Lett. 2022, 24, 8406–8411. [Google Scholar] [CrossRef]
- He, C.; Ma, F.; Zhang, W.; Tong, R. Reinvestigating FeBr3-Catalyzed Alcohol Oxidation with H2O2: Is a High-Valent Iron Species (HIS) or a Reactive Brominating Species (RBS) Responsible for Alcohol Oxidation? Org. Lett. 2022, 24, 3499–3503. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, C.; Wang, Z.; Kato, T.; Liu, Y.; Maruoka, K. Fe-Catalyzed Three-Component Coupling Reaction of α, β, γ, δ-Unsaturated Carbonyl Compounds and Conjugate Dienes with Alkylsilyl Peroxides and Nucleophiles. J. Org. Chem. 2022, 87, 8824–8834. [Google Scholar] [CrossRef]
- Qiu, Y.-F.; Chen, S.-P.; Cao, J.-H.; Li, M.; Quan, Z.-J.; Wang, X.-C.; Liang, Y.-M. Iron(II)-Catalyzed Bisphosphorylation Cascade Cycloisomerization of γ-Hydroxyl Ynones and Diphenylphosphine Oxides: Synthesis of Highly Substituted Bisphosphorylated Dihydrofuran Derivatives. Org. Lett. 2022, 24, 2264–2268. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.-J.; Kessler, S.N.; Wu, H.; Bäckvall, J.-E. Iron-Catalyzed Cross-Coupling of α-Allenyl Esters with Grignard Reagents for the Synthesis of 1,3-Dienes. Org. Lett. 2023, 25, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qian, J.; Wang, M.; Huang, Y.; Hu, P. Visible-Light-Induced Decarboxylative Fluorination of Aliphatic Carboxylic Acids Catalyzed by Iron. Org. Lett. 2022, 24, 5972–5976. [Google Scholar] [CrossRef] [PubMed]
- Bisz, E.; Szostak, M. Iron-Catalyzed C(sp2)–C(sp3) Cross-Coupling of Aryl Chlorobenzoates with Alkyl Grignard Reagents. Molecules 2020, 25, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Wendt, B.; Möller, K.; Junge, K.; Beller, M. Two iron catalysts are better than one: A general and convenient reduction of aromatic and aliphatic primary amides. Angew. Chem. Int. Ed. 2012, 51, 1662–1666. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y. Light-Induced Iron-Catalyzed Trifluoromethylative Thiolation of Alkenes. Org. Lett. 2022, 24, 8057–8061. [Google Scholar] [CrossRef]
- Emayavaramban, B.; Chakraborty, P.; Dahiya, P.; Sundararaju, B. Iron-Catalyzed α-Methylation of Ketones Using Methanol as the C1 Source under Photoirradiation. Org. Lett. 2022, 24, 6219–6223. [Google Scholar] [CrossRef]
- Gudmundsson, A.; Schlipköter, K.E.; Bäckvall, J.-E. Iron (II)-Catalyzed Biomimetic Aerobic Oxidation of Alcohols. Angew. Chem. Int. Ed. 2020, 59, 5403–5406. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, T.; Okuzono, C.; Adak, L.; Jin, M.; Nakamura, M. Iron-catalysed enantioselective Suzuki–Miyaura coupling of racemic alkyl bromides. Chem. Commun. 2019, 55, 1128–1131. [Google Scholar] [CrossRef]
- Vicens, L.; Olivo, G.; Costas, M. Rational design of bioinspired catalysts for selective oxidations. ACS Catal. 2020, 10, 8611–8631. [Google Scholar] [CrossRef]
