


Jumping in the Chiral Pool: Asymmetric Hydroaminations with Early Metals
Abstract
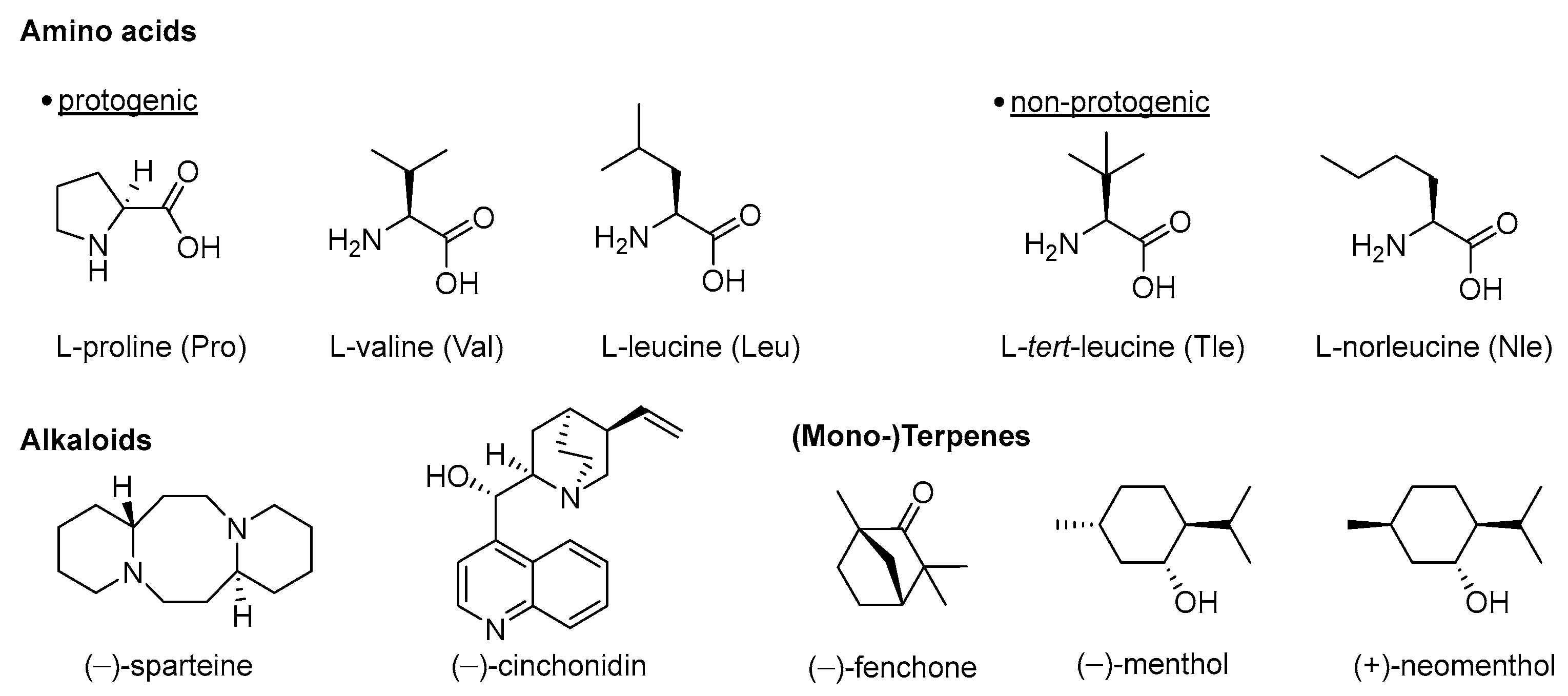
:1. Introduction
2. Chiral Pool-Based Catalysts for Asymmetric Hydroamination Reactions
2.1. Late Transition Metals
2.2. Early Transition Metals
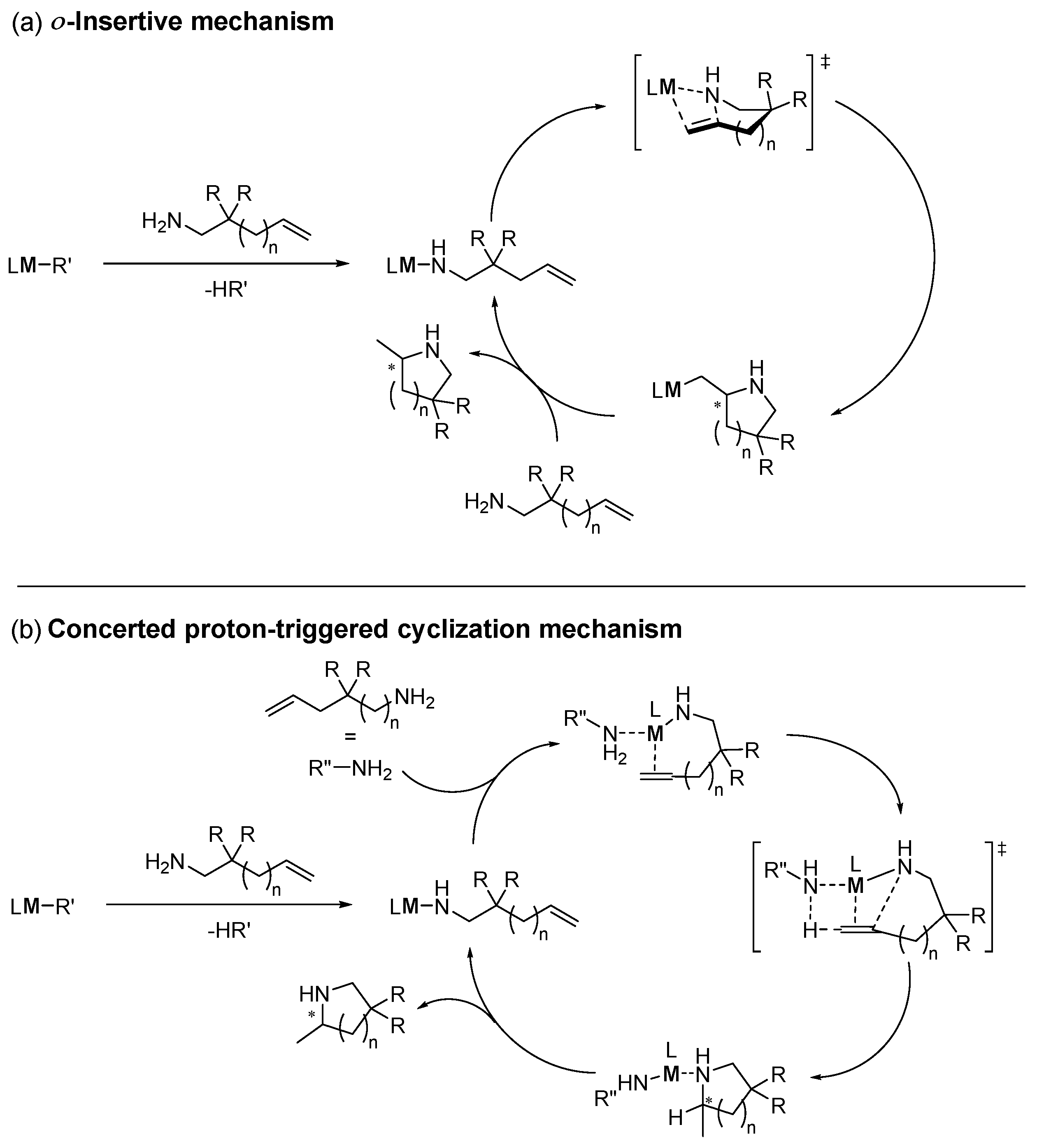
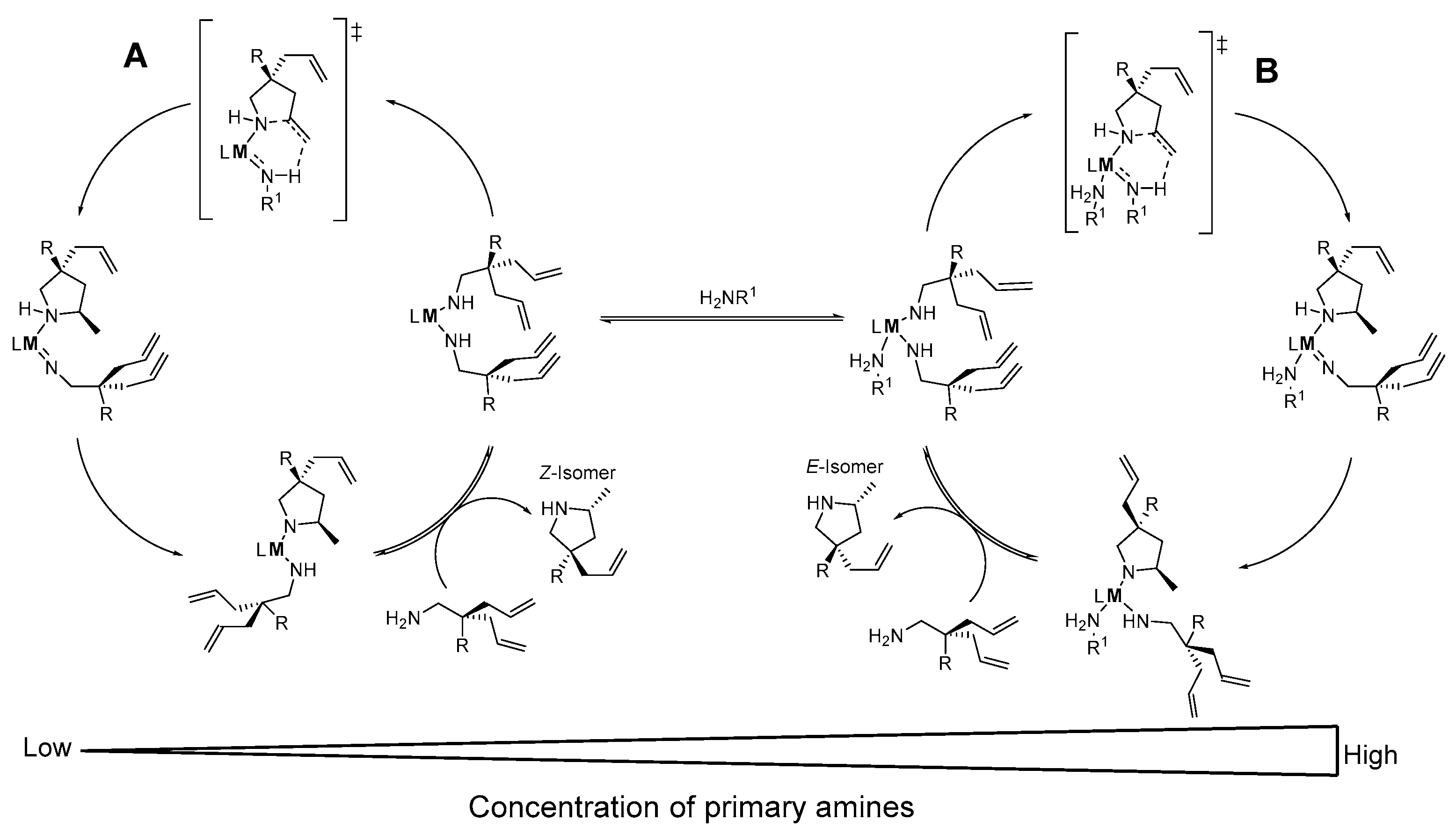
2.2.1. Rare Earth Metals

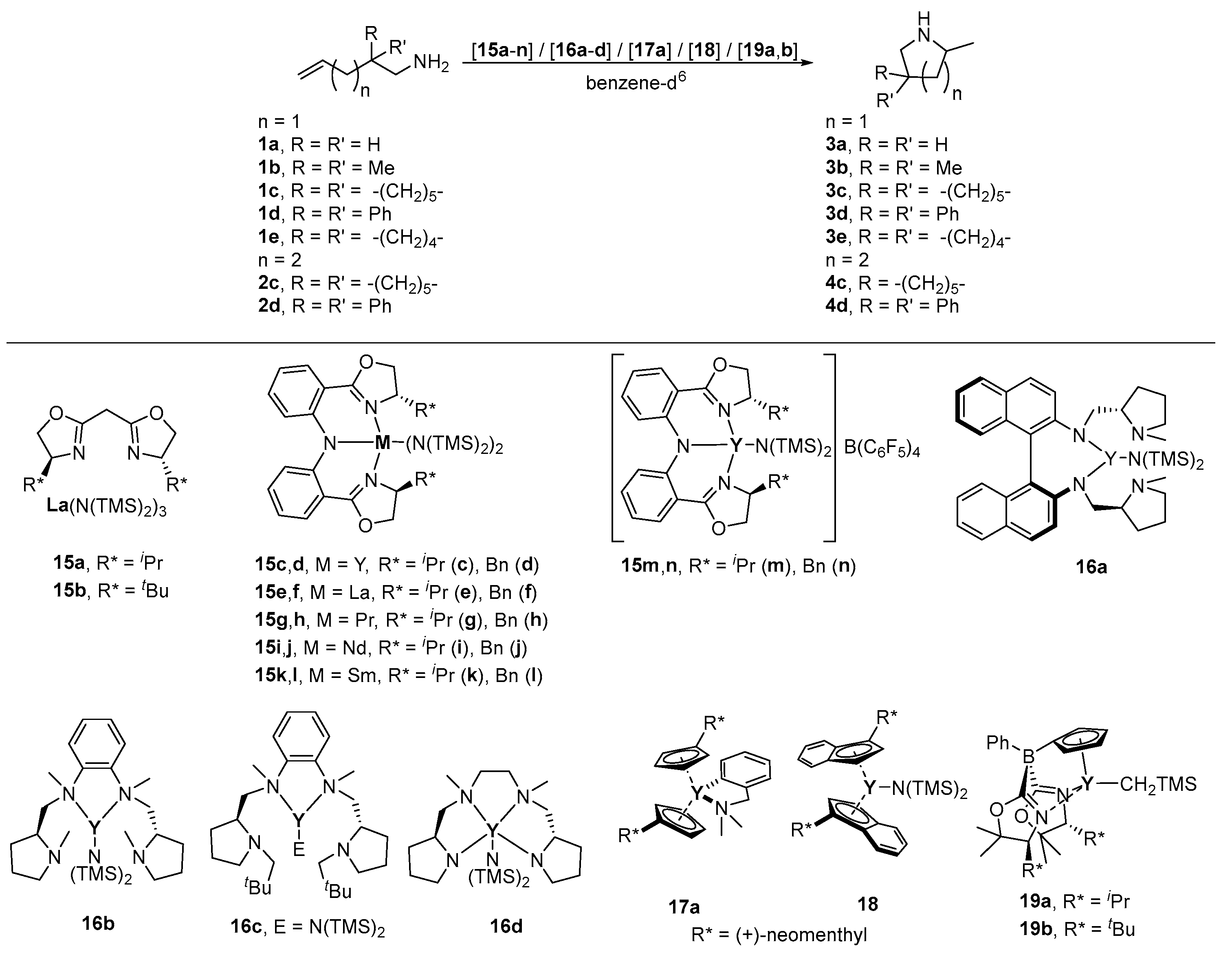
| Entry | Cat. | R* | [cat] [mol-%] | Substr. | Prod. | T [°C] | t [h] | Conv. [%] | ee [%] a | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 15a | iPr | 5 b | 1b | 3b | 23 b | n.a. | >98 | 6 (R) | [75] |
| 2 | 15b | tBu | 5 b | 1b | 3b | 23 b | n.a. | >98 | 39 (R) | [75] |
| 3 | 15c | iPr | 10 | 1d | 3d | 22 | 0.25 | >99 | 43 c | [76] |
| 4 | 15d | Bn | 10 | 1d | 3d | 22 | 0.25 | >99 | 30 c | [76] |
| 5 | 15e | iPr | 10 | 1b | 3b | 30 | 72 | >99 | 7 c | [76] |
| 6 | 10 | 1d | 3d | 22 | 0.25 | >99 | 5 c | [76] | ||
| 7 | 15f | Bn | 10 | 1b | 3b | 30 | 72 | >99 | 6 c | [76] |
| 8 | 10 | 1d | 3d | 22 | 0.25 | >99 | 6 c | [76] | ||
| 9 | 15g | iPr | 10 | 1b | 3b | 22 | 12 | >99 | 14 c | [76] |
| 10 | 10 | 1d | 3d | 22 | 1 | >99 | 16 c | [76] | ||
| 11 | 15h | Bn | 10 | 1b | 3b | 22 | 12 | >99 | 10 c | [76] |
| 12 | 10 | 1d | 3d | 22 | 1 | >99 | 42 c | [76] | ||
| 13 | 15i | iPr | 10 | 1b | 3b | 22 | 168 | - | - | [76] |
| 14 | 10 | 1d | 3d | 22 d | 12 | >99 | 36 c | [76] | ||
| 15 | 15j | Bn | 10 | 1b | 3b | 22 | 168 | - | - | [76] |
| 16 | 10 | 1d | 3d | 22 d | 12 | >99 | 46 c | [76] | ||
| 17 | 15k | iPr | 10 | 1b | 3b | 22 | 12 | >99 | 14 c | [76] |
| 18 | 10 | 1d | 3d | 22 d | 12 | >99 | 30 c | [76] | ||
| 19 | 15l | Bn | 10 | 1b | 3b | 22 | 12 | >99 | 12 c | [76] |
| 20 | 10 | 1d | 3d | 22 d | 12 | >99 | 30 c | [76] | ||
| 21 | 15m | iPr | 10 | 1d | 3d | 22 e | 0.25 | >99 | 38 c | [76] |
| 22 | 15n | Bn | 10 | 1d | 3d | 22 e | 0.25 | >99 | 32 c | [76] |
| 23 | 16a | - | 5 | 1b | 3b | 60 | 5.5 | 95 | 2 | [77] |
| 24 | 16b | - | 5 | 1b | 3b | 10 | 168 | 95 | 66 | [77] |
| 25 | 16c | - | 5 | 1b | 3b | 25 | 288 | 95 | 5 | [77] |
| 26 | 16d | - | 7 | 1b | 3b | 25 | 8 | 95 | 11 c | [78] |
| 27 | 10 | 1c | 3c | 25 | 0.5 | 100 | 11 c | [78] | ||
| 28 | 8 | 2c | 4c | 25 | 64 | 100 | 5 c | [78] | ||
| 29 | 17a | (+)-neomenthyl | 4 | 1a | 3a | 65 | 65 | 96 | 22 (R) | [79] |
| 30 | 3 | 1b | 3b | 25 | 12.7 | 96 | 21 (R) | [79] | ||
| 31 | 18 | (−)-menthyl | 3 | 1b | 3b | 25 | 6.25 | 80 | 11 (S) | [79] |
| 32 | 19a | iPr | 5 | 1c | 3c | r.t. | 0.17 | 100 | 34 (S) | [56] |
| 33 | 5 | 2d | 4d | r.t. | 0.83 | 100 | 22 (S) | [56] | ||
| 34 | 19b | tBu | 5 | 1c | 3c | r.t. | 0.17 | 100 | 93 (S) | [56] |
| 35 | 5 | 1d | 3d | r.t. | 0.17 | 100 | 94 (S) | [56] | ||
| 36 | 5 | 1e | 3e | r.t. | 3 | 95 | 89 (S) | [56] |

2.2.2. Group IV and V Metals
2.3. Early Main-Group Elements
2.3.1. Alkaline Metals
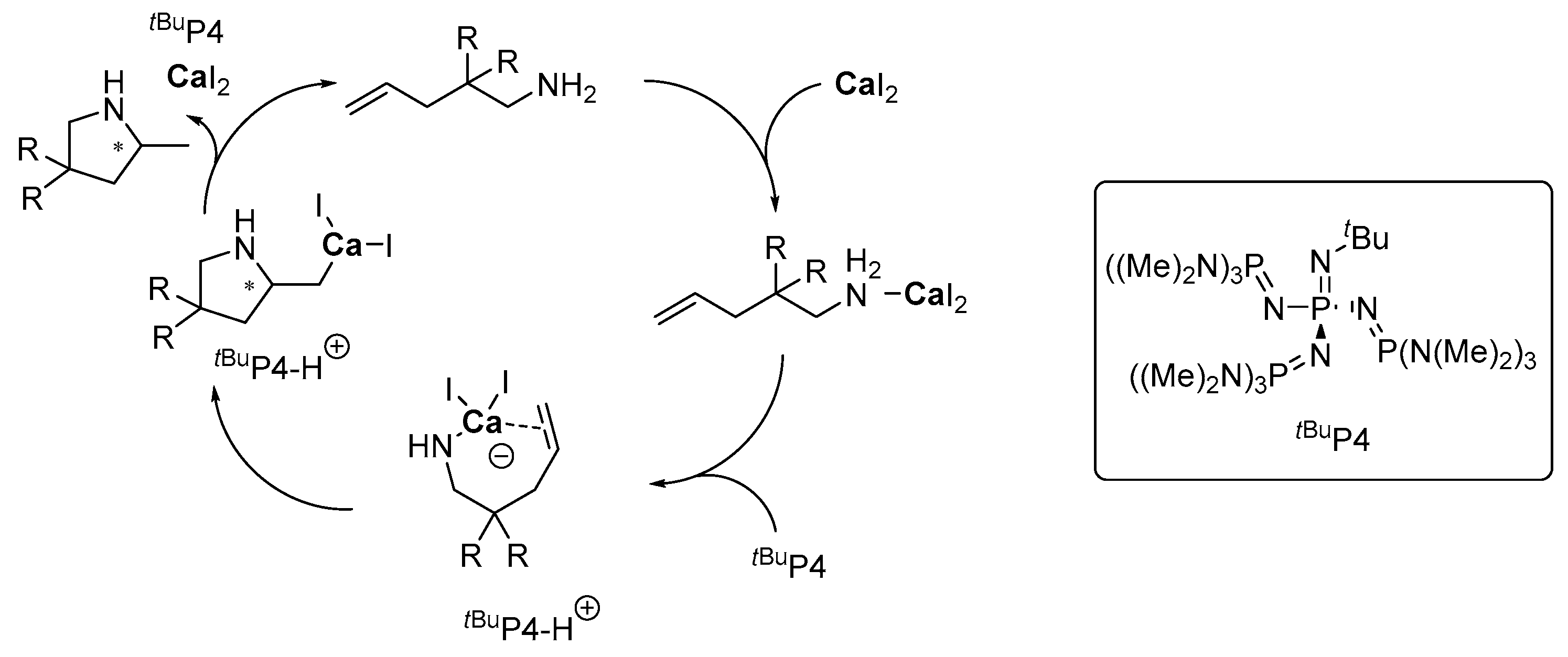
2.3.2. Alkaline Earth Metals
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, D.; Kumar Jain, S. A Comprehensive Review of N-Heterocycles as Cytotoxic Agents. Curr. Med. Chem. 2016, 23, 4338–4394. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, P.; Bezzenine, S.; Gil, R.; Hannedouche, J. Alkene Hydroamination via Earth-Abundant Transition Metal (Iron, Cobalt, Copper and Zinc) Catalysis: A Mechanistic Overview. Adv. Synth. Catal. 2020, 362, 1550–1563. [Google Scholar] [CrossRef]
- Hannedouche, J.; Collin, J.; Trifonov, A.; Schulz, E. Intramolecular Enantioselective Hydroamination Catalyzed by Rare Earth Binaphthylamides. J. Organomet. Chem. 2011, 696, 255–262. [Google Scholar] [CrossRef]
- Müller, T.E.; Hultzsch, K.C.; Yus, M.; Foubelo, F.; Tada, M. Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev. 2008, 108, 3795–3892. [Google Scholar] [CrossRef]
- Bestgen, S.; Roesky, P.W. Inramolecular Hydroamination of Alkenes. In Early Main Group Metal Catalysis; Harder, S., Ed.; Wiley-VCH: Weinheim, Germany, 2020; ISBN 9783527344482. [Google Scholar]
- Huang, L.; Arndt, M.; Gooßen, K.; Heydt, H.; Gooßen, L.J. Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev. 2015, 115, 2596–2697. [Google Scholar] [CrossRef]
- Seayad, J.; Tillack, A.; Hartung, C.G.; Beller, M. Base-Catalyzed Hydroamination of Olefins: An Environmentally Friendly Route to Amines. Adv. Synth. Catal. 2002, 344, 795–813. [Google Scholar] [CrossRef]
- Ye, Y.; Cao, J.; Oblinsky, D.G.; Verma, D.; Prier, C.K.; Scholes, G.D.; Hyster, T.K. Using Enzymes to Tame Nitrogen-Centered Radicals for Enantioselective Hydroamination. Nat. Chem. 2022, 15, 206–212. [Google Scholar] [CrossRef]
- He, Y.; Chen, J.; Jiang, X.; Zhu, S. Enantioselective NiH-Catalyzed Reductive Hydrofunctionalization of Alkenes. Chin. J. Chem. 2022, 40, 651–661. [Google Scholar] [CrossRef]
- Nuñez Bahena, E.; Schafer, L.L. From Stoichiometric to Catalytic E-H Functionalization by Non-Metallocene Zirconium Complexes-Recent Advances and Mechanistic Insights. ACS Catal. 2022, 12, 14934–14953. [Google Scholar] [CrossRef]
- Brunet, J.J.; Neibecker, D. Hydroamination of unsaturated Carbon bonds. In Catalytic Heterofunctionalization: From Hydroanimation to Hydrozirconation, 1st ed.; Togni, A., Grützmacher, H., Eds.; Wiley-VCH: Weinheim, Germany, 2001; ISBN 3527302344. [Google Scholar]
- Müller, T.E.; Beller, M. Metal-Initiated Amination of Alkenes and Alkynes. Chem. Rev. 1998, 98, 675–703. [Google Scholar] [CrossRef] [PubMed]
- Giofrè, S.; Molteni, L.; Beccalli, E.M. Asymmetric Pd(II)-Catalyzed C−O, C−N, C−C Bond Formation Using Alkenes as Substrates: Insight into Recent Enantioselective Developments. Eur. J. Org. Chem. 2022, 26, e202200976. [Google Scholar] [CrossRef]
- Jia, S.M.; Huang, Y.H.; Wang, F. Aminium-Radical-Mediated Intermolecular Hydroamination of Nonactivated Olefins. Synlett 2022, 34, 93–100. [Google Scholar] [CrossRef]
- Bernoud, E.; Lepori, C.; Mellah, M.; Schulz, E.; Hannedouche, J. Recent Advances in Metal Free- and Late Transition Metal-Catalysed Hydroamination of Unactivated Alkenes. Catal. Sci. Technol. 2015, 5, 2017–2037. [Google Scholar] [CrossRef]
- Beesley, R.M.; Ingold, C.K.; Thorpe, J.F. The Formation and Stability of Spiro-Compounds. Part I. Spiro-Compounds from Cyclo-Hexane. J. Chem. Soc. Trans. 1915, 107, 1080–1106. [Google Scholar] [CrossRef] [Green Version]
- Brunet, J.; Neibecker, D.; Niedercorn, F. Functionalisation of Alkenes: Catalytic Amination of Monoolefins. J. Mol. Catal. 1989, 49, 235–259. [Google Scholar] [CrossRef]
- Johns, A.M.; Sakai, N.; Ridder, A.; Hartwig, J.F. Direct Measurement of the Thermodynamics of Vinylarene Hydroamination. J. Am. Chem. Soc. 2006, 128, 9306–9307. [Google Scholar] [CrossRef]
- Hickinbottom, W.J. Reactions of Unsaturated Compounds. Part I. Addition of Arylamines to CycloHexene and 1:4- Dihydronaphthalene. J. Chem. Soc. 1932, 2646–2654. [Google Scholar] [CrossRef]
- Kozlov, N.S.; Gimpelevich, E. Catalytic Condensation of Acetylene with Aromatic Amines. IV. Condensation of Acetylene with Aniline and p-Toluidine in the Presence of Silver Nitrate. J. Gen. Chem. USSR 1936, 6, 1341–1345. [Google Scholar]
- Kozlov, N.S.; Bogdanovskaya, R. Catalytic Condensation of Acetylene with Aromatic Amines. V. Condensation of Acetylene with o- and p-Anisidine in the Presence of Cu2Cl2 and HgCl2. J. Gen. Chem. USSR 1936, 6, 1346–1348. [Google Scholar]
- Coulson, D.R. Catalytic Addition of Secondary Amines to Ethylene. Tetrahedron Lett. 1971, 12, 429–430. [Google Scholar] [CrossRef]
- Nobis, M.; Drießen-Hölscher, B. Recent developments in transition metal catalyzed intermolecular hydroamination reactions—A breakthrough? Angew. Chemie Int. Ed. 2001, 40, 3983–3985. [Google Scholar] [CrossRef]
- Hong, S.; Marks, T.J. Highly Stereoselective Intramolecular Hydroamination/Cyclization of Conjugated Aminodienes Catalyzed by Organolanthanides. J. Am. Chem. Soc. 2002, 124, 7886–7887. [Google Scholar] [CrossRef] [PubMed]
