
Structural Insights into the Ligand–LsrK Kinase Binding Mode: A Step Forward in the Discovery of Novel Antimicrobial Agents
 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
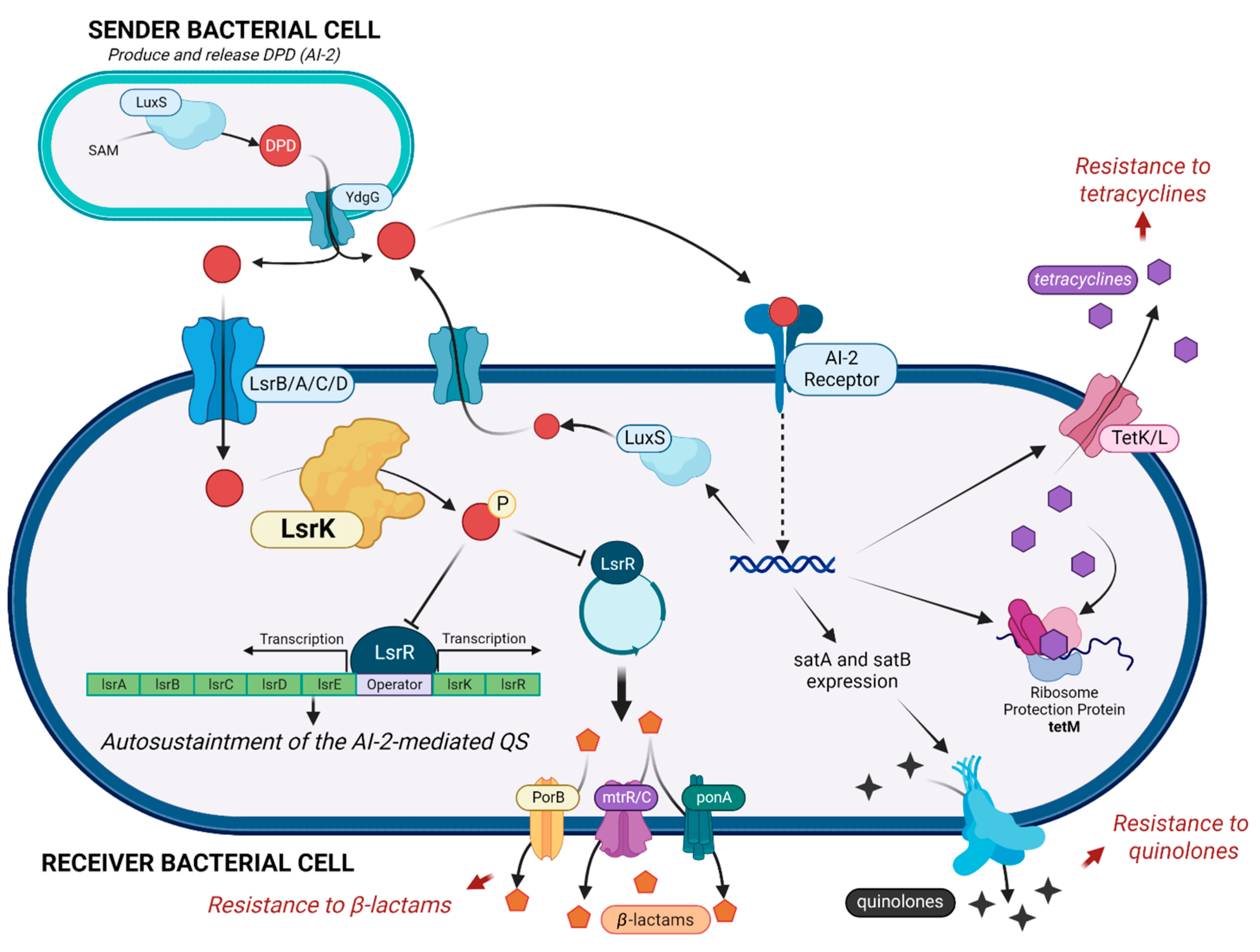