- Gérard, E.F.; Yadav, V.; Goldberg, D.P.; de Visser, S.P. What Drives Radical Halogenation versus Hydroxylation in Mononuclear Nonheme Iron Complexes? A Combined Experimental and Computational Study. J. Am. Chem. Soc. 2022, 144, 10752–10767. [Google Scholar] [CrossRef]
- Olivo, G.; Capocasa, G.; Ticconi, B.; Lanzalunga, O.; Di Stefano, S.; Costas, M. Predictable selectivity in remote C− H oxidation of steroids: Analysis of substrate binding mode. Angew. Chem. Int. Ed 2020, 59, 12703–12708. [Google Scholar] [CrossRef]
- Olivo, G.; Capocasa, G.; Lanzalunga, O.; Di Stefano, S.; Costas, M. Enzyme-like substrate-selectivity in C–H oxidation enabled by recognition. Chem. Commun. 2019, 55, 917–920. [Google Scholar] [CrossRef]
- Olivo, G.; Farinelli, G.; Barbieri, A.; Lanzalunga, O.; Di Stefano, S.; Costas, M. Supramolecular Recognition Allows Remote, Site-Selective C− H Oxidation of Methylenic Sites in Linear Amines. Angew. Chem. Int. Ed. 2017, 129, 16565–16569. [Google Scholar] [CrossRef]
- Lauzon, S.; Schouwey, L.; Ollevier, T. C2-Symmetric 2,2′-Bipyridine-α,α′-1-adamantyl-diol Ligand: Bulky Iron Complexes in Asymmetric Catalysis. Org. Lett. 2022, 24, 1116–1120. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective iron-catalyzed transformations. Coord. Chem. Rev. 2019, 386, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Ollevier, T. Iron bis(oxazoline) complexes in asymmetric catalysis. Catal. Sci. Technol. 2016, 6, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Lenze, M.; Bauer, E.B. Chemoselective, iron (II)-catalyzed oxidation of a variety of secondary alcohols over primary alcohols utilizing H2O2 as the oxidant. Chem. Commun. 2013, 49, 5889–5891. [Google Scholar] [CrossRef]
- Lenze, M.; Martin, E.T.; Rath, N.P.; Bauer, E.B. Iron (II) α-Aminopyridine Complexes and Their Catalytic Activity in Oxidation Reactions: A Comparative Study of Activity and Ligand Decomposition. ChemPlusChem 2013, 78, 101–116. [Google Scholar] [CrossRef]
- Lenze, M.; Sedinkin, S.L.; Bauer, E.B. Polydentate pyridyl ligands and the catalytic activity of their iron (II) complexes in oxidation reactions utilizing peroxides as the oxidants. J. Mol. Catal. A 2013, 373, 161–171. [Google Scholar] [CrossRef]
- Shejwalkar, P.; Rath, N.P.; Bauer, E.B. New iron (II) α-iminopyridine complexes and their catalytic activity in the oxidation of activated methylene groups and secondary alcohols to ketones. Dalton Trans. 2011, 40, 7617–7631. [Google Scholar] [CrossRef] [PubMed]
- Shejwalkar, P.; Rath, N.P.; Bauer, E.B. New chiral phosphoramidite complexes of iron as catalytic precursors in the oxidation of activated methylene groups. Molecules 2010, 15, 2631–2650. [Google Scholar] [CrossRef] [PubMed]
- Lenze, M.; Sedinkin, S.L.; Rath, N.P.; Bauer, E.B. New indenyl phosphinooxazoline complexes of iron and their catalytic activity in the Mukaiyama aldol reaction. Tetrahedron Lett. 2010, 51, 2855–2858. [Google Scholar] [CrossRef]