- Wixey, J.S.; Ward, B.D. Chiral Calcium Catalysts for Asymmetric Hydroamination/Cyclisation. Chem. Commun. 2011, 47, 5449–5451. [Google Scholar] [CrossRef]
- Stegner, P.C.; Fischer, C.A.; Nguyen, D.T.; Rösch, A.; Penafiel, J.; Langer, J.; Wiesinger, M.; Harder, S. Intramolecular Alkene Hydroamination with Hybrid Catalysts Consisting of a Metal Salt and a Neutral Organic Base. Eur. J. Inorg. Chem. 2020, 2020, 3387–3394. [Google Scholar] [CrossRef]
- Stegner, P.C.; Eyselein, J.; Ballmann, G.M.; Langer, J.; Schmidt, J.; Harder, S. Calcium Catalyzed Enantioselective Intramolecular Alkene Hydroamination with Chiral C2-Symmetric Bis-Amide Ligands. Dalt. Trans. 2021, 50, 3178–3185. [Google Scholar] [CrossRef]
- Zhang, X.; Tobisch, S.; Hultzsch, K.C. σ-Insertive Mechanism versus Concerted Non-Insertive Mechanism in the Intramolecular Hydroamination of Aminoalkenes Catalyzed by Phenoxyamine Magnesium Complexes: A Synthetic and Computational Study. Chem. Eur. J. 2015, 21, 7841–7857. [Google Scholar] [CrossRef]
- Michon, C.; Abadie, M.A.; Medina, F.; Agbossou-Niedercorn, F. Recent Metal-Catalysed Asymmetric Hydroaminations of Alkenes. J. Organomet. Chem. 2017, 847, 13–27. [Google Scholar] [CrossRef]
- McGrane, P.L.; Jensen, M.; Livinghouse, T. Intramolecular [2 + 2] Cycloadditions of Group IV Metal-Imido Complexes. Applications to the Synthesis of Dihydropyrrole and Tetrahydropyridine Derivatives. J. Am. Chem. Soc. 1992, 114, 5459–5460. [Google Scholar] [CrossRef]
- Walsh, P.J.; Baranger, A.M.; Bergman, R.G. Stoichiometric and Catalytic Hydroamination of Alkynes and Allene by Zirconium Bisamides Cp2Zr(NHR)2. J. Am. Chem. Soc. 1992, 114, 1708–1719. [Google Scholar] [CrossRef]
- Gagné, M.R.; Marks, T.J. Organolanthanide-Catalyzed Hydroamination. Facile, Regiospecific Cyclization of Unprotected Amino Olefins. J. Am. Chem. Soc. 1989, 111, 4108–4109. [Google Scholar] [CrossRef]
- Crimmin, M.R.; Casely, I.J.; Hill, M.S. Calcium-Mediated Intramolecular Hydroamination Catalysis. J. Am. Chem. Soc. 2005, 127, 2042–2043. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.K. BINOL: A Versatile Chiral Reagent. Synlett 2006, 2006, 3366–3367. [Google Scholar] [CrossRef] [Green Version]
- Berthod, M.; Mignani, G.; Woodward, G.; Lemaire, M. Modified BINAP: The How and the Why. Chem. Rev. 2005, 105, 1801–1836. [Google Scholar] [CrossRef]
- Blaser, H.U. The Chiral Pool as a Source of Enantioselective Catalysts and Auxiliaries. Chem. Rev. 1992, 92, 935–952. [Google Scholar] [CrossRef]
- Casiraghi, G.; Zanardi, F.; Rassu, G.; Spanu, P. Stereoselective Approaches to Bioactive Carbohydrates and Alkaloids-with a Focus on Recent Syntheses Drawing from the Chiral Pool. Chem. Rev. 1995, 95, 1677–1716. [Google Scholar] [CrossRef]
- Brill, Z.G.; Condakes, M.L.; Ting, C.P.; Maimone, T.J. Navigating the Chiral Pool in the Total Synthesis of Complex Terpene Natural Products. Chem. Rev. 2017, 117, 11753–11795. [Google Scholar] [CrossRef]
- Stout, C.N.; Renata, H. Reinvigorating the Chiral Pool: Chemoenzymatic Approaches to Complex Peptides and Terpenoids. Acc. Chem. Res. 2021, 54, 1143–1156. [Google Scholar] [CrossRef]
- Paek, S.M.; Jeong, M.; Jo, J.; Heo, Y.M.; Han, Y.T.; Yun, H. Recent Advances in Substrate-Controlled Asymmetric Induction Derived from Chiral Pool α-Amino Acids for Natural Product Synthesis. Molecules 2016, 21, 951. [Google Scholar] [CrossRef] [Green Version]
- Money, T.; Wong, M.K.C. The Use of Cyclic Monoterpenoids as Enantiopure Starting Materials in Natural Product Synthesis. Stud. Nat. Prod. Chem. 1995, 16, 123–288. [Google Scholar] [CrossRef]
- Koskinen, A.M.P. Chirospecific Synthesis: Catalysis and Chiral Pool Hand in Hand. Pure Appl. Chem. 2011, 83, 435–443. [Google Scholar] [CrossRef]
- Li, F.; Renata, H. A Chiral-Pool-Based Strategy to Access Trans-Syn-Fused Drimane Meroterpenoids: Chemoenzymatic Total Syntheses of Polysin, N-Acetyl-Polyveoline and the Chrodrimanins. J. Am. Chem. Soc. 2021, 143, 18280–18286. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, R.; Rana, R.; Maurya, S.K. Organocatalyzed C−N Bond-Forming Reactions for the Synthesis of Amines and Amides. ChemCatChem 2021, 13, 1867–1897. [Google Scholar] [CrossRef]
- Amadji, M.; Vadecard, J.; Plaquevent, J.; Duhamel, L.; Duhamel, P. First Catalytic Enantioselective Proton Abstraction. J. Am. Chem. Soc. 1996, 118, 12483–12484. [Google Scholar] [CrossRef]
- Krix, G.; Bommarius, A.S.; Drauz, K.; Kottenhahn, M.; Schwarm, M.; Kula, M.R. Enzymatic Reduction of α-Keto Acids Leading to L-Amino Acids, D- or L-Hydroxy Acids. J. Biotechnol. 1997, 53, 29–39. [Google Scholar] [CrossRef]
- Hannedouche, J.; Schulz, E. Hydroamination and Hydroaminoalkylation of Alkenes by Group 3-5 Elements: Recent Developments and Comparison with Late Transition Metals. Organometallics 2018, 37, 4313–4326. [Google Scholar] [CrossRef]
- Chen, Q.A.; Chen, Z.; Dong, V.M. Rhodium-Catalyzed Enantioselective Hydroamination of Alkynes with Indolines. J. Am. Chem. Soc. 2015, 137, 8392–8395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, M.; Yokoyama, H.; Endo, K.; Shibata, T. Ru-Catalyzed β-Selective and Enantioselective Addition of Amines to Styrenes Initiated by Direct Arene-Exchange. Org. Biomol. Chem. 2012, 10, 3815–3818. [Google Scholar] [CrossRef]
- Flaget, A.; Zhang, C.; Mazet, C. Ni-Catalyzed Enantioselective Hydrofunctionalizations of 1,3-Dienes. ACS Catal. 2022, 12, 15638–15647. [Google Scholar] [CrossRef]
- Rocard, L.; Chen, D.; Stadler, A.; Zhang, H.; Gil, R.; Bezzenine, S.; Hannedouche, J. Earth-Abundant 3d TransitionMetal Catalysts for Hydroalkoxylation and Hydroamination of Unactivated Alkenes. Catalysts 2021, 11, 674. [Google Scholar] [CrossRef]
- Li, Y.; Marks, T.J. Organolanthanide-Catalyzed Intramolecular Hydroamination/Cyclization of Aminoalkynes. J. Am. Chem. Soc. 1996, 7863, 9295–9306. [Google Scholar] [CrossRef]
- Gagné, M.R.; Brard, L.; Conticello, V.P.; Giardello, M.A.; Stern, C.L.; Marks, T.J. Stereoselection Effects In the Catalytic Hydroaminatlon/Cyclization of Aminoolefins at Chiral Organolanthanide Centers. Organometallics 1992, 11, 2003–2005. [Google Scholar] [CrossRef]
- Li, Y.; Fu, P.; Marks, T.J. Organolanthanide-Catalyzed Carbon-Heteroatom Bond Formation. Observations on the Facile, Regiospecific Cyclization of Aminoalkynes. Organometallics 1994, 13, 4349–4440. [Google Scholar] [CrossRef]
- Manna, K.; Kruse, M.L.; Sadow, A.D. Concerted C-N/C-H Bond Formation in Highly Enantioselective Yttrium(III)-Catalyzed Hydroamination. ACS Catal. 2011, 1, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Manna, K.; Xu, S.; Sadow, A.D. A Highly Enantioselective Zirconium Catalyst for Intramolecular Alkene Hydroamination: Significant Isotope Effects on Rate and Stereoselectivity. Angew. Chem. 2011, 123, 1905–1908. [Google Scholar] [CrossRef]
- Manna, K.; Everett, W.C.; Schoendorff, G.; Ellern, A.; Windus, T.L.; Sadow, A.D. Highly Enantioselective Zirconium-Catalyzed Cyclization of Aminoalkenes. J. Am. Chem. Soc. 2013, 135, 7235–7250. [Google Scholar] [CrossRef] [Green Version]
- Gagné, M.R.; Nolan, S.P.; Marks, T.J. Organolanthanide-Centered Hydroamination/Cyclizatlon of Aminoolefins. Expedient Oxidative Access to Catalytic Cycles. Organometallics 1990, 9, 1716–1718. [Google Scholar] [CrossRef]
- Douglass, M.R.; Ogasawara, M.; Hong, S.; Metz, M.V.; Marks, T.J. “Widening the Roof”: Synthesis and Characterization of New Chiral C1-Symmetric Octahydrofluorenyl Organolanthanide Catalysts and Their Implementation in the Stereoselective Cyclizations of Aminoalkenes and Phosphinoalkenes. Organometallics 2002, 21, 283–292. [Google Scholar] [CrossRef]
- Hong, S.; Kawaoka, A.M.; Marks, T.J. Intramolecular Hydroamination/Cyclization of Conjugated Aminodienes Catalyzed by Organolanthanide Complexes. Scope, Diastereo- and Enantioselectivity, and Reaction Mechanism. J. Am. Chem. Soc. 2003, 125, 15878–15892. [Google Scholar] [CrossRef]