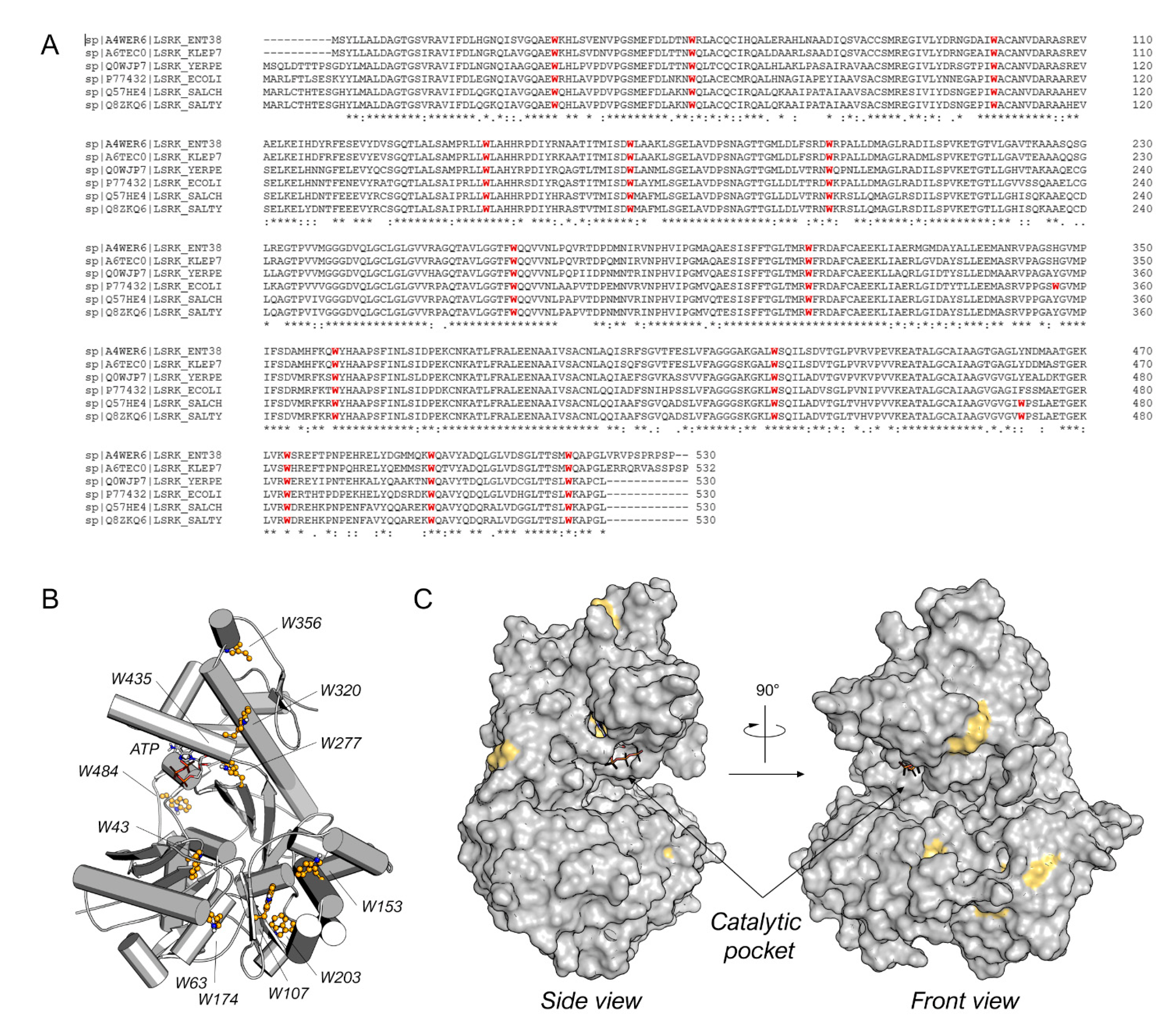
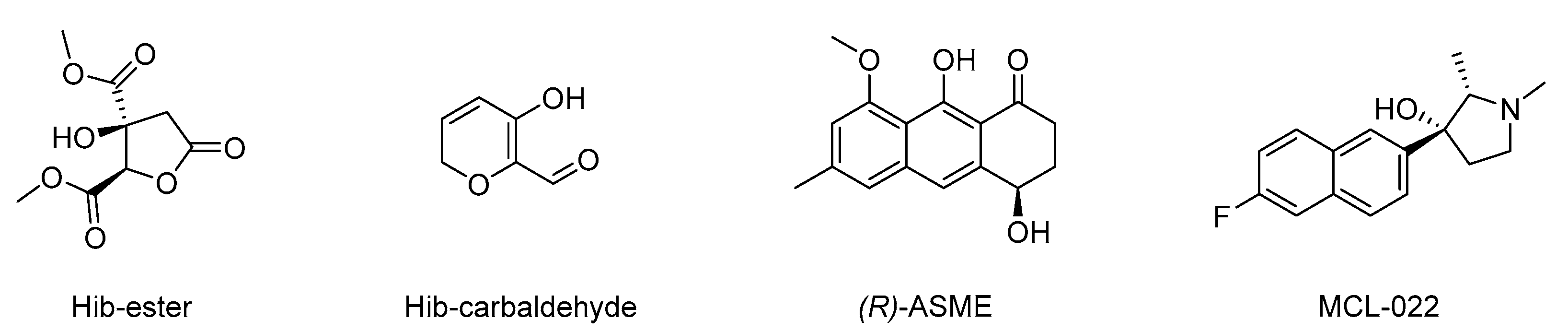
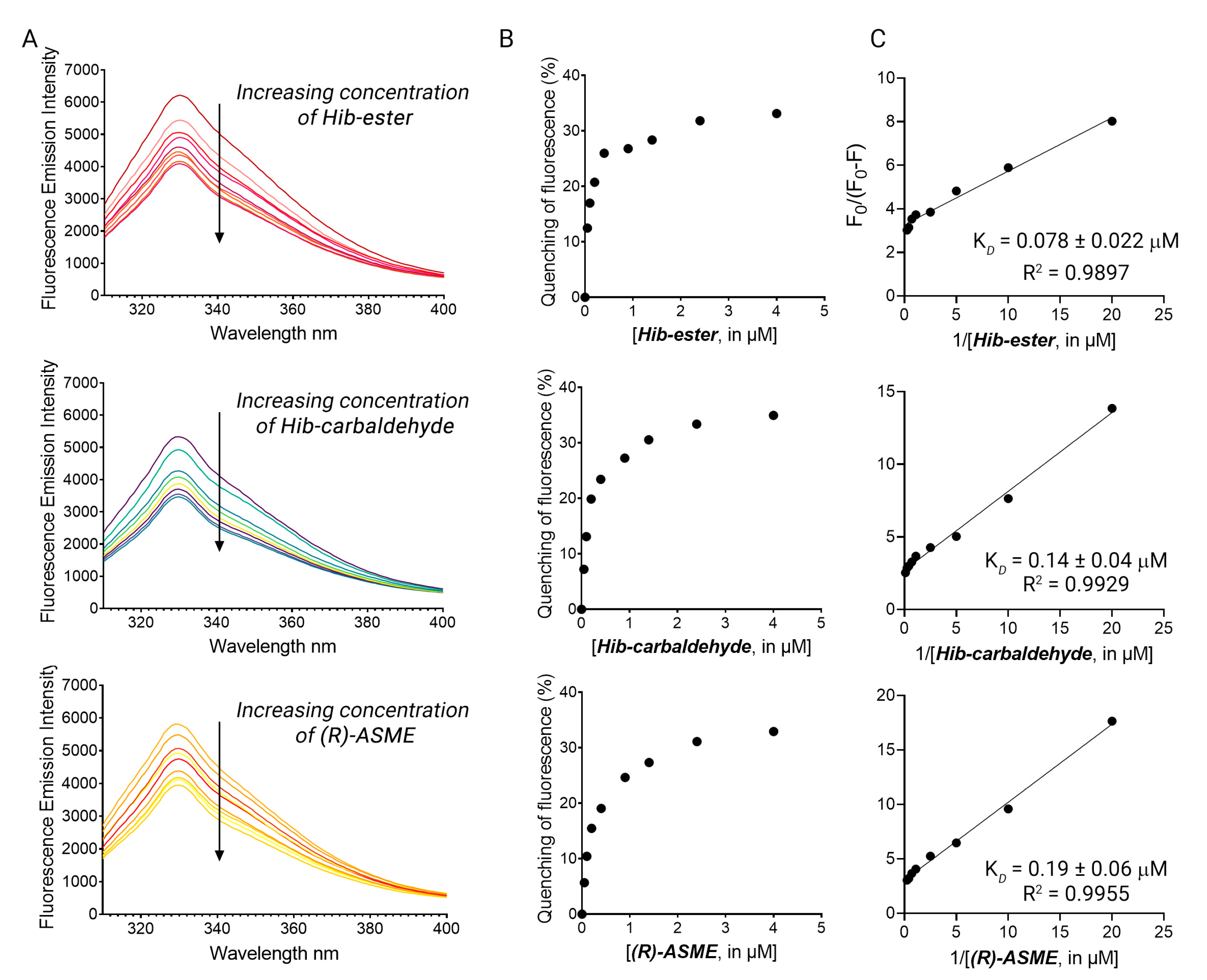