- Lenze, M.; Bauer, E.B. Oxidation of activated methylene groups to ketones catalyzed by new iron phosphinooxazoline complexes and by iron (II) triflate. J. Mol. Catal. A 2009, 309, 117–123. [Google Scholar] [CrossRef]
- Talasila, D.S.; Queensen, M.J.; Barnes-Flaspoler, M.; Jurkowski, K.; Stephenson, E.; Rabus, J.M.; Bauer, E.B. Ferrocenium Cations as Catalysts for the Etherification of Cyclopropyl-Substituted Propargylic Alcohols: Ene-yne Formation and Mechanistic Insights. Eur. J. Org. Chem. 2019, 2019, 7348–7358. [Google Scholar] [CrossRef]
- Queensen, M.Q.; Rabus, J.M.; Bauer, E.B. Ferrocenium hexafluorophosphate as an inexpensive, mild catalyst for the etherification of propargylic alcohols. J. Mol. Cat. A: Chem 2015, 407, 221–229. [Google Scholar] [CrossRef]
- Toma, Š.; Šebesta, R. Applications of ferrocenium salts in organic synthesis. Synthesis 2015, 47, 1683–1695. [Google Scholar] [CrossRef] [Green Version]
- Ang, H.T.; Rygus, J.P.G.; Hall, D.G. Two-component boronic acid catalysis for increased reactivity in challenging Friedel–Crafts alkylations with deactivated benzylic alcohols. Org. Biomol. Chem. 2019, 17, 6007–6014. [Google Scholar] [CrossRef]
- Mo, X.; Yakiwchuk, J.; Dansereau, J.; McCubbin, J.A.; Hall, D.G. Unsymmetrical Diarylmethanes by Ferroceniumboronic Acid Catalyzed Direct Friedel–Crafts Reactions with Deactivated Benzylic Alcohols: Enhanced Reactivity due to Ion-Pairing Effects. J. Am. Chem. Soc. 2015, 137, 9694–9703. [Google Scholar] [CrossRef]
- Mo, X.; Hall, D.G. Dual catalysis using boronic acid and chiral amine: Acyclic quaternary carbons via enantioselective alkylation of branched aldehydes with allylic alcohols. J. Am. Chem. Soc. 2016, 138, 10762–10765. [Google Scholar] [CrossRef]
- Deb, M.; Hazra, S.; Dolui, P.; Elias, A.J. Ferrocenium Promoted Oxidation of Benzyl Amines to Imines Using Water as the Solvent and Air as the Oxidant. ACS Sustainable Chem. Eng. 2019, 7, 479–486. [Google Scholar] [CrossRef]
- Shul’pina, L.S.; Kudinov, A.R.; Mandelli, D.; Carvalho, W.A.; Kozlov, Y.N.; Vinogradov, M.M.; Ikonnikov, N.S.; Shul’pin, G.B. Oxidation of alkanes and benzene with hydrogen peroxide catalyzed by ferrocene in the presence of acids. J. Organomet. Chem. 2015, 793, 217–231. [Google Scholar] [CrossRef]
- Khan, N.-u.H.; Agrawal, S.; Kureshy, R.I.; Abdi, S.H.R.; Singh, S.; Jasra, R.V. Fe(Cp)2PF6: An efficient catalyst for cyanosilylation of carbonyl compounds under solvent free condition. J. Organomet. Chem. 2007, 692, 4361–4366. [Google Scholar] [CrossRef]
- Argouarch, G.; Grelaud, G.; Roisnel, T.; Humphrey, M.G.; Paul, F. [Fp*Fc][PF6]: A remarkable non-symmetric dinuclear cation in a very stable mixed-valent state. J. Organomet. Chem. 2017, 847, 218–223. [Google Scholar] [CrossRef]
- Yadav, G.D.; Chauhan, M.M.S.; Singh, S. Fe(Cp)2BF4: An efficient Lewis acid catalyst for the aminolysis of epoxides. Synthesis 2014, 46, 629–634. [Google Scholar]