- Molander, G.A.; Dowdy, E.D. Catalytic Intramolecular Hydroamination of Hindered Alkenes Using Organolanthanide Complexes. J. Org. Chem. 1998, 63, 8983–8988. [Google Scholar] [CrossRef]
- Molander, G.A.; Dowdy, E.D. Lanthanide-Catalyzed Hydroamination of Hindered Alkenes in Synthesis: Rapid Access to 10,11-Dihydro-5H-Dibenzo[a,d]Cyclohepten-5,10-Imines. J. Org. Chem. 1999, 64, 6515–6517. [Google Scholar] [CrossRef]
- Tobisch, S. Organolanthanide-Mediated Intermolecular Hydroamination of 1,3-Dienes: Mechanistic Insights from a Computational Exploration of Diverse Mechanistic Pathways for the Stereoselective Hydroamination of 1,3-Butadiene with a Primary Amine Supported by an Ansa-Neodymocene-Based Catalyst. Chem. Eur. J. 2005, 11, 6372–6385. [Google Scholar] [CrossRef]
- Tobisch, S. Mechanism and Exo-Regioselectivity of Organolanthanide-Mediated Intramolecular Hydroamination/Cyclization of 1,3-Disubstituted Aminoallenes: A Computational Study. Chem. Eur. J. 2006, 12, 2520–2531. [Google Scholar] [CrossRef] [PubMed]
- Motta, A.; Fragalà, I.L.; Marks, T.J. Energetics and Mechanism of Organolanthanide-Mediated Phosphinoalkene Hydrophosphination/Cyclization. A Density Functional Theory Analysis. Organometallics 2005, 24, 4995–5003. [Google Scholar] [CrossRef]
- Motta, A.; Fragalà, I.L.; Marks, T.J. Organolanthanide-Catalyzed Hydroamination/Cyclization Reactions of Aminoalkynes. Computational Investigation of Mechanism, Lanthanide Identity, and Substituent Effects for a Very Exothermic C-N Bond-Forming Process. Organometallics 2006, 25, 5533–5539. [Google Scholar] [CrossRef]
- Tobisch, S. Computational Mechanistic Elucidation of the Intramolecular Aminoalkene Hydroamination Catalysed by Iminoanilide Alkaline-Earth Compounds. Chem. Eur. J. 2015, 21, 6765–6779. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Crimmin, M.R.; Barrett, A.G.M.; Hill, M.S.; Kociok-Köhn, G.; Procopiou, P.A. Cation Charge Density and Precatalyst Selection in Group 2-Catalyzed Aminoalkene Hydroamination. Organometallics 2011, 30, 1493–1506. [Google Scholar] [CrossRef]
- Ryu, J.S.; Marks, T.J.; McDonald, F.E. Organolanthanide-Catalyzed Intramolecular Hydroamination/Cyclization/Bicyclization of Sterically Encumbered Substrates. Scope, Selectivity, and Catalyst Thermal Stability for Amine-Tethered Unactivated 1,2-Disubstituted Alkenes. J. Org. Chem. 2004, 69, 1038–1052. [Google Scholar] [CrossRef]
- Gagné, M.R.; Stern, C.L.; Marks, T.J. Organolanthanide-Catalyzed Hydroamination. A Kinetic, Mechanistic, and Diastereoselectivity Study of the Cyclization of N-Unprotected Amino Olefins. J. Am. Chem. Soc. 1992, 114, 275–294. [Google Scholar] [CrossRef]
- Tobisch, S. Intermolecular Hydroamination of Vinylarenes by Iminoanilide Alkaline-Earth Catalysts: A Computational Scrutiny of Mechanistic Pathways. Chem. Eur. J. 2014, 20, 8988–9001. [Google Scholar] [CrossRef]
- Giardello, M.A.; Conticello, V.P.; Brard, L.; Gagné, M.R.; Marks, T.J. Chiral Organolanthanides Designed for Asymmetric Catalysis. J. Am. Chem. Soc. 1994, 116, 10241–10254. [Google Scholar] [CrossRef]
- Conticello, V.P.; Brard, L.; Giardello, M.A.; Tsuji, Y.; Sabat, M.; Stern, C.L.; Marks, T.J. Chiral Organolanthanide Complexes for Enantioselective Olefin Hydrogenation. J. Am. Chem. Soc. 1992, 114, 2761–2762. [Google Scholar] [CrossRef]
- Hong, S.; Tian, S.; Metz, M.V.; Marks, T.J. C2-Symmetric Bis (Oxazolinato) Lanthanide Catalysts for Enantioselective Intramolecular Hydroamination/Cyclization. J. Am. Chem. Soc. 2003, 125, 14768–14783. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.D.; Core, B.A.; Blake, M.P.; Pope, S.J.A.; Mountford, P.; Ward, B.D. Chiral Lanthanide Complexes: Coordination Chemistry, Spectroscopy, and Catalysis. Dalt. Trans. 2014, 43, 5871–5885. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, Y.K.; Shim, J.H.; Kim, M.; Han, M.; Livinghouse, T.; Lee, P.H. Internal Alkene Hydroaminations Catalyzed by Zirconium(IV) Complexes and Asymmetric Alkene Hydroaminations Catalyzed by Yttrium(III) Complexes. Adv. Synth. Catal. 2006, 348, 2609–2618. [Google Scholar] [CrossRef]
- Heck, R.; Schulz, E.; Collin, J.; Carpentier, J.F. Group 3 Metal Complexes Based on a Chiral Tetradentate Diamine-Diamide Ligand: Synthesis and Use in Polymerization of (d,l)-Lactide and Intramolecular Alkene Hydroamination Catalysis. J. Mol. Catal. A Chem. 2007, 268, 163–168. [Google Scholar] [CrossRef]
- Vitanova, D.V.; Hampel, F.; Hultzsch, K.C. (+)-Neomenthyl- and (-)-Phenylmenthyl-Substituted Cyclopentadienyl and Indenyl Yttrocenes as Catalysts in Asymmetric Hydroamination/Cyclization of Aminoalkenes (AHA). J. Organomet. Chem. 2007, 692, 4690–4701. [Google Scholar] [CrossRef]
- Huynh, K.; Anderson, B.K.; Livinghouse, T. Enantioselective Hydroamination/Cyclization of Aminoalkenes by (Bis)-C2 Symmetric and (Mono)-C2 Symmetric Anionic Tetraamide Complexes of La(III). Tetrahedron Lett. 2015, 56, 3658–3661. [Google Scholar] [CrossRef] [Green Version]
- Reznichenko, A.L.; Hultzsch, K.C. C2-Symmetric Zirconium Bis(Amidate) Complexes with Enhanced Reactivity in Aminoalkene Hydroamination. Organometallics 2010, 29, 24–27. [Google Scholar] [CrossRef]
- Chapurina, Y.; Guillot, R.; Lyubov, D.; Trifonov, A.; Hannedouche, J.; Schulz, E. LiCl-Effect on Asymmetric Intramolecular Hydroamination Catalyzed by Binaphthylamido Yttrium Complexes. J. Chem. Soc. Dalt. Trans. 2013, 42, 507–520. [Google Scholar] [CrossRef]
- Chapurina, Y.; Ibrahim, H.; Guillot, R.; Kolodziej, E.; Collin, J.; Trifonov, A.; Schulz, E.; Hannedouche, J. Catalytic, Enantioselective Intramolecular Hydroamination of Primary Amines Tethered to Di- and Trisubstituted Alkenes. J. Org. Chem. 2011, 76, 10163–10172. [Google Scholar] [CrossRef] [PubMed]
- Yonson, N.; Yim, J.C.H.; Schafer, L.L. Alkene Hydroamination with a Chiral Zirconium Catalyst. Connecting Ligand Design, Precatalyst Structure and Reactivity Trends. Inorganica Chim. Acta 2014, 422, 14–20. [Google Scholar] [CrossRef]
- Reznichenko, A.L.; Hultzsch, K.C. C1-Symmetric Rare-Earth-Metal Aminodiolate Complexes for Intra- and Intermolecular Asymmetric Hydroamination of Alkenes. Organometallics 2013, 32, 1394–1408. [Google Scholar] [CrossRef]
- Reznichenko, A.L.; Emge, T.J.; Audörsch, S.; Klauber, E.G.; Hultzsch, K.C.; Schmidt, B. Group 5 Metal Binaphtholate Complexes for Catalytic Asymmetric Hydroaminoalkylation and Hydroamination/Cyclization. Organometallics 2011, 30, 921–924. [Google Scholar] [CrossRef]
- Teng, H.L.; Luo, Y.; Wang, B.; Zhang, L.; Nishiura, M.; Hou, Z. Synthesis of Chiral Aminocyclopropanes by Rare-Earth-Metal-Catalyzed Cyclopropene Hydroamination. Angew. Chem. 2016, 128, 15632–15636. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Lee, H.; Audörsch, S.; Reznichenko, A.L.; Nawara-Hultzsch, A.J.; Schmidt, B.; Hultzsch, K.C. Asymmetric Intra- and Intermolecular Hydroamination Catalyzed by 3,3′-Bis(Trisarylsilyl)- and 3,3′-Bis(Arylalkylsilyl)-Substituted Binaphtholate Rare-Earth-Metal Complexes. Organometallics 2018, 37, 4358–4379. [Google Scholar] [CrossRef]
- Chai, Z.; Hua, D.; Li, K.; Chu, J.; Yang, G. A Novel Chiral Yttrium Complex with a Tridentate Linked Amido-Indenyl Ligand for Intramolecular Hydroamination. Chem. Commun. 2014, 50, 177–179. [Google Scholar] [CrossRef]
- Hoover, J.M.; Petersen, J.R.; Pikul, J.H.; Johnson, A.R. Catalytic Intramolecular Hydroamination of Substituted Aminoallenes by Chiral Titanium Amino-Alcohol Complexes. Organometallics 2004, 23, 4614–4620. [Google Scholar] [CrossRef]
- Hickman, A.J.; Hughs, L.D.; Jones, C.M.; Li, H.; Redford, J.E.; Sobelman, S.J.; Kouzelos, J.A.; Johnson, A.R. Sterically Encumbered Chiral Amino Alcohols for Titanium-Catalyzed Asymmetric Intramolecular Hydroamination of Aminoallenes. Tetrahedron Asymmetry 2009, 20, 1279–1285. [Google Scholar] [CrossRef]