- Yadav, G.D.; Singh, S. Ring opening of epoxides with alcohols using Fe(Cp)2BF4 as catalyst. Tetrahedron Lett. 2014, 55, 3979–3983. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Kitagawa, H.; Matsuo, J.-i. Aromatic iodination with iodine monochloride by using a catalytic amount of ferrocenium tetrakis (3, 5-bis (trifluoromethyl) phenyl) borate. Tetrahedron Lett. 2000, 41, 9383–9386. [Google Scholar] [CrossRef]
- Noor-ul, K.H.; Agrawal, S.; Kureshy, R.I.; Abdi, S.H.R.; Singh, S.; Suresh, E.; Jasra, R.V. Fe(Cp)2PF6 catalyzed efficient Strecker reactions of ketones and aldehydes under solvent-free conditions. Tetrahedron Lett. 2008, 49, 640–644. [Google Scholar]
- Kureshy, R.I.; Agrawal, S.; Saravanan, S.; Khan, N.-u.H.; Shah, A.K.; Abdi, S.H.R.; Bajaj, H.C.; Suresh, E. Direct Mannich reaction mediated by Fe(Cp)2PF6 under solvent-free conditions. Tetrahedron Lett. 2010, 51, 489–494. [Google Scholar] [CrossRef]
- Peña, L.A.; Seidl, A.J.; Cohen, L.R.; Hoggard, P.E. Ferrocene/ferrocenium ion as a catalyst for the photodecomposition of chloroform. Transition Met. Chem. 2009, 34, 135–141. [Google Scholar] [CrossRef]
- Neumann, A.; Prehn Junquera, A.; Wismach, C.; Jones, P.G.; Streubel, R. Synthesis of functional Δ3-1,3,5-oxazaphospholene and 2H-1,4,2-diazaphosphole complexes via catalytic ring expansion reactions of a 2H-azaphosphirene complex. Tetrahedron 2003, 59, 6213–6220. [Google Scholar] [CrossRef]
- Zhang, J.; Campolo, C.; Dumur, F.; Xiao, P.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. The carbazole-bound ferrocenium salt as a specific cationic photoinitiator upon near-UV and visible LEDs (365–405 nm). Polym. Bull. 2016, 73, 493–507. [Google Scholar] [CrossRef]
- Li, M.; Chen, Y.; Zhang, H.; Wang, T. A novel ferrocenium salt as visible light photoinitiator for cationic and radical photopolymerization. Prog. Org. Coat. 2010, 68, 234–239. [Google Scholar] [CrossRef]
- Wang, T.; Chen, J.W.; Li, Z.Q.; Wan, P.Y. Several ferrocenium salts as efficient photoinitiators and thermal initiators for cationic epoxy polymerization. J. Photochem. Photobiol. A Chem. 2007, 187, 389–394. [Google Scholar] [CrossRef]
- Wang, T.; Li, B.S.; Zhang, L.X. Carbazole-bound ferrocenium salt as an efficient cationic photoinitiator for epoxy polymerization. Polym. Int. 2005, 54, 1251–1255. [Google Scholar] [CrossRef]
- Görmen, M.; Pigeon, P.; Hillard, E.A.; Vessières, A.; Huché, M.; Richard, M.-A.; McGlinchey, M.J.; Top, S.; Jaouen, G. Synthesis and antiproliferative effects of [3] ferrocenophane transposition products and pinacols obtained from McMurry cross-coupling reactions. Organometallics 2012, 31, 5856–5866. [Google Scholar] [CrossRef]
- Singh, A.; Roy Chowdhury, D.; Paul, A. A kinetic study of ferrocenium cation decomposition utilizing an integrated electrochemical methodology composed of cyclic voltammetry and amperometry. Analyst 2014, 139, 5747–5754. [Google Scholar] [CrossRef]
- Prins, R.; Korswagen, A.R.; Kortbeek, A.G.T.G. Decomposition of the ferricenium cation by nucleophilic reagents. J. Organomet. Chem. 1972, 39, 335–344. [Google Scholar] [CrossRef]
- Zotti, G.; Schiavon, G.; Zecchin, S.; Favretto, D. Dioxygen-decomposition of ferrocenium molecules in acetonitrile: The nature of the electrode-fouling films during ferrocene electrochemistry. J. Electroanal. Chem. 1998, 456, 217–221. [Google Scholar] [CrossRef]
- Hurvois, J.P.; Moinet, C. Reactivity of ferrocenium cations with molecular oxygen in polar organic solvents: Decomposition, redox reactions and stabilization. J. Organomet. Chem. 2005, 690, 1829–1839. [Google Scholar] [CrossRef]
- Lorans, J.; Pierre, F.; Toupet, L.; Moinet, C. Anodic co-oxidation of urazole and ferrocenes: First trapping ofcyclopentadienols. Chem. Commun. 1997, 1279–1280. [Google Scholar] [CrossRef]
- Allen, S.K.; Lathrop, T.E.; Patel, S.B.; Harrell Moody, D.M.; Sommer, R.D.; Coombs, T.C. Synthesis of 7-norbornenols via Diels–Alder cycloadditions of cyclopentadienol generated by decomposition of ferrocenium cation. Tetrahedron Lett. 2015, 56, 6038–6042. [Google Scholar] [CrossRef]
- Heo, R.W.; Randall Lee, T. Ferrocenophanes with all carbon bridges. J. Organomet. Chem. 1999, 578, 31–42. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Jones, D.; Turner, M.L.; Adams, H. First synthesis and X-ray crystal structure of 1, 2-(1, 1′-ferrocenediyl) ethene. J. Organomet. Chem. 1996, 524, 263–266. [Google Scholar] [CrossRef]
- Werner, I.; Heinisch, S.L.; Nowik, I.; Herber, R.H.; Butenschön, H. 1,16-Di-tert-butyl-1,16-diphospha[5.5]ferrocenophane: Synthesis, Reactions and Mössbauer Spectroscopy. ChemistrySelect 2018, 3, 13132–13139. [Google Scholar] [CrossRef]
- Braunschweig, H.; Krummenacher, I.; Lichtenberg, C.; Mattock, J.D.; Schäfer, M.; Schmidt, U.; Schneider, C.; Steffenhagen, T.; Ullrich, S.; Vargas, A. Dibora[2]ferrocenophane: A Carbene-Stabilized Diborene in a Strained cis-Configuration. Angew. Chem. Int. Ed. 2017, 56, 889–892. [Google Scholar] [CrossRef] [Green Version]
- Buchowicz, W.; Guńka, P.A.; Buchalski, P.; Piszcz, M.; Buś, S.; Mrozowicz, M.; Mazur, M.; Wasilewski, R. E/Z Switchable Ring-Closing Metathesis in 1,1′-Bis(but-3-enyl)ferrocenes: Synthesis and Characterization of Axially Chiral ansa[6]-Ferrocenes. Organometallics 2022, 41, 1968–1976. [Google Scholar] [CrossRef]
- Musgrave, R.A.; Russell, A.D.; Manners, I. Strained ferrocenophanes. Organometallics 2013, 32, 5654–5667. [Google Scholar] [CrossRef]
- Turbitt, T.D.; Watts, W.E. Bridged ferrocenes: XII. The synthesis of [3] ferrocenophan-1-one from ferrocene by a novel one-step annelation reaction. J. Organomet. Chem. 1972, 46, 109–117. [Google Scholar] [CrossRef]