- Hansen, M.C.; Heusser, C.A.; Narayan, T.C.; Fong, K.E.; Hara, N.; Kohn, A.W.; Venning, A.R.; Rheingold, A.L.; Johnson, A.R. Asymmetric Catalytic Intramolecular Hydroamination of Aminoallenes by Tantalum Amidoalkoxide Complexes. Organometallics 2011, 30, 4616–4623. [Google Scholar] [CrossRef]
- Near, K.E.; Chapin, B.M.; McAnnally-Linz, D.C.; Johnson, A.R. Asymmetric Hydroamination of Aminoallenes Catalyzed by Titanium and Tantalum Complexes of Chiral Sulfonamide Alcohol Ligands. J. Organomet. Chem. 2011, 696, 81–86. [Google Scholar] [CrossRef]
- Sha, F.; Mitchell, B.S.; Ye, C.Z.; Abelson, C.S.; Reinheimer, E.W.; Lemagueres, P.; Ferrara, J.D.; Takase, M.K.; Johnson, A.R. Catalytic Intramolecular Hydroamination of Aminoallenes Using Titanium Complexes of Chiral, Tridentate, Dianionic Imine-Diol Ligands. Dalt. Trans. 2019, 48, 9603–9616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, F.; Shimizu, E.A.; Slocumb, H.S.; Towell, S.E.; Zhen, Y.; Porter, H.Z.; Takase, M.K.; Johnson, A.R. Catalytic Intramolecular Hydroamination of Aminoallenes Using Titanium and Tantalum Complexes of Sterically Encumbered Chiral Sulfonamides. Dalt. Trans. 2020, 49, 12418–12431. [Google Scholar] [CrossRef] [PubMed]
- Manna, K.; Eedugurala, N.; Sadow, A.D. Zirconium-Catalyzed Desymmetrization of Aminodialkenes and Aminodialkynes through Enantioselective Hydroamination. J. Am. Chem. Soc. 2015, 137, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fok, E.Y.; Show, V.L.; Johnson, A.R. Intramolecular Hydroamination of Trisubstituted Aminoallenes Catalyzed by Titanium Complexes of Diaryl Substituted Tridentate Imine-Diols. Polyhedron 2021, 198, 115070. [Google Scholar] [CrossRef]
- Zhou, X.; Wei, B.; Sun, X.L.; Tang, Y.; Xie, Z. Asymmetric Hydroamination Catalyzed by a New Chiral Zirconium System: Reaction Scope and Mechanism. Chem. Commun. 2015, 51, 5751–5753. [Google Scholar] [CrossRef] [Green Version]
- Hussein, L.; Purkait, N.; Biyikal, M.; Tausch, E.; Roesky, P.W.; Blechert, S. Highly Enantioselective Hydroamination to Six-Membered Rings by Heterobimetallic Catalysts. Chem. Commun. 2014, 50, 3862–3864. [Google Scholar] [CrossRef] [Green Version]
- Wood, M.C.; Leitch, D.C.; Yeung, C.S.; Kozak, J.A.; Schafer, L.L. Chiral Neutral Zirconium Amidate Complexes for the Asymmetric Hydroamination of Alkenes. Angew. Chem. 2007, 119, 358–362. [Google Scholar] [CrossRef]
- Eisenberger, P.; Schafer, L.L. Catalytic Synthesis of Amines and N-Containing Heterocycles: Amidate Complexes for Selective C-N and C-C Bond-Forming Reactions. Pure Appl. Chem. 2010, 82, 1503–1515. [Google Scholar] [CrossRef]
- Wang, Q.; Song, H.; Zi, G. Synthesis, Structure, and Catalytic Activity of Group 4 Complexes with New Chiral Biaryl-Based NO2 Ligands. J. Organomet. Chem. 2010, 695, 1583–1591. [Google Scholar] [CrossRef]
- Zhang, F.; Song, H.; Zi, G. Synthesis and Catalytic Activity of Group 5 Metal Amides with Chiral Biaryldiamine-Based Ligands. Dalt. Trans. 2011, 40, 1547–1566. [Google Scholar] [CrossRef] [PubMed]
- Begouin, J.M.; Niggemann, M. Calcium-Based Lewis Acid Catalysts. Chem. Eur. J. 2013, 19, 8030–8041. [Google Scholar] [CrossRef] [PubMed]
- Ates, A.; Quinet, C. Efficient Intramolecular Hydroamination of Unactivated Alkenes Catalysed by Butyllithium. Eur. J. Org. Chem. 2003, 2003, 1623–1626. [Google Scholar] [CrossRef]
- Peng, X.; Kaga, A.; Hirao, H.; Chiba, S. Hydroamination of Alkenyl: N-Arylhydrazones Mediated by t -BuOK for the Synthesis of Nitrogen Heterocycles. Org. Chem. Front. 2016, 3, 609–613. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Wu, L.Y.; Fang, H.S.; Zhang, T.; Mao, Z.F.; Zou, Y.; Zhang, X.J.; Yan, M. Intramolecular Hydroamidation of Ortho-Vinyl Benzamides Promoted by Potassium Tert-Butoxide/N,N-Dimethylformamide. Adv. Synth. Catal. 2017, 359, 3894–3899. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Guo, S.; Fan, J.; Fan, X. T - BuOK-Catalyzed Regio- and Stereoselective Intramolecular Hydroamination Reaction Leading to Phthalazinoquinazolinone Derivatives. J. Org. Chem. 2023, 8, 1282–1291. [Google Scholar] [CrossRef]
- Horrillo-Martínez, P.; Hultzsch, K.C.; Gil, A.; Branchadell, V. Base-Catalyzed Anti-Markovnikov Hydroamination of Vinylarenes—Scope, Limitations and Computational Studies. Eur. J. Org. Chem. 2007, 2007, 3311–3325. [Google Scholar] [CrossRef]
- Martínez, P.H.; Hultzsch, K.C.; Hampel, F. Base-Catalysed Asymmetric Hydroamination/Cyclisation of Aminoalkenes Utilising a Dimeric Chiral Diamidobinaphthyl Dilithium Salt. Chem. Commun. 2006, 37, 2221–2223. [Google Scholar] [CrossRef]
- Deschamp, J.; Olier, C.; Schulz, E.; Guillot, R.; Hannedouche, J.; Collin, J. Simple Chiral Diaminobinaphthyl Dilithium Salts for Intramolecular Catalytic Asymmetric Hydroamination of Amino-1,3-Dienes. Adv. Synth. Catal. 2010, 352, 2171–2176. [Google Scholar] [CrossRef]
- Deschamp, J.; Collin, J.; Hannedouche, J.; Schulz, E. Easy Routes towards Chiral Lithium Binaphthylamido Catalysts for the Asymmetric Hydroamination of Amino-1,3-Dienes and Aminoalkenes. Eur. J. Org. Chem. 2011, 2011, 3329–3338. [Google Scholar] [CrossRef]
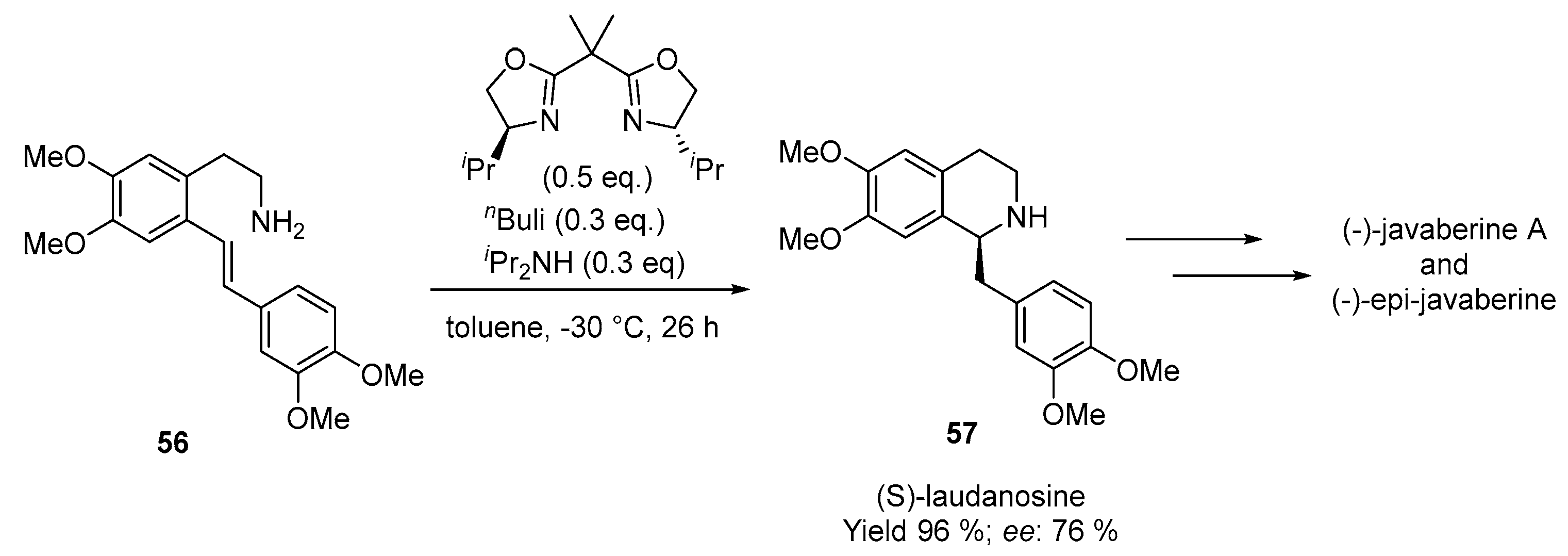
- Ogata, T.; Ujihara, A.; Tsuchida, S.; Shimizu, T.; Kaneshige, A.; Tomioka, K. Catalytic Asymmetric Intramolecular Hydroamination of Aminoalkenes. Tetrahedron Lett. 2007, 48, 6648–6650. [Google Scholar] [CrossRef]
- Ogata, T.; Kimachi, T.; Yamada, K.I.; Yamamoto, Y.; Tomioka, K. Catalytic Asymmetric Synthesis of (S)-Laudanosine by Hydroamination. Heterocycles 2012, 86, 469–485. [Google Scholar] [CrossRef]
- Uenishi, S.; Kakigi, R.; Hideshima, K.; Miyawaki, A.; Matsuoka, J.; Ogata, T.; Tomioka, K.; Yamamoto, Y. Asymmetric Total Synthesis of (−)-Javaberine A and (−)-Epi-Javaberine A Based on Catalytic Intramolecular Hydroamination of N-Methyl-2-(2-Styrylaryl)Ethylamine. Tetrahedron 2021, 90, 132165. [Google Scholar] [CrossRef]
- Horrillo-Martínez, P.; Hultzsch, K.C. Intramolecular Hydroamination/Cyclization of Aminoalkenes Catalyzed by Diamidobinaphthyl Magnesium- and Zinc-Complexes. Tetrahedron Lett. 2009, 50, 2054–2056. [Google Scholar] [CrossRef]
- Buch, F.; Harder, S. A Study on Chiral Organocalcium Complexes: Attempts in Enantioselective Catalytic Hydrosilylation and Intramolecular Hydroamination of Alkenes. Z. Naturforsch. B 2008, 63, 169–177. [Google Scholar] [CrossRef]
- Liu, B.; Roisnel, T.; Carpentier, J.F.; Sarazin, Y. Cyclohydroamination of Aminoalkenes Catalyzed by Disilazide Alkaline-Earth Metal Complexes: Reactivity Patterns and Deactivation Pathways. Chem. Eur. J. 2013, 19, 2784–2802. [Google Scholar] [CrossRef]
- Schlenk, W.; Schlenk, W. Über Die Konstitution Der Grignardschen Magnesiumverbindungen. Berichte Dtsch. Chem. Gesellschaft 1929, 62, 920–924. [Google Scholar] [CrossRef]
- Neal, S.R.; Ellern, A.; Sadow, A.D. Optically Active, Bulky Tris(Oxazolinyl)Borato Magnesium and Calcium Compounds for Asymmetric Hydroamination/Cyclization. J. Organomet. Chem. 2011, 696, 228–234. [Google Scholar] [CrossRef]
- Zhang, X.; Emge, T.J.; Hultzsch, K.C. A Chiral Phenoxyamine Magnesium Catalyst for the Enantioselective Hydroamination/Cyclization of Aminoalkenes and Intermolecular Hydroamination of Vinyl Arenes. Angew. Chemie Int. Ed. 2012, 51, 394–398. [Google Scholar] [CrossRef]
- Wixey, J.S.; Ward, B.D. Modular Ligand Variation in Calcium Bisimidazoline Complexes: Effects on Ligand Redistribution and Hydroamination Catalysis. Dalt. Trans. 2011, 40, 7693–7696. [Google Scholar] [CrossRef]
- Nixon, T.D.; Ward, B.D. Calcium Amido-Bisoxazoline Complexes in Asymmetric Hydroamination/Cyclisation Catalysis. Chem. Commun. 2012, 48, 11790–11792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notz, S. Ph.D. Thesis, TU Chemnitz: Chemnitz, Germany, ongoing.