- Dong, T.-Y.; Lee, S.-H. The effects of an interannular bridge on the electronic structure of ferrocenium cations. J. Organomet. Chem. 1995, 487, 77–88. [Google Scholar] [CrossRef]
- Carty, P.; Dove, M.F.A. The reaction of some ferrocenyl ketones with anhydrous silver tetrafluoroborate, a new route to substituted ferricenium salts. J. Organomet. Chem. 1971, 28, 125–132. [Google Scholar] [CrossRef]
- Watanabe, M.; Sato, K.; Motoyama, I.; Sano, H. Mössbauer Studies of Bridged Ferrocenophane Derivative’s Polyiodides. Chemistry Letters 1983, 12, 1775–1778. [Google Scholar] [CrossRef]
- Duggani, D.M.; Hendrickson, D.N. Electronic structure of various ferricenium systems as inferred from Raman, infrared, low-temperature electronic absorption, and electron paramagnetic resonance measurements. Inorg. Chem. 1975, 14, 955–970. [Google Scholar] [CrossRef]
- Khozeimeh Sarbisheh, E.; Bhattacharjee, H.; Cao, M.P.T.; Zhu, J.; Müller, J. How strained are [1] ferrocenophanes? Organometallics 2017, 36, 614–621. [Google Scholar] [CrossRef]
- Althoff, A.; Jutzi, P.; Lenze, N.; Neumann, B.; Stammler, A.; Stammler, H.-G. A Digalla[1.1]ferrocenophane and Its Coordination Chemistry: Synthesis and Structure of [{Fe(η5-C5H4)2}2{GaMe}2] and of the Adducts [{Fe(η5-C5H4)2}2{GaMe(D)}2] (D = Monodentate Donor) and [{Fe(η5-C5H4)2}2{GaMe}2D] (D = Bidentate Donor). Organometallics 2003, 22, 2766–2774. [Google Scholar] [CrossRef]
- Bhattacharjee, H.; Dey, S.; Zhu, J.; Sun, W.; Müller, J. Strained azabora [2] ferrocenophanes. Chem. Commun. 2018, 54, 5562–5565. [Google Scholar] [CrossRef]
- Herbert, D.E.; Mayer, U.F.J.; Manners, I. Strained Metallocenophanes and Related Organometallic Rings Containing π-Hydrocarbon Ligands and Transition-Metal Centers. Angew. Chem. Int. Ed. 2007, 46, 5060–5081. [Google Scholar] [CrossRef]
- Reichert, A.; Bolte, M.; Lerner, H.-W.; Wagner, M. Mono- and di-borylated [3]ferrocenophanes. J. Organomet. Chem. 2013, 744, 15–23. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical redox agents for organometallic chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef]
- Elgrishi, N.; Rountree, K.J.; McCarthy, B.D.; Rountree, E.S.; Eisenhart, T.T.; Dempsey, J.L. A practical beginner’s guide to cyclic voltammetry. J. Chem. Educ. 2018, 95, 197–206. [Google Scholar] [CrossRef]
- Korb, M.; Lehrich, S.W.; Lang, H. Reactivity of ferrocenyl Phosphates bearing (hetero-) aromatics and [3] ferrocenophanes toward anionic phospho-fries rearrangements. J. Org. Chem. 2017, 82, 3102–3124. [Google Scholar] [CrossRef]
- Frenzel, P.; Lehrich, S.W.; Korb, M.; Hildebrandt, A.; Lang, H. Ferrocenyloxysilanes: Synthesis, characterization and electrochemical investigations. J. Organomet. Chem. 2017, 845, 98–106. [Google Scholar] [CrossRef]
- Plażuk, D.; Vessiéres, A.; Hillard, E.A.; Buriez, O.; Labbé, E.; Pigeon, P.; Plamont, M.-A.; Amatore, C.; Zakrzewski, J.; Jaouen, G. A [3] ferrocenophane polyphenol showing a remarkable antiproliferative activity on breast and prostate cancer cell lines. J. Med. Chem. 2009, 52, 4964–4967. [Google Scholar] [CrossRef] [Green Version]
- Rao, W.; Zhang, X.; Sze, E.M.L.; Chan, P.W.H. Ytterbium(III) Triflate-Catalyzed Amination of 1-Cyclopropylprop-2-yn-1-ols as an Expedient Route to Conjugated Enynes. J. Org. Chem. 2009, 74, 1740–1743. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Onodera, G.; Sakata, K.; Yuki, M.; Miyake, Y.; Uemura, S.; Nishibayashi, Y. Ruthenium-Catalyzed Reactions of 1-Cyclopropyl-2-propyn-1-ols with Anilines and Water via Allenylidene Intermediates: Selective Preparation of Tri- and Tetrasubstituted Conjugated Enynes. J. Am. Chem. Soc. 2007, 129, 5175–5179. [Google Scholar] [CrossRef]
- Bruker. APEX3, SAINT-Plus and SADABS; Bruker AXS Inc.: Madison, WC, USA, 2016. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta. Cryst. 2015, C71, 3–8. [Google Scholar]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Cryst. 2020, E76, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2009, 41, 466–470. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Complex 1+SbF6 |
|---|---|
| Empirical formula | C13H12F6FeOSb |
| Formula weight | 475.83 |
| Temperature (K) | 296.15 |
| Wavelength (Å) | 1.54178 |
| Crystal system, space group | Monoclinic, P21/c |
| Unit cell dimensions a (Å) | 7.2292(2) |
| b (Å) | 20.6452(5) |
| c (Å) | 10.7792(3) |
| α (°) | 90 |
| β (°) | 107.127(2) |
| γ (°) | 90 |
| Volume (Å3) | 1537.43(7) |
| Z, Calculated density (Mg/m3) | 4, 2.056 |
| Absorption coefficient (mm–1) | 22.107 |
| F(000) | 916.0 |
| Crystal size (mm) | 0.08 × 0.050 × 0.020 |
| Theta range for data collection (°) | 4.883 to 68.48 deg. |
| Limiting indices | –8 ≤ h ≤ 8, –24 ≤ k ≤ 24, –12 ≤ l ≤ 12 |
| Reflections collected/unique | 15701/2821 [R(int) = 0.0815] |
| Completeness to theta = 68.48 (%) | 100.0 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 2821/0/199 |
| Goodness-of-fit on F2 | 1.068 |
| Final R indices [I>2sigma(I)] | R1 = 0.0520, wR2 = 0.1482 |
| R indices (all data) | R1 = 0.0706, wR2 = 0.1603 |
| Largest diff. peak and hole (eÅ−3) | 1.12→nd −1.08 |
| Entry | Compound | E1/2/V | ΔE/mV | ic/ia | Solvent |
|---|---|---|---|---|---|
| 1 | Ferrocene | 0.460 ± 37 | 143 ± 26 | 0.97 ± 0.03 | CH2Cl2 |
| 2 | Keto-Ferrocenophane (1) | 0.604 ± 20 | 115 ± 12 | 0.86 ± 0.04 | CH2Cl2 |
| 3 | Ferrocenophane (2) | 0.318 ± 63 | 137 ± 21 | 0.87 ± 0.04 | CH2Cl2 |
| 4 | Ferrocene | 0.400 ± 94 | 184 ± 40 | 0.98 ± 0.02 | CH3CN |
| 5 | Keto-Ferrocenophane (1) | 0.658 ± 70 | 327 ± 53 | 0.89 ± 0.06 | CH3CN |
| 6 | Ferrocenophane (2) | 0.395 ± 38 | 106 ± 10 | 0.94 ± 0.03 | CH3CN |
| Entry | Compound | Nucleophile | E1/2/V | ΔE/mV | ic/ia | Solvent |
|---|---|---|---|---|---|---|
| Ferrocene | ||||||