- Penafiel, J.; Maron, L.; Harder, S. Early Main Group Metal Catalysis: How Important Is the Metal? Angew. Chemie 2015, 54, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Jenter, J.; Köppe, R.; Roesky, P.W. 2,5-Bis{N-(2,6-Diisopropylphenyl)Iminomethyl}pyrrolyl Complexes of the Heavy Alkaline Earth Metals: Synthesis, Structures, and Hydroamination Catalysis. Organometallics 2011, 30, 1404–1413. [Google Scholar] [CrossRef]
- Romero, N.; Roşca, S.C.; Sarazin, Y.; Carpentier, J.F.; Vendier, L.; Mallet-Ladeira, S.; Dinoi, C.; Etienne, M. Highly Fluorinated Tris(Indazolyl)Borate Silylamido Complexes of the Heavier Alkaline Earth Metals: Synthesis, Characterization, and Efficient Catalytic Intramolecular Hydroamination. Chem. Eur. J. 2014, 21, 4115–4125. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




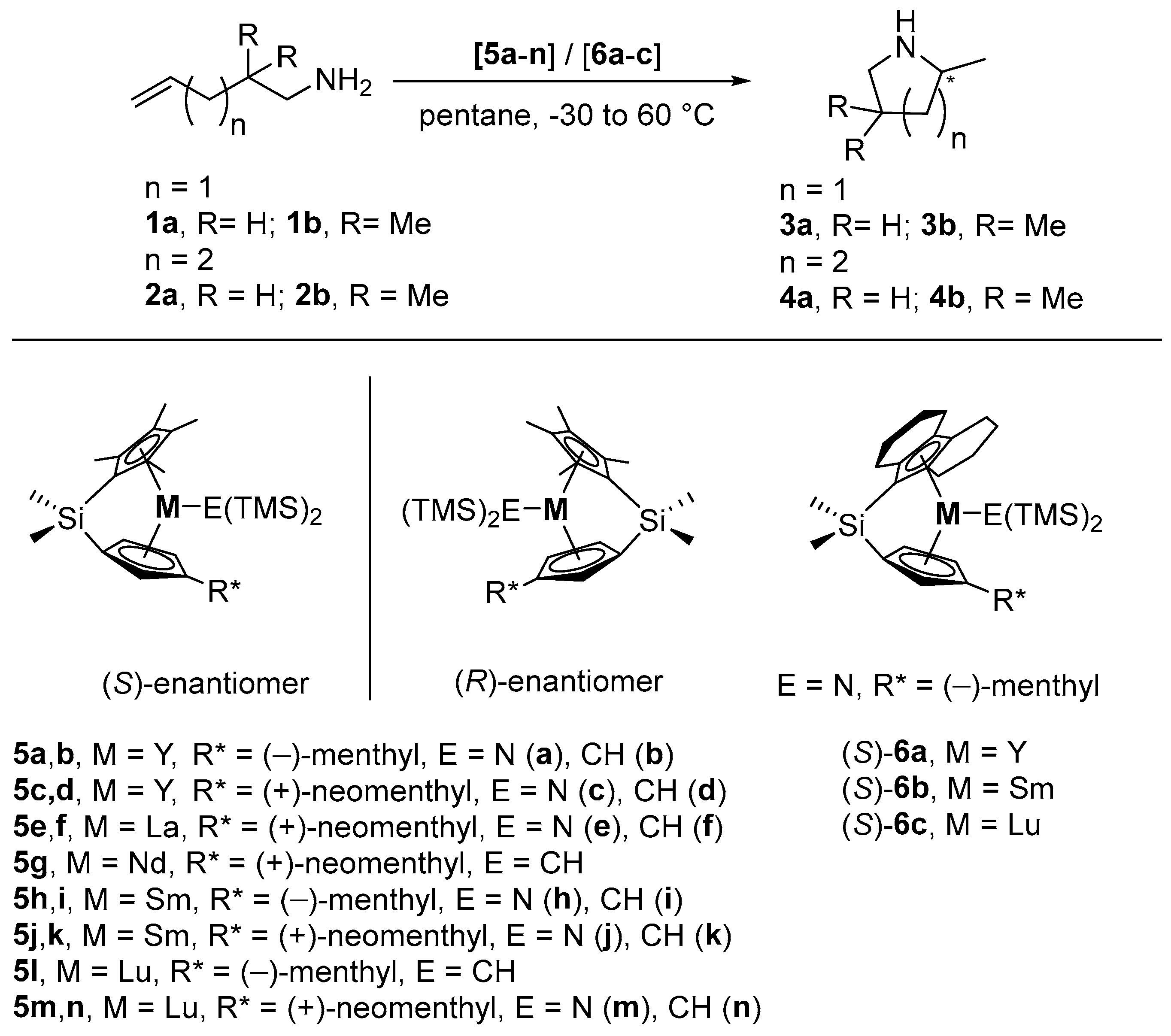
| Entry | Cat. | M | E | R* | Substr. | Prod. | T a [°C] | ee [%] b,c | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | (R)-5a,b d | Y | N,CH b | (−)-menthyl | 1a | 3a | 25 | 69 (+) | [73] |
| 2 | 1b | 3b | 25 | 43 (+) | [73] | ||||
| 3 | (R)-5c | Y | N | (+)-neomenthyl | 1a | 3a | 25 | 50 (−) | [73] |
| 4 | 1b | 3b | 25 | 40 (−) | [73] | ||||
| 5 | (R/S)-5d e | Y | CH | (+)-neomenthyl | 1a | 3a | 25 | 47 (−) | [73] |
| 6 | 1b | 3b | 25 | 36 (−) | [73] | ||||
| 7 | (R)-5e | La | N | (+)-neomenthyl | 1a | 3a | 25 | 31 (−) | [54,73] |
| 8 | 1b | 3b | 25 | 14 (−) | [54,73] | ||||
| 9 | (R,S)-5f | La | CH | (+)-neomenthyl | 1a | 3a | 25 | 36 (−) | [73] |
| 10 | (R/S)-5g e | Nd | CH | (+)-neomenthyl | 1a | 3a | 25 | 55 (−) | [73] |
| 11 | 0 | 64 (−) | [73] | ||||||
| 12 | 1b | 3b | −20 | 61 (−) | [73] | ||||
| 13 | (S)-5h,i d | Sm | N,CH b | (−)-menthyl | 1a | 3a | 25 | 62 (+) | [54,73] |
| 14 | 0 | 72 (+) | [54,73] | ||||||
| 15 | 1b | 3b | 25 | 53 (+) | [54,73] | ||||
| 16 | 0 | 61 (+) | [54,73] | ||||||
| 17 | −30 | 74 (+) | [54,73] | ||||||
| 18 | 2b | 4b | 25 | 15 (−) | [54,73] | ||||
| 19 | (R)-5h | Sm | N | (−)-menthyl | 1a | 3a | 25 | 60 (+) | [73] |
| 20 | (S)-5j | Sm | N | (+)-neomenthyl | 1a | 3a | 25 | 55 (−) | [73] |
| 21 | (R)-5j,k d | Sm | N,CH b | (+)-neomenthyl | 1a | 3a | 25 | 52 (−) | [54,73] |
| 22 | 0 | 58 (−) | [54,73] | ||||||
| 23 | 1b | 3b | 25 | 51 (−) | [54,73] | ||||
| 24 | 0 | 54 (−) | [54,73] | ||||||
| 25 | −30 | 64 (−) | [54,73] | ||||||
| 26 | 2b | 4b | 25 | 17 (−) | [54,73] | ||||
| 27 | (R/S)-5k e | Sm | CH | (+)-neomenthyl | 1a | 3a | 25 | 61 (−) | [73] |
| 28 | (R)-5l | Lu | CH | (−)-menthyl | 1b | 3b | 25 | 29 (−) | [73] |
| 29 | (R)-5m | Lu | N | (+)-neomenthyl | 1b | 3b | 25 | 40(+) | [73] |
| 30 | (R/S)-5n e | Lu | CH | (+)-neomenthyl | 1a | 3a | 25 | 29 (+) | [73] |
| 31 | 1b | 3b | 25 | 36 (+) | [73] | ||||
| 32 | (S)-6a | Y | N | (−)-menthyl | 1a | 3a | 60 | 5 (+) | [60] |
| 33 | 1b | 3b | 25 | 17 (+) | [60] | ||||
| 34 | 2a | 4a | 60 | 3 (+) | [60] | ||||
| 35 | 2b | 4b | 60 | 54 (+) | [60] | ||||
| 36 | 25 | 67 (+) | [60] | ||||||
| 37 | (S)-6b | Sm | N | (−)-menthyl | 1a | 3a | 60 | 37 (+) | [60] |
| 38 | 25 | 46 (+) | [60] | ||||||
| 39 | 1b | 3b | 25 | 32 (+) | [60] | ||||
| 40 | 2a | 4a | 60 | 10 (+) | [60] | ||||
| 41 | 2b | 4b | 60 | 43 (+) | [60] | ||||
| 42 | 40 | 41 (+) | [60] | ||||||
| 43 | 25 | 41 (+) | [60] | ||||||
| 44 | (S)-6c | Lu | N | (−)-menthyl | 1a | 3a | 60 | 16 (−) | [60] |
| 45 | 1b | 3b | 25 | 2 (+) | [60] | ||||
| 46 | 2b | 4b | 60 | 15 (+) | [60] |

| Entry | Cat. | M | R* | Substr. | Prod. | T [°C] | TOF [h−1] | ee [%] b | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | (S)-5h | Sm | (−)-menthyl | E-7a | 9a | 80 | 0.26 | 28 (+) | [70] |
| 2 | E-7b | 9b | 80 | 0.15 | 32 (+) | [70] | |||
| 3 | Z-8 | 10 | 80 | 0.16 | 16 (+) | [70] | |||
| 3 | (S)-6a | Y | (+)-neomenthyl | E-7a | 9a | 100 | 0.07 | 26 (+) | [70] |
| 4 | E-7b | 9b | 100 | 0.06 | 28 (+) | [70] | |||
| 5 | Z-8 | 10 | 100 c | 0.30 | 58 (−) | [70] | |||
| 6 | 80 d | 0.16 | 64 (−) | [70] | |||||
| 7 | 60 c | 0.03 | 68 (−) | [70] | |||||
| 8 | (S)-6b | Sm | (+)-neomenthyl | E-7a | 9a | 80 | 0.18 | 24 (+) | [70] |
| 9 | E-7b | 9b | 80 | 0.06 | 22 (+) | [70] | |||
| 10 | Z-8 | 10 | 80 | 0.11 | 16 (+) | [70] |

| Entry | Cat. | Substr. | Prod. | T [°C] | Ratio E/Z b | ee [%] c | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | (S)-5a,b | 11 | E/Z-13 | 25 | 98:2 | 41 | [61] |
| 2 | (S)-5h | 11 | E/Z-13 | 23 | 98:2 | 25 d | [61] |
| 3 | 12a | E/Z-14a | 25 | 98:2 | 37 (R) | [61] | |
| 4 | (S)-6b | 11 | E/Z-13 | 25 | 93:7 | 23 | [25,61] |
| 5 | 12a | E/Z-14a | 25 | 97:3 | 63 (R) | [25,61] | |
| 6 | 0 e | 96:4 | 64 (R) | [61] | |||
| 7 | 25 f | 96:4 | 64 (R) | [61] | |||
| 8 | 0 f | 95:5 | 69 (R) | [25,61] | |||
| 9 | 25 g | 97:3 | 64 (R) | [61] | |||
| 10 | 0 g | 97:3 | 71 (R) | [61] | |||
| 11 | 25 h | 97:3 | 65 (R) | [61] | |||
| 12 | 12b | E/Z-14b | 25 | 96:4 | 19 | [61] | |
| 13 | 0 f | 93:7 | 24 | [61] |

| Entry | Cat. | R’ a | t [h] b | 35 [%] c | Z-36 [%] c | ee [%] d | E-36 [%] c | ee [%] d | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 21a | iPr | 48 | 20 | 41 | 1 | 39 | 4 | [90] |
| 2 | 21b | cC6H11 | 91 | 19 | 41 | 0 | 41 | 5 | [90] |
| 3 | 21c | 2-Ad | 94 | 24 | 40 | 0 | 36 | 5 | [90] |
| 4 | 22a | iPr | 22 | 33 | 34 | 6 | 33 | 4 | [90] |
| 5 | 22b | cC6H11 | 22 | 32 | 33 | 8 | 35 | 5 | [90] |
| 6 | 22c | 2-Ad | 43 | 22 | 42 | 7 | 36 | 16 | [90] |

| Entry | Cat. | M | n | R* | R b | R’ | t [h] | Conv. [%] | ee [%] c | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 21a | Ti | 4 | iPr | iPr | H | 18 | >95 | 4 (+) | [90,91] |
| 2 | 21b | Ti | 4 | iPr | cC6H11 | H | 18 | >95 | 4 (+) | [90,91] |
| 3 | 21c | Ti | 4 | iPr | 2-Ad | H | 17 | >95 | 5 (+) | [90,91] |
| 4 | 22a | Ti | 4 | Bn | iPr | H | 16 | >95 | 2 (+) | [90,91] |
| 5 | 22b | Ti | 4 | Bn | cC6H11 | H | 16 | >95 | 6 (+) | [90,91] |
| 6 | 22c | Ti | 4 | Bn | 2-Ad | H | 20 | >95 | 15 (+) | [90,91] |
| 7 | 23a | Ti | 4 | iPr | iPr | Me | 18 | 100 | 2 (−) | [91] |
| 8 | 23b | Ti | 4 | iPr | iPr | nBu | 18 | 100 | 5 (+) | [91] |
| 9 | 23c d | Ti | 4 | iPr | iPr | Ph | 18 | 100 | n.a. | [91] |
| 10 | 23d | Ti | 4 | iPr | cC6H11 | Me | 18 | 100 | 1 (−) | [91] |
| 11 | 23e | Ti | 4 | iPr | cC6H11 | nBu | 18 | 100 | 1 (+) | [91] |
| 12 | 23f | Ti | 4 | iPr | cC6H11 | Ph | 18 | 100 | 5 (+) | [91] |
| 13 | 23g | Ti | 4 | iPr | 2-Ad | Me | 18 | 100 | 2 (+) | [91] |
| 14 | 23h | Ti | 4 | iPr | 2-Ad | nBu | 18 | 100 | 10 (+) | [91] |
| 15 | 23i | Ti | 4 | iPr | 2-Ad | Ph | 18 | 100 | 0 | [91] |
| 16 | 24a | Ta | 5 | iPr | iPr | H | 162 | 78 | 10 (−) | [92] |