| 1 | MeOH | 0.324 ± 0.043 | 75 ± 30 | 0.94 ± 4 | CH3CN | |
| 2 | n-BuOH | 0.301 ± 0.047 | 121 ± 20 | 0.78 ± 4 | CH3CN | |
| 3 | i-PrOH | 0.339 ± 0.021 | 83 ± 5 | 0.96 ± 4 | CH3CN | |
| 4 | Propargylic alcohol 3 | 0.410 ± 0.010 | 117 ± 31 | 0.88 ± 5 | CH3CN | |
| 5 | n-BuOH | 0.404 ± 0.062 | 128 ± 26 | 0.94 ± 4 | CH2Cl2 | |
| 6 | MeOH | 0.352 ± 0.041 | 143 ± 19 | 0.94 ± 3 | CH2Cl2 | |
| 7 | i-PrOH | 0.302 ± 0.080 | 138 ± 8 | 0.95 ± 2 | CH2Cl2 | |
| 8 | Propargylic alcohol 3 | 0.344 ± 0.047 | 145 ± 12 | 0.94 ± 2 | CH2Cl2 | |
| Keto-Ferroceno-phane (1) | ||||||
| 9 | MeOH | 0.560 ± 0.032 | 76 ± 24 | 0.81 ± 0.04 | CH3CN | |
| 10 | n-BuOH | 0.610 ± 0.050 | 73 ± 20 | 0.81 ± 0.04 | CH3CN | |
| 11 | i-PrOH | 0.590 ± 0.040 | 72 ± 22 | 0.82 ± 0.05 | CH3CN | |
| 12 | Propargylic alcohol 3 | CH3CN | ||||
| 13 | MeOH | 0.553 ± 0.078 | 105 ± 7 | 0.84 ± 0.06 | CH2Cl2 | |
| 14 | n-BuOH | 0.616 ± 0.046 | 123 ± 23 | 0.84 ± 0.03 | CH2Cl2 | |
| 15 | i-PrOH | 0.432 ± 0.042 | 117 ± 22 | 0.87 ± 0.05 | CH2Cl2 | |
| 16 | Propargylic alcohol 3 | 0.648 ± 0.015 | 109 ± 12 | 0.75 ± 0.09 | CH2Cl2 | |
| Ferroceno-phane (2) | ||||||
| 17 | MeOH | 0.233 ± 0.022 | 75 ± 20 | 0.97 ± 0.03 | CH3CN | |
| 18 | n-BuOH | 0.263 ± 0.048 | 96 ± 26 | 0.96 ± 0.04 | CH3CN | |
| 19 | MeOH | 0.288 ± 0.010 | 108 ± 13 | 0.96 ± 0.04 | CH2Cl2 | |
| 20 | n-BuOH | 0.336 ± 0.007 | 137 ± 23 | 0.91 ± 0.02 | CH2Cl2 |
| Entry | Sweep Rate mV/s | ic/ia Ferrocene and n-BuOH | ic/ia Ketoferroceno-phane 1 and n-BuOH | ic/ia Ferroceno-phane 2 and n-BuOH | |||
|---|---|---|---|---|---|---|---|
| CH3CN | CH2Cl2 | CH3CN | CH2Cl2 | CH3CN | CH2Cl2 | ||
| 1 | 50 | 55 ± 4 | 92 ± 4 | 90 ± 2 | 83 ± 2 | 92 ± 6 | 95 ± 2 |
| 2 | 100 | 71 ± 5 | 95 ± 2 | 92 ± 4 | 83 ± 3 | 96 ± 2 | 91 ± 2 |
| 3 | 200 | 66 ± 6 | 92 ± 2 | 90 ± 2 | 79 ± 5 | 92 ± 6 | 88 ± 1 |
| 4 | 300 | 71 ± 2 | 90 ± 2 | 91 ± 4 | 77 ± 6 | 95 ± 4 | 87 ± 2 |
| 5 | 400 | 77 ± 4 | 90 ± 3 | 91 ± 6 | 75 ± 5 | 96 ± 2 | 86 ± 4 |
| 6 | 500 | 78 ± 3 | 89 ± 3 | 91 ± 2 | 70 ± 6 | 96 ± 2 | 91 ± 2 |
 | ||||
|---|---|---|---|---|
| Substrate | Product 1 | Isolated Yield Fc+ | Isolated Yield 1+ | Isolated Yield 2+ |
 |  | 74% 2 | 0% | 0% |
 |  | 37% 3 | 13% | 44% |
 |  | 44% 3 | - | 6% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezawada, S.A.; Ušto, N.; Wilke, C.; Barnes-Flaspoler, M.; Jagan, R.; Bauer, E.B. Ferrocenophanium Stability and Catalysis. Molecules 2023, 28, 2729. https://doi.org/10.3390/molecules28062729
Bezawada SA, Ušto N, Wilke C, Barnes-Flaspoler M, Jagan R, Bauer EB. Ferrocenophanium Stability and Catalysis. Molecules. 2023; 28(6):2729. https://doi.org/10.3390/molecules28062729
Chicago/Turabian StyleBezawada, Sai Anvesh, Neira Ušto, Chloe Wilke, Michael Barnes-Flaspoler, Rajamoni Jagan, and Eike B. Bauer. 2023. "Ferrocenophanium Stability and Catalysis" Molecules 28, no. 6: 2729. https://doi.org/10.3390/molecules28062729