| 17 | 24b | Ta | 5 | iPr | iPr | Me | 23 | 100 | 2 (−) | [92] |
| 18 | 24c | Ta | 5 | iPr | iPr | nBu | 98 | 39 | 24 (−) | [92] |
| 19 | 24d d | Ta | 5 | iPr | iPr | Ph | 15 | 100 | 29 (−) | [92] |
| 20 | 24e | Ta | 5 | iPr | cC6H11 | H | 286 | 64 | 7 (−) | [92] |
| 21 | 24f | Ta | 5 | iPr | cC6H11 | Me | 46 | 90 | 3 (−) | [92] |
| 22 | 24g | Ta | 5 | iPr | cC6H11 | nBu | 43 | 100 | 8 (−) | [92] |
| 23 | 24h | Ta | 5 | iPr | cC6H11 | Ph | 18 | 100 | 74 (−) | [92] |
| 24 | 24i | Ta | 5 | iPr | 2-Ad | H | 336 | 65 | 2 (−) | [92] |
| 25 | 24j | Ta | 5 | iPr | 2-Ad | Me | 334 | 44 | 3 (−) | [92] |
| 26 | 24k | Ta | 5 | iPr | 2-Ad | nBu | 134 | 100 | 6 (−) | [92] |
| 27 | 24l | Ta | 5 | iPr | 2-Ad | Ph | 42 | 91 | 37 (−) | [92] |
| 28 | 25a | Ti | 4 | Bn | iPr | Me | 18 | 100 | 4 (−) | [91] |
| 29 | 25b | Ti | 4 | Bn | iPr | nBu | 18 | 100 | 3 (+) | [91] |
| 30 | 25c | Ti | 4 | Bn | iPr | Ph | 18 | 100 | 16 (+) | [91] |
| 31 | 25d | Ti | 4 | Bn | cC6H11 | Me | 18 | 100 | 1 (−) | [91] |
| 32 | 25e | Ti | 4 | Bn | cC6H11 | nBu | 18 | 100 | 5 (−) | [91] |
| 33 | 25f | Ti | 4 | Bn | cC6H11 | Ph | 18 | 100 | 16 (+) | [91] |
| 34 | 25g | Ti | 4 | Bn | 2-Ad | Me | 18 | 100 | 2 (−) | [91] |
| 35 | 25h | Ti | 4 | Bn | 2-Ad | nBu | 18 | 100 | 1 (+) | [91] |
| 36 | 25i | Ti | 4 | Bn | 2-Ad | Ph | 18 | 100 | 7 (+) | [91] |
| 37 | 26a | Ta | 5 | Bn | iPr | H | 69 | 24 | 13 (−) | [92] |
| 38 | 26b | Ta | 5 | Bn | iPr | Me | 15 | 100 | 46 (−) | [92] |
| 39 | 26c | Ta | 5 | Bn | iPr | nBu | 23 | 100 | 1 (−) | [92] |
| 40 | 26d d | Ta | 5 | Bn | iPr | Ph | 18 | 100 | 65 (−) | [92] |
| 41 | 26e | Ta | 5 | Bn | cC6H11 | H | 50 | 35 | 13 (−) | [92] |
| 42 | 26f | Ta | 5 | Bn | cC6H11 | Me | 65 | 100 | 35 (−) | [92] |
| 43 | 26g | Ta | 5 | Bn | cC6H11 | nBu | 23 | 100 | 32 (−) | [92] |
| 44 | 26h | Ta | 5 | Bn | cC6H11 | Ph | 16 | 100 | 80 (−) | [92] |
| 45 | 26i | Ta | 5 | Bn | 2-Ad | H | 116 | 33 | 6 (−) | [92] |
| 46 | 26j | Ta | 5 | Bn | 2-Ad | Me | 24 | 100 | 2 (−) | [92] |
| 47 | 26k | Ta | 5 | Bn | 2-Ad | nBu | 115 | 100 | 24 (−) | [92] |
| 48 | 26l | Ta | 5 | Bn | 2-Ad | Ph | 230 | 28 | 25 (−) | [92] |

| Entry | Cat. | M | n | RAr | R | T a [°C] | t [h] | Conv. [%] | ee b [%] | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 27a | Ti | 4 | 4-CH3 | H | 125 c | 51 | 38 | 11 (+) | [93] |
| 2 | 135 | 40 | 65 | 3 (+) | [93] | |||||
| 3 | 27b | Ti | 4 | 4-CF3 | H | 125 c | 51 | 61 | 9 (+) | [93] |
| 4 | 135 c | 18 | 73 | 4 (+) | [93] | |||||
| 5 | 27c | Ti | 4 | 3,5-di-CF3 | H | 125 | 49 | 29 | 0 | [93] |
| 6 | 135 | 15 | 85 | 7 (+) | [93] | |||||
| 7 | 27d | Ti | 4 | 4-CH3 | Me | 125 | 40 | 33 | 4 (+) | [93] |
| 8 | 135 | 18 | 95 | 6 (+) | [93] | |||||
| 9 | 27e | Ti | 4 | 4-CF3 | Me | 125 c | 49 | 55 | 8 (+) | [93] |
| 10 | 135 c | 40 | 61 | 7 (+) | [93] | |||||
| 11 | 27f | Ti | 4 | 3,5-di-CF3 | Me | 125 | 27 | 45 | 5 (+) | [93] |
| 12 | 135 | 37 | 80 | 2 (+) | [93] | |||||
| 13 | 27g | Ti | 4 | 4-CH3 | Ph | 135 | 18 | 82 | 18 (−) | [95] |
| 14 | 27h | Ti | 4 | 4-CF3 | Ph | 135 | 32 | 95 | 24 (−) | [95] |
| 15 | 27i | Ti | 4 | 3,5-di-CF3 | Ph | 135 | 106 | 18 | 41 (−) | [95] |
| 16 | Ph | 135 c | 20 | 100 | 21 (−) | [95] | ||||
| 17 | 27j | Ti | 4 | 2,4,6-tri-CH3 | Ph | 135 | 75 | 71 | 27 (−) | [95] |
| 18 | 28a | Ta | 5 | 4-CH3 | H | 125 | 132 | 20 | 24 (−) | [93] |
| 19 | 135 | 124 | 60 | 21 (−) | [93] | |||||
| 20 | 28b | Ta | 5 | 4-CF3 | H | 125 | 115 | 85 | 28 (−) | [93] |
| 21 | 135 | 69 | 32 | 5 (−) | [93] | |||||
| 22 | 28c | Ta | 5 | 3,5-(CF3)2 | H | 125 | 71 | 100 | 34 (−) | [93] |
| 23 | 135 | 69 | 34 | 17 (−) | [93] | |||||
| 24 | 28d | Ta | 5 | 4-CH3 | Me | 125 | 17 | 88 | 24 (−) | [93] |
| 25 | 135 | 18 | 100 | 15 (−) | [93] | |||||
| 26 | 28e | Ta | 5 | 4-CF3 | Me | 125 | 15 | 100 | 20 (−) | [93] |
| 27 | 135 | 18 | 100 | 26 (−) | [93] | |||||
| 28 | 28f | Ta | 5 | 3,5-(CF3)2 | Me | 125 | 15 | 100 | 23 (−) | [93] |
| 29 | 135 | 19 | 100 | 7 (−) | [93] | |||||
| 30 | 28g | Ta | 5 | 4-CH3 | Ph | 135 | 57 | 100 | 37 (−) | [95] |
| 31 | 28h | Ta | 5 | 4-CF3 | Ph | 135 | 49 | 100 | 33 (−) | [95] |
| 32 | 28i | Ta | 5 | 3,5-di-CF3 | Ph | 135 | 23 | 100 | 35 (−) | [95] |
| 33 | 28j | Ta | 5 | 2,4,6-tri-CH3 | Ph | 135 | 47 | 100 | 39 (−) | [95] |

| Entry | Cat. | M | n | R* | RAr | R | t [h] | Conv. [%] | 35 [%] | E-36 [%] | ee [%] b | Z-36 [%] | ee [%] b | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 27g | Ti | 4 | Bn | 4-CH3 | Ph | 64 | 84 | 60 | 6 | 17 | 18 | 5 | [95] |
| 2 | 27h | Ti | 4 | Bn | 4-CF3 | Ph | 40 | 95 | 73 | 4 | 46 | 18 | 20 | [95] |
| 3 | 27i | Ti | 4 | Bn | 3,5-di-CF3 | Ph | 23 | 98 | 82 | 5 | 55 | 10 | 45 | [95] |
| 4 | 27j | Ti | 4 | Bn | 2,4,6-tri-CH3 | Ph | 30 | 83 | 59 | 4 | 19 | 20 | 8 | [95] |
| 5 | 28g | Ta | 5 | Bn | 4-CH3 | Ph | 30 | 96 | 49 | 10 | 40 | 37 | 17 | [95] |
| 6 | 28h | Ta | 5 | Bn | 4-CF3 | Ph | 30 | 97 | 50 | 10 | 25 | 36 | 16 | [95] |
| 7 | 28i | Ta | 5 | Bn | 3,5-di-CF3 | Ph | 23 | 98 | 48 | 18 | 8 | 32 | 21 | [95] |
| 8 | 28j | Ta | 5 | Bn | 2,4,6-tri-CH3 | Ph | 23 | 95 | 49 | 10 | 40 | 36 | 15 | [95] |
| 9 | 29a | Ti | - | Bn | H | Me | 43 | 76 | 46 | 17 | 9 | 22 | 8 | [94] |
| 10 | 29b | Ti | - | Bn | H | Ph | 31 | 87 | 40 | 14 | 1 | 36 | 2 | [94] |
| 11 | 29c | Ti | - | Bn | 3,5-di-tBu | Ph | 54 | 96 | 72 | 8 | 3 | 17 | 7 | [94] |
| 12 | 29d | Ti | - | Bn | 5-F | Ph | 54 | 91 | 44 | 13 | 2 | 38 | 1 | [94] |
| 13 | 29e | Ti | - | iPr | 5-F | Ph | 54 | 88 | 42 | 13 | 2 | 39 | 2 | [94] |

| Entry | Cat. | R* | RAr | R | [cat.] [%] | t [h] | Conv. [%] | ee [%] b | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 29a | Bn | H | Me | 5 | 67 | 7 | 11 (−) | [94] |
| 2 | 29b | Bn | H | Ph | 5 | 43 | 86 | 6 (−) | [94] |
| 3 | 29c | Bn | 3,5-di-tBu | Ph | 5 | 34 | 87 | 17 (−) | [94] |
| 4 | 29d | Bn | 5-F | Ph | 5 | 81 | 73 | 1 (−) | [94] |
| 5 | 29e | iPr | 5-F | Ph | 5 | 65 | 74 | 3 (−) | [94] |
| 6 | 29f | Bn | 3,5-di-C6H5 | Ph | 20 | 18 | 100 | 8 (−) | [97] |
| 7 | 10 | 66 | 25 | 15 (−) | [97] | ||||
| 8 | 20 | 18 c | 100 | 21 (−) | [97] | ||||
| 9 | 20 | 66 d | 100 | 19 (−) | [97] | ||||
| 10 | 20 | 18 e | 100 | 22 (−) | [97] | ||||
| 11 | 29g | Bn | 3,5-di-(4-C6H4(CF3)) | Ph | 20 | 13 | 100 | 8 (−) | [97] |
| 12 | 10 | 13 | 100 | 6 (−) | [97] | ||||
| 13 | 20 | 21 c | 100 | 6 (−) | [97] | ||||
| 14 | 20 | 19 d | 100 | 7 (−) | [97] | ||||
| 15 | 20 | 19 e | 100 | 8 (−) | [97] | ||||
| 16 | 29h | Bn | 3,5-di-(3,5-C6H3(CF3)2) | Ph | 20 | 18 | 100 | 6 (−) | [97] |
| 17 | 10 | 18 | 40 | 12 (−) | [97] | ||||
| 18 | 20 | 18 c | 100 | 5 (−) | [97] | ||||
| 19 | 10 | 18 c | 25 | 18 (−) | [97] | ||||
| 20 | 20 | 18 d | 100 | 7 (−) | [97] | ||||
| 21 | 10 | 66 d | 63 | 21 (−) | [97] | ||||
| 22 | 20 | 18 e | 100 | 6 (−) | [97] |
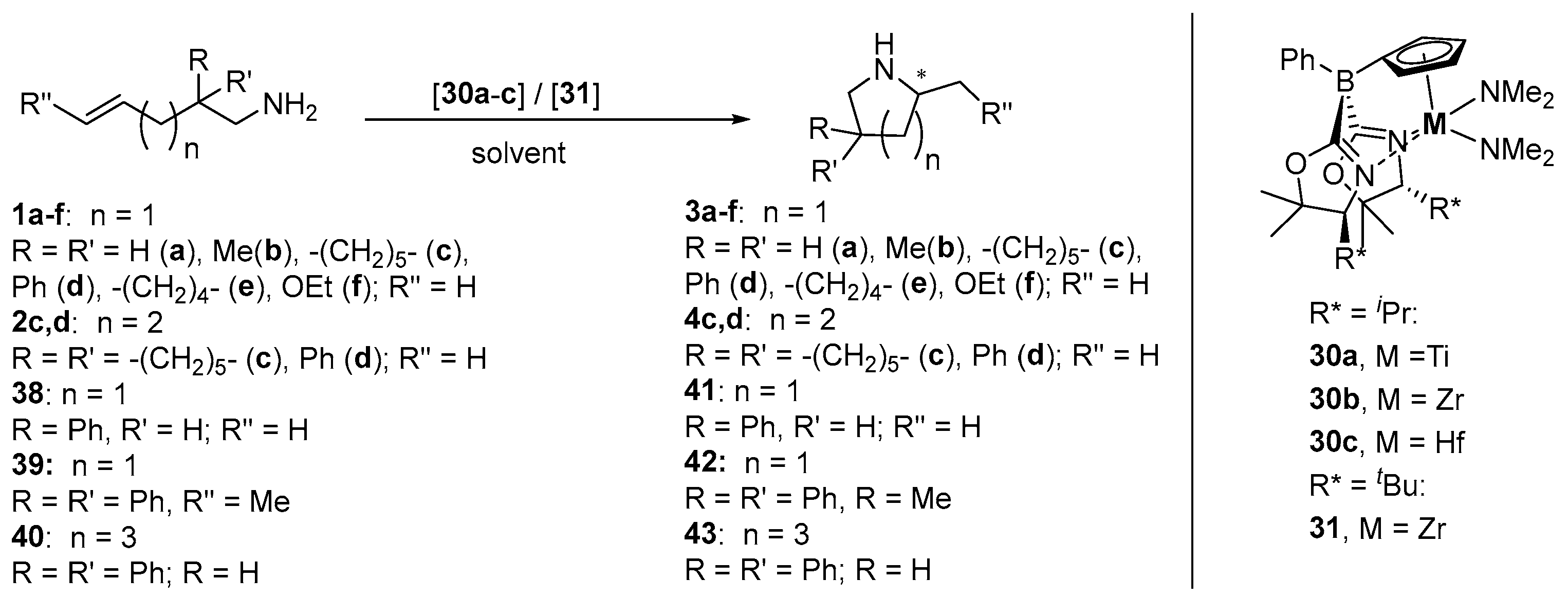
| Entry | Cat. | M | R* | Substr. | Prod. | Solvent b | T [°C] | t [h] | Conv. [%] | ee [%] c | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 30a | Ti | iPr | 1c | 3c | benzene-d6 | 25 | 120 | 75 | 83 (R) | [58] |
| 2 | 1d | 3d | benzene-d6 | 25 | 120 | 93 | 76 (R) | [58] | |||
| 3 | 30b | Zr | iPr | 1a | 3a | benzene-d6 | 110 | 15 | 24 | n.d. | [57,58] |
| 4 | 1b | 3b | benzene-d6 | 25 | 7 | 89 | 89 (R) | [57] | |||
| 5 | benzene | 25 | 7 | 89 | 89 (R) | [58] | |||||
| 6 | toluene-d8 | −30 | 192 | 95 | 93 (R) | [58] | |||||
| 7 | 1c | 3c | benzene-d6 | 25 | 1.25 | >95 | 90 (R) | [57] | |||
| 8 | benzene | 25 | 1.25 | 96 | 90 (R) | [58] | |||||
| 9 | toluene-d8 | 25 d | 6.5 | >95 | 90 (R) | [57] | |||||
| 10 | thf-d8 | 0 | 11 | 93 | 94 (R) | [57,58] | |||||
| 11 | 1d | 3d | benzene-d6 | 25 | 1.25 | >95 | 93 (R) | [57] | |||
| 12 | benzene | 25 | 1.25 | 95 | 93 (R) | [58] | |||||
| 13 | benzene-d6 | 25 d | 6 | 96 | 93 (R) | [57] | |||||
| 14 | toluene | −30 | 120 | 98 | 98 (R) e | [58] | |||||
| 15 | dcm-d2 | 25 | 5 | >95 | 94 (R) | [57,58] | |||||
| 16 | thf-d8 | 25 | 5 | >95 | 95 (R) | [57,58] | |||||
| 17 | thf-d8 | 0 | 12 | >95 | 96 (R) e | [57] | |||||
| 18 | thf-d8 | −30 | 120 | >95 | 98 (R) e | [57] | |||||
| 19 | 1e | 3e | benzene-d6 | 25 | 4 | 88 | 92 (R) | [57] | |||
| 20 | benzene | 25 | 4 | 88 | 92 (R) | [58] | |||||
| 21 | 1f | 3f | benzene-d6 | 25 | 30 | 90 | 97 (R) | [58] | |||
| 22 | 2c | 4c | benzene-d6 | 25 | 40 | 48 | 31 (R) e | [57,58] | |||
| 23 | 2d | 4d | benzene-d6 | 25 | 96 | 65 | 46 (R) | [57,58] | |||
| 24 | 38 | 41 | benzene-d6 | 25 | 15 | 89 | 66/57 f | [58] | |||
| 25 | 39 | 42 | benzene-d6 | 25 | 96 | 85 | 89 (R) | [58] | |||
| 26 | 40 | 43 | benzene | 25 | 120 | 73 | 91 (R) | [58] | |||
| 27 | 30c | Hf | iPr | 1b | 3b | benzene-d6 | 25 | 20 | 90 | 87 (R) | [58] |
| 28 | 1c | 3c | benzene | 25 | 5 | 95 | 93 (R) | [58] | |||
| 29 | 1d | 3d | toluene | 0 | 15 | 98 | 97 (R) e | [58] | |||
| 30 | 1e | 3e | toluene-d8 | 0 | 8 | 85 | 95 (R) e | [58] | |||
| 31 | 1f | 3f | benzene | 25 | 120 | 90 | 96 (R) | [58] | |||
| 32 | 2c | 4c | benzene | 85 | 30 | 85 | 26 (R) e | [58] | |||
| 33 | 2d | 4d | benzene | 85 | 20 | 89 | 18 (R) | [58] | |||
| 34 | 38 | 41 | benzene | 25 | 24 | 90 | 65/58 g | [58] | |||
| 35 | 31 | Zr | tBu | 1c | 3c | benzene | 25 | 30 | 92 | 87 (R) | [56] |
| 36 | 1d | 3d | benzene | 25 | 18 | 95 | 93 (R) | [56] | |||
| 37 | 1e | 3e | benzene | 25 | 48 | 77 | 88 (R) | [56] | |||
| 38 | 2c | 4c | benzene | 25 | 48 | 80 | 29 (R) e | [58] |
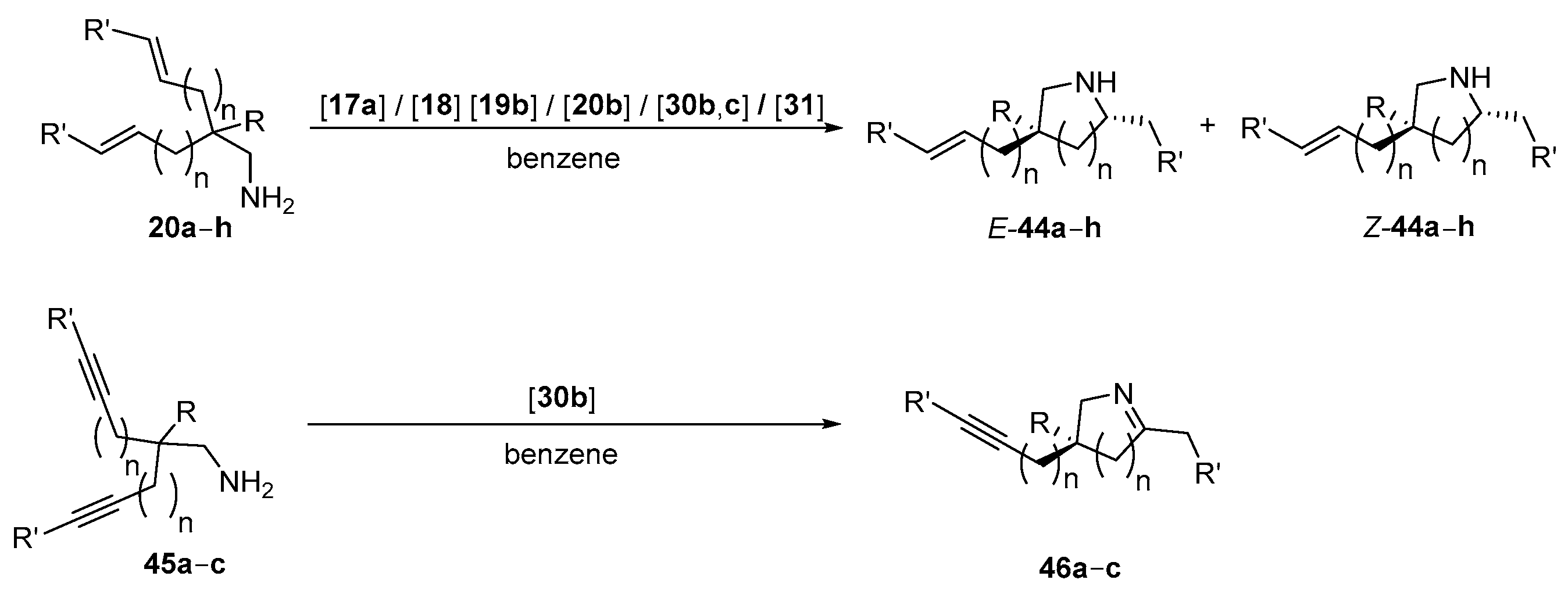
| Entry | Cat. | R | R’ | n | Substr. | Prod. | t [h] | Conv. [%] | dr b | ee [%] c | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 17a | Me | H | 1 | 20a | 44a | 2.6 d | 100 | 1.36:1 e | 5/21 (R) | [79] |
| 2 | 18 | Me | H | 1 | 20a | 44a | 0.5 f | 96 | 1.55:1 e | 38/25 (S) | [79] |
| 3 | 19b | Me | H | 1 | 20a | 44a | 0.25 | 100 | 1.2:1 e | 95/95 | [56] |
| 4 | Ph | H | 1 | 20b | 44b | 0.25 | 100 | 1.2:1 e | 95/96 | [56] | |
| 5 | 4-C6H4Br | H | 1 | 20c | 44c | 0.25 | 100 | 1.9:1 e | 95/92 | [56] | |
| 6 | ethenyl | H | 1 | 20d | 44d | 0.17 | 100 | - | 96 (S) | [56] | |
| 7 | 30b | Me | H | 1 | 20a | 44a | 0.5 | 100 | 1.1:1 | 93/92 | [96] |
| 8 | 48 g | 100 | 4.2:1 | 93/92 | [96] | ||||||
| 9 | 144 h,i | 100 | 1:6.5 | 96/97 | [96] | ||||||
| 10 | Ph | H | 1 | 20b | 44b | 0.5 | 100 | 3.3:1 | 96/96 | [96] | |
| 11 | 6 g | 100 | 8.9:1 | 96/95 | [96] | ||||||
| 12 | 55 j | 100 | 2:1 | 96/96 | [96] | ||||||
| 13 | 96 g,k | 95 | 1:1.1 | 99/99 | [96] | ||||||
| 14 | 96 h | 95 | 1:4.5 | 99/99 | [96] | ||||||
| 15 | 144 h,i,k | 100 | 1:6 | 99/99 | [96] | ||||||
| 16 | 4-C6H4Br | H | 1 | 20c | 44c | 0.5 | 100 | 4:1 | 97/95 | [96] | |
| 17 | 3 g | 100 | 8:1 | 95 (cis) | [96] | ||||||
| 18 | ethenyl | H | 1 | 20d | 44d | 0.75 | 98 | - | 92 (R) | [58] | |
| 19 | OMe | H | 1 | 20e | 44e | 48 | 90 | >20:1 | 97 (cis) | [96] | |
| 20 | 48 h | 90 | 10:1 | 97 (cis) | [96] | ||||||
| 21 | Ph | Me | 1 | 20f | 44f | 96 | 87 | 2:1 | 93/95 | [96] | |
| 22 | 192 l | 91 | 8:1 | 92 (cis) | [96] | ||||||
| 23 | Ph | H | 2 | 20g | 44g | 72 | 90 | 2.4:1 | 33/12 | [96] | |
| 24 | 96 | 90 | 6.6:1 | 32/12 | [96] | ||||||
| 25 | Ph | H | 3 | 20h | 44h | 96 | 90 | 2.8:1 | 89/92 | [96] | |
| 26 | 144 m | 86 | 7:1 | 89/91 | [96] | ||||||
| 27 | Ph | Me | 1 | 45a | 46a | 0.6 | 100 | - | 87 | [96] | |
| 28 | 2 g | 100 | - | 91 | [96] | ||||||
| 29 | Ph | Me | 2 | 45b | 46b | 20 | 100 | - | 71 | [96] | |
| 30 | 48 g | 100 | - | 77 | [96] | ||||||
| 31 | Ph | Me | 3 | 45c | 46c | 72 g | 100 | - | 89 | [96] | |
| 32 | 30c | Me | H | 1 | 20a | 44a | 20 | 81 | 1.4:1 e | 87/63 | [58] |
| 33 | 4-C6H4Br | H | 1 | 20c | 44c | 3 | 84 | 2:1 e | 93/96 | [58] | |
| 34 | ethenyl | H | 1 | 20d | 44d | 20 n | 90 | - | 96 (R) | [58] | |
| 35 | 31 | Me | H | 1 | 20a | 44a | 30 | 85 | 1.2:1 e | 92/91 | [56,58] |
| 36 | Ph | H | 1 | 20b | 44b | 48 | 93 | 2.5:1 e | 88/92 | [56,58] | |
| 37 | 4-C6H4Br | H | 1 | 20c | 44c | 48 | 90 | 1.2:1 e | 96/98 | [56,58] | |
| 38 | ethenyl | H | 1 | 20d | 44d | 30 | 90 | - | 88 (R) | [56,58] |
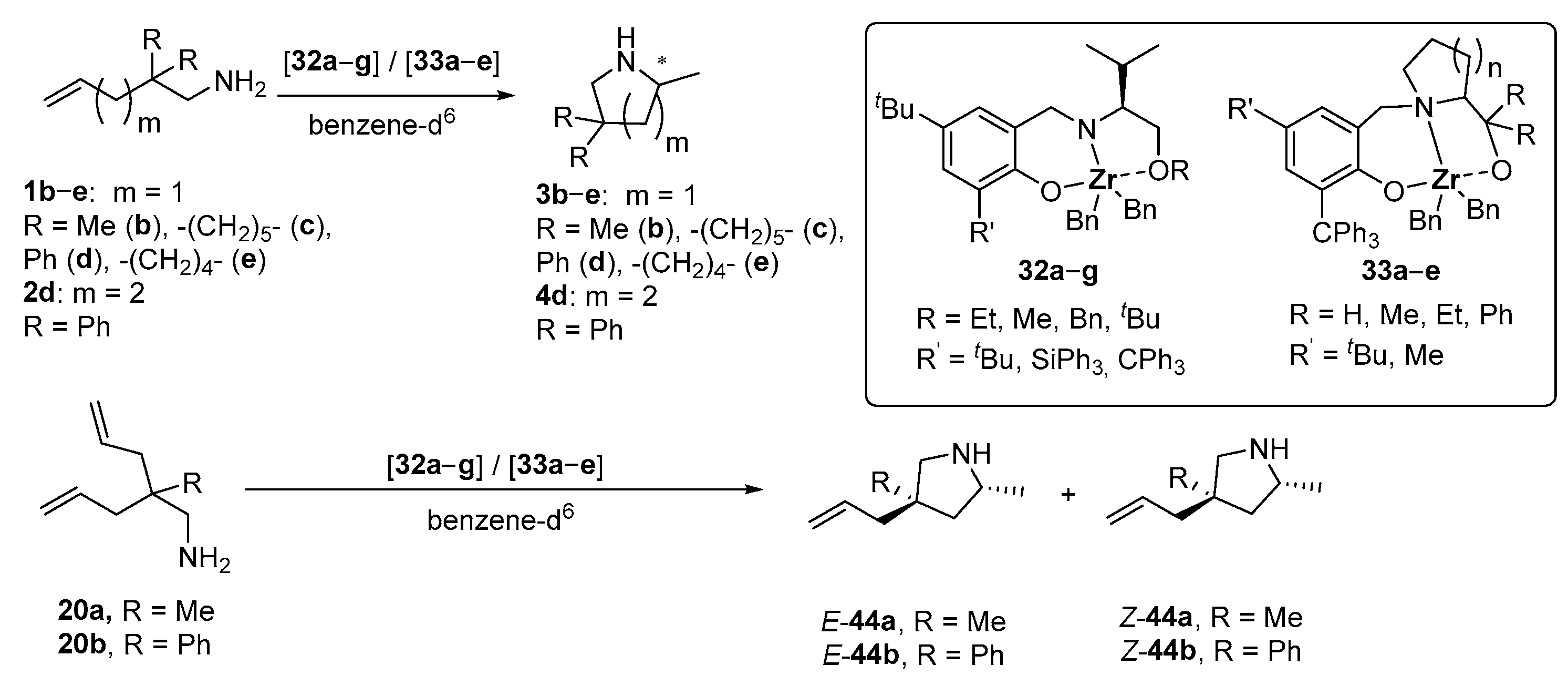
| Entry | Cat. | R | R’ | n | Substr. | Prod. | t [h] | T [°C] | Conversion [%] | ee [%] b | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 32a | Et | tBu | - | 1d | 3d | 4 | 100 | >95 | 27 | [98] |
| 2 | 32b | Et | 2-Ad | - | 1d | 3d | 8.5 | 100 | >95 | 28 | [98] |
| 3 | 32c | Et | Ph3Si | - | 1d | 3d | 4 | 100 | >95 | 38 | [98] |
| 4 | 32d | Et | Ph3C | - | 1d | 3d | 2 | 100 | >95 | 56 | [98] |
| 5 | 32e | Me | Ph3C | - | 1d | 3d | 3 | 100 | >95 | 49 | [98] |
| 6 | 32f | Bn | Ph3C | - | 1d | 3d | 1.5 | 100 | >95 | 40 | [98] |
| 7 | 32g | tBu | Ph3C | - | 1d | 3d | 2 | 100 | >95 | 1 | [98] |
| 8 | 33a | H | tBu | 1 | 1d | 3d | 2.5 | 100 | 93 | 74 | [98] |
| 9 | 33b | Me | tBu | 1 | 1b | 3b | 120 | 85 | 96 | 89 | [98] |
| 10 | 1c | 3c | 21 | 85 | 97 | 93 | [98] | ||||
| 11 | 1d | 3d | 4.5 | 115 | 95 | 84 | [98] | ||||
| 12 | 4.5 | 100 | 94 | 89 | [98] | ||||||
| 13 | 11 | 85 | 92 | 92 | [98] | ||||||
| 14 | 14 | 80 | 90 | 93 | [98] | ||||||
| 15 | 19 | 70 | 95 | 94 | [98] | ||||||
| 16 | 29 | 55 | 93 | 94 | [98] | ||||||
| 17 | 1e | 3e | 72 | 85 | 91 | 87 | [98] | ||||
| 18 | 2d | 4d | 2 | 85 | 91 | 66 | [94] | ||||
| 19 | 20a | E/Z-44a | 89 | 85 | 89 c | 90/93 | [98] | ||||
| 20 | 20b | E/Z-44b | 24 | 85 | 91 d | 88/92 | [98] | ||||
| 21 | 33c | Et | tBu | 1 | 1d | 3d | 11 | 100 | 96 | 68 | [98] |
| 22 | 33d | Ph | tBu | 1 | 1d | 3d | 59.5 | 100 | 85 | −13 | [98] |
| 23 | 33e | H | Me | 2 | 1d | 3d | 4 | 100 | 94 | 77 | [98] |
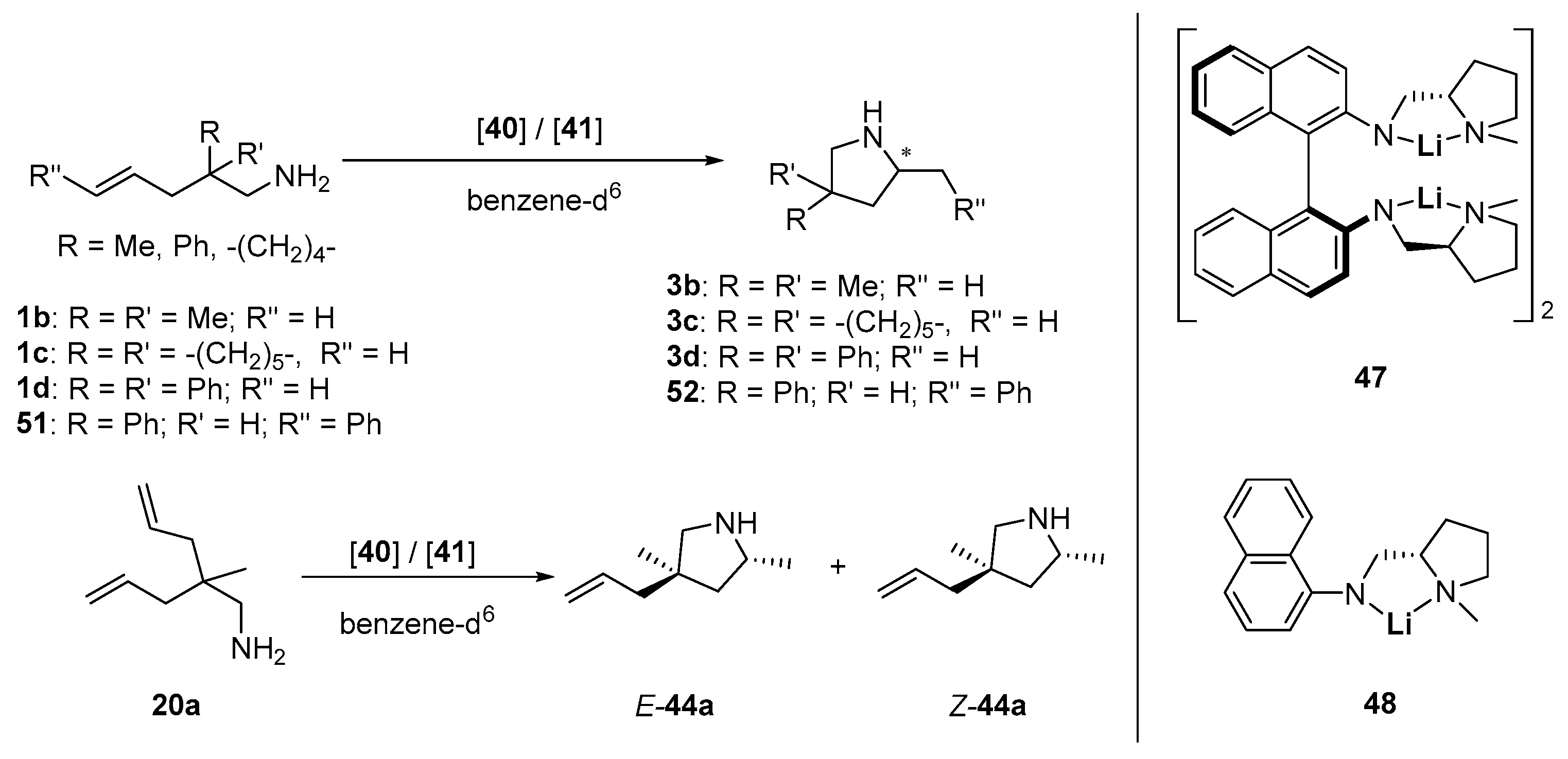
| Entry | Cat. | Substr. | Prod. | [cat.] [mol-%] b | t a [h] | T [°C] | Conv. [%] | ee [%] c | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 47 | 1b | 3b | 7.5 | 9 | 22 | 96 | 64 (S) | [110] |
| 2 | 5 | 42 | 22 | 96 | 68 (S) | [110] | |||
| 3 | 2.5 | 45 | 22 | 93 | 67 (S) | [110] | |||
| 4 | 5 d | 407 | 80 | 66 | 53 (S) | [110] | |||
| 5 | 1c | 3c | 2.5 | 1.1 | 22 | 91 | 75 (S) | [110] | |
| 6 | 5 e | 2 | 20 | 98 | 74 (S) | [110] | |||
| 7 | 5 d | 91 | 60 | 64 | 69 (S) | [110] | |||
| 8 | 1d | 3d | 5 | 0.8 | 22 | 97 | 31 (S) | [110] | |
| 9 | 5 d | 27 | 80 | 70 | 24 (S) | [110] | |||
| 10 | 51 | 52 | 5 | 0.08 | 22 | 98 | 17 | [110] | |
| 11 | 20a | 44a | 5 | 2 | 22 | 98 | 64/72 f | [110] | |
| 12 | 48 | 1b | 3b | 10 | 333 | 120 | 56 | 2 | [110] |
| 13 | 1d | 3d | 10 | 140 | 120 | 0 | - | [110] |

| Entry | Cat. | R* | R | R’ | R’’ | n | Substr. | Prod. | t [h] | Yield [%] b | ee [%] | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 50a | iPr | Me | Ph | Me | 2 | 53a | 54a | 5 | 99(0) | 71 (S) | [113] |
| 2 | Ph | Me | 1 | 53b | 54b | 5 | 99(0) | 84 (S) | [113] | |||
| 3 | Ph | 4-C6H4OMe | 2 | 53c | 54c | 31 c | 94(0) | 11 d | [114] | |||
| 4 | Ph | 2-propenyl | 1 | 53d | 54d | 2 e | 98(0) | 83 (S) | [114] | |||
| 5 | Ph | 2-propenyl | 1 | 53e f | 54e f | 21 e | 90(0) | 18 d | [114] | |||
| 6 | H | Me | 1 | 53f | 54f | 1 g | 33(7) | 43 d | [114] | |||
| 7 | 50b | tBu | Me | Ph | Me | 2 | 53a | 54a | 5 h | 99(0) | 31 (S) | [113] |
| 8 | Ph | Me | 1 | 53b | 54b | 5 | 89(0) | 19 (S) | [113] | |||
| 9 | 50c | iPr | Et | Ph | Me | 2 | 53a | 54a | 27 | 99(0) | 62 (S) | [113] |
| 10 | Ph | Me | 1 | 53b | 54b | 5 | 99(0) | 84 (S) | [113] | |||
| 11 | 50d | iPrCH2 | Me | Ph | Me | 2 | 53a | 54a | 5 | 97(0) | 84 (S) | [113] |
| 12 | Ph | Me | 1 | 53b | 54b | 5 | 99(0) | 79 (S) | [113] | |||
| 13 | 50e | secBu | Me | Ph | Me | 2 | 53a | 54a | 5 | 25(0) | 81 (S) | [113] |
| 14 | Ph | Me | 1 | 53b | 54b | 5 | 99(0) | 66 (S) | [113] | |||
| 15 | 50f | - | Me | Ph | Me | 2 | 53a | 54a | 22 | 54(0) | 62 (R) | [113] |
| 16 | Ph | Me | 1 | 53b | 54b | 5 | 98(0) | 86 (R) | [113] |

| Entry | Cat. | Additive | Vinylarene | Amine | Prod. | t a [h] | Yield [%] b | ee [%] | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 49a | (−)-sparteine | 58a | 59a | 60a | 37 | 57 | - | [109] |
| 2 | 58a | 59b | 60b | 65 | 40 | 7 | [109] | ||
| 3 | 58b | 59a | 60c | 13.5 | 71 | - | [109] | ||
| 4 | 58b | 59b | 60d | 18 | 60 | 14 | [109] | ||
| 5 | 49b | α-isosparteine | 58a | 58b | 60b | 70 | 38 | - | [109] |
| Entry | Cat. | Substr. | Prod. | [cat] [mol-%] | T [°C] | t a [h] | Conv. [%] | ee [%] b | Ref. |
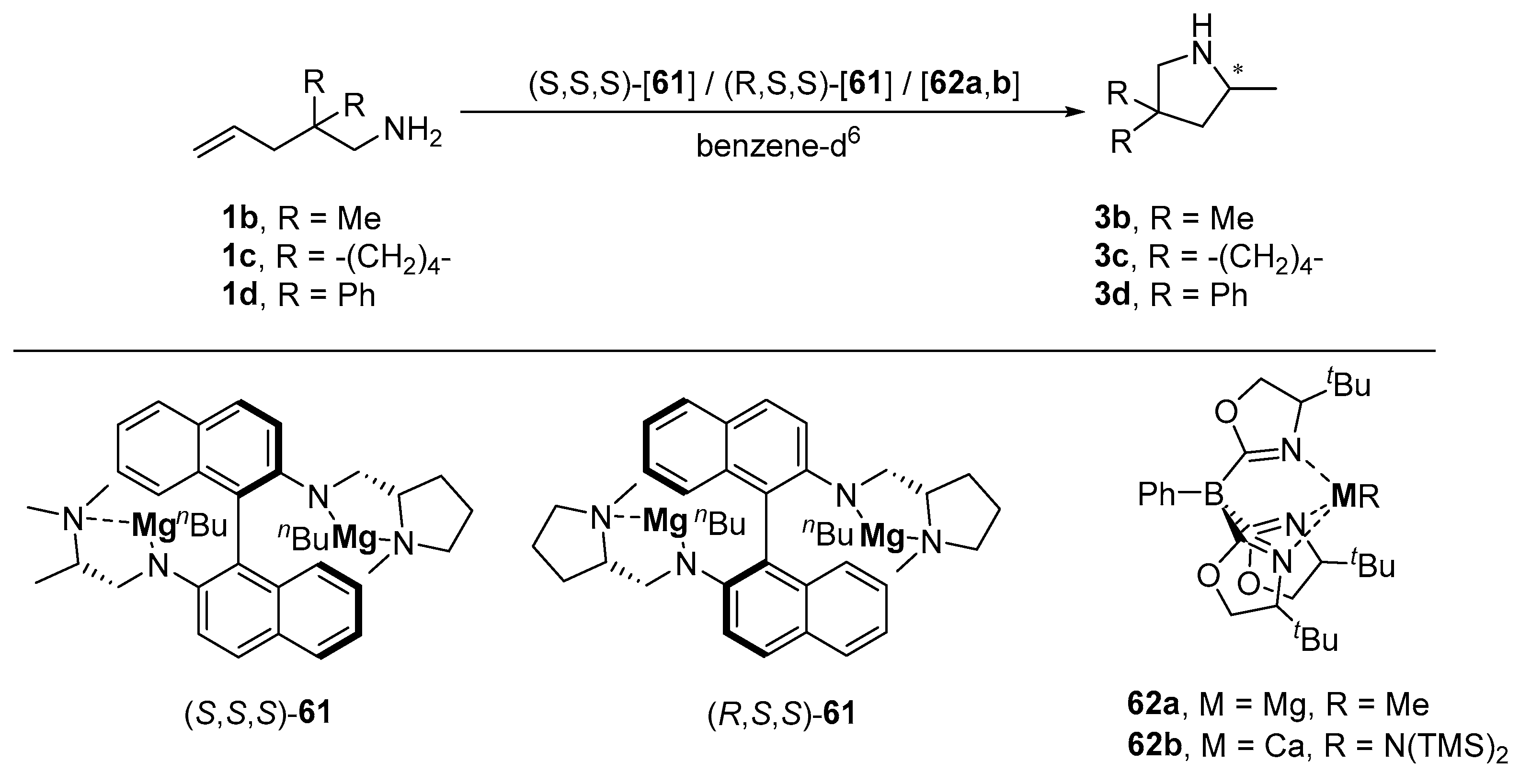
|---|---|---|---|---|---|---|---|---|---|
| 1 | (S,S,S)-61 | 1b | 3b | 5 | 100 | 22 | ≥99 | 4 (S) | [116] |
| 2 | 1c | 3c | 5 | 22 | 22 | 94 | 0 | [116] | |
| 3 | 1d | 3d | 4 | 22 | 0.33 | ≥99 | 6 (R) | [116] | |
| 4 | (R,S,S)-61 | 1b | 3b | 10 | 100 | 21 | ≥99 | 0 | [116] |
| 5 | 1c | 3c | 10 | 22 | 3.5 | 80 | 6 (R | [116] | |
| 6 | 1d | 3d | 10 | 22 | 0.17 | ≥99 | 14 (R) | [116] | |
| 7 | 62a | 1b | 3b | 10 | r.t. | 168 | 0 | - | [120] |
| 8 | 10 | 80 | 120 | 80 | 27 (R) | [120] | |||
| 9 | 1c | 3c | 10 | r.t. | 24 | 0 | - | [120] | |
| 10 | 10 | 60 | 26 | 93 | 36 (R) | [120] | |||
| 11 | 1d | 3d | 10 | r.t. | 24 | 89 | 0 | [120] | |
| 12 | 10 | 60 | 12 | ≥99 | 0 | [120] | |||
| 13 | 62b | 1b | 3b | 10 | r.t. | 0.08 | 100 | 18 (S) | [120] |
| 14 | 1c | 3c | 10 | r.t. | 0.08 | ≥99 | 18 (S) | [120] | |
| 15 | 1 | 80 | 168 | ≤10 | n.d. | [120] | |||
| 16 | 1d | 3d | 10 | r.t. | 0.08 | ≥99 | 0 | [120] |
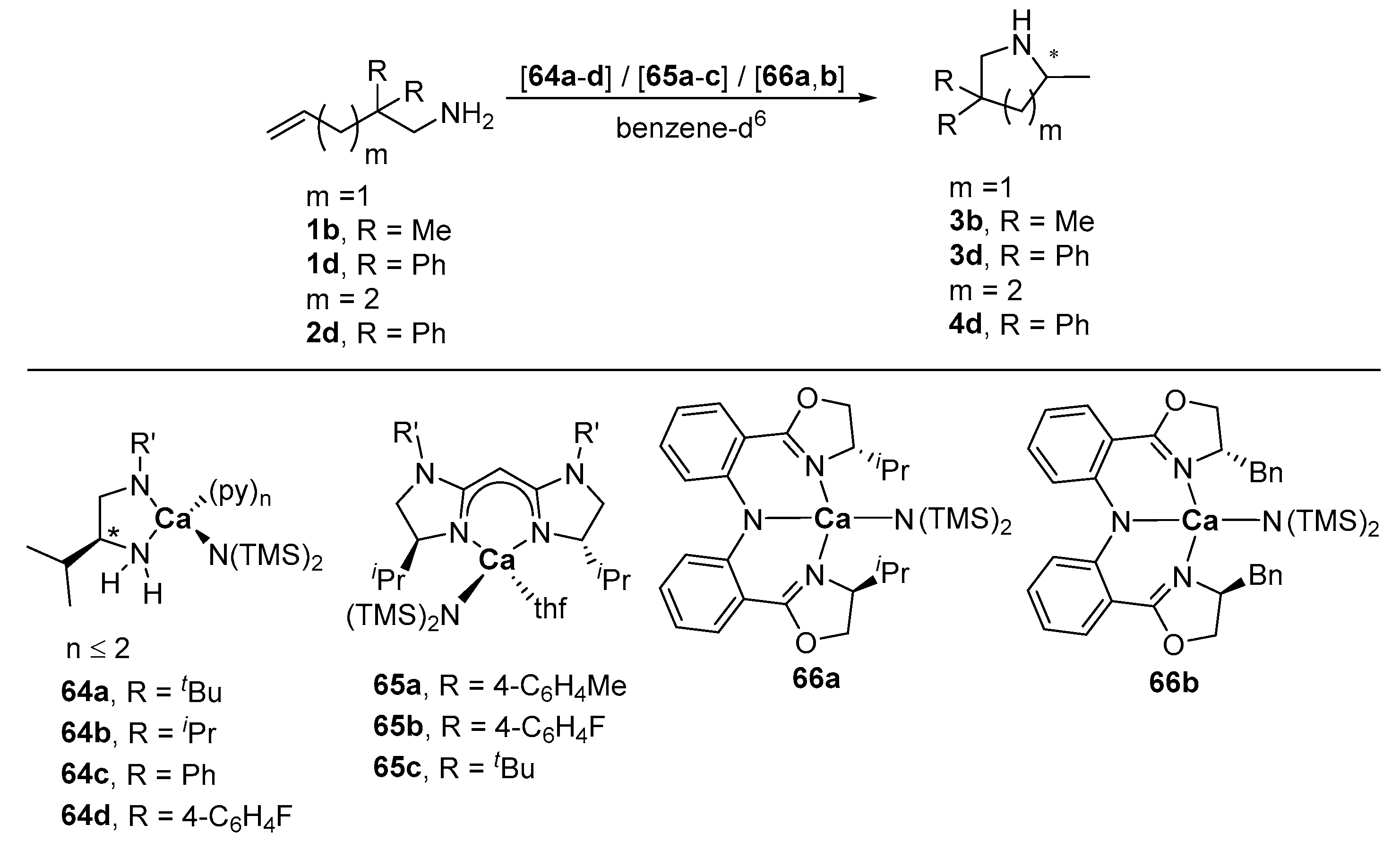
| Entry | Cat. | R’ | Substr. | Prod. | T [°C] | t [h] | Conv. [%] b | ee [%] c | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 64a | tBu | 1b | 3b | r.t. | 168 | 90 | 0 | [26] |
| 2 | 1d | 3d | r.t. | 24 | ≥99 | 6 | [26] | ||
| 3 | 64b | iPr | 1b | 3b | r.t. | 120 | ≥99 | 12 | [26] |
| 4 | 1d | 3d | r.t. | 1 | ≥99 | 5 | [26] | ||
| 5 | 64c | Ph | 1b | 3b | r.t. | 504 | 0 | - | [26] |
| 6 | 1d | 3d | r.t. | 72 | 80 | 26 | [26] | ||
| 7 | 64d | 4-C6H4F | 1b | 3b | r.t. | 336 | 0 | - | [26] |
| 8 | 1d | 3d | r.t. | 336 | 0 | - | [26] | ||
| 9 | 65a | 4-C6H4Me | 1b | 3b | r.t. | n/a d | 8 | 5 | [122] |
| 10 | 1d | 3d | r.t. | n/a d | 95 | 0 | [122] | ||
| 11 | 65b | 4-C6H4F | 1b | 3b | r.t. | n/a d | 3 | 9 | [122] |
| 12 | 1d | 3d | r.t. | n/a d | >99 | 9 | [122] | ||
| 13 | 65c | tBu | 1b | 3b | r.t. | n/a d | 18 | 12 | [122] |
| 14 | 1d | 3d | r.t. | n/a d | ≥99 | 12 | [122] | ||
| 15 | 66a | - | 1d | 3d | 30 | 24 | 51 | 14 | [123] |
| 16 | 40 | 24 | ≥99 | 22 | [123] | ||||
| 17 | 50 | 72 | 82 | 24 | [123] | ||||
| 18 | 2d | 4d | 80 | 120 | 14 | 0 | [123] | ||
| 19 | 50 e | 24 | 26 | 8 | [123] | ||||
| 20 | 80 e | 120 | 83 | 6 | [123] | ||||
| 21 | 66b | - | 1d | 3d | 21 | 24 | ≥99 | 25 | [123] |
| 22 | 30 | 24 | ≥99 | 26 | [123] | ||||
| 23 | 40 | 24 | 88 | 20 | [123] | ||||
| 24 | 2d | 4d | 80 | 120 | trace | - | [123] | ||
| 25 | 50 | 24 | 0 | - | [123] | ||||
| 26 | 80 | 120 | 14 | 16 | [123] |

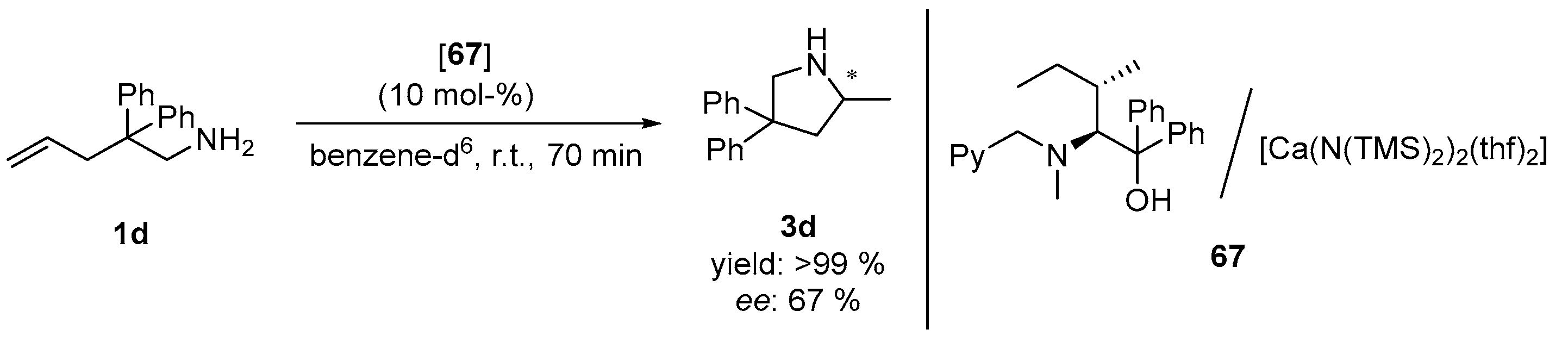
| Entry | Cat. | Substr. | Prod. | T [°C] | t [h] | Conv. [%] b | ee [%] c | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | 68 | 1b | 3b | 90 | 18 | >98 | 8 | [27] |
| 2 | 1c | 3c | 60 | 5 | >98 | 8 | [27] | |
| 3 | 1d | 3d | 20 | 5 | >99 | 15 | [27] | |
| 4 | 60 | 1 | >99 | 15 | [27] | |||
| 5 | (S)-69 | 1b | 3b | 90 | 24 | >97 | 26 | [27] |
| 6 | 1c | 3c | 60 | 5 | >98 | 23 | [27] | |
| 7 | 1d | 3d | 20 | 5 | >99 | 33 | [27] | |
| 8 | 60 | 1 | >99 | 33 | [27] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Notz, S.; Scharf, S.; Lang, H. Jumping in the Chiral Pool: Asymmetric Hydroaminations with Early Metals. Molecules 2023, 28, 2702. https://doi.org/10.3390/molecules28062702
Notz S, Scharf S, Lang H. Jumping in the Chiral Pool: Asymmetric Hydroaminations with Early Metals. Molecules. 2023; 28(6):2702. https://doi.org/10.3390/molecules28062702
Chicago/Turabian StyleNotz, Sebastian, Sebastian Scharf, and Heinrich Lang. 2023. "Jumping in the Chiral Pool: Asymmetric Hydroaminations with Early Metals" Molecules 28, no. 6: 2702. https://doi.org/10.3390/molecules28062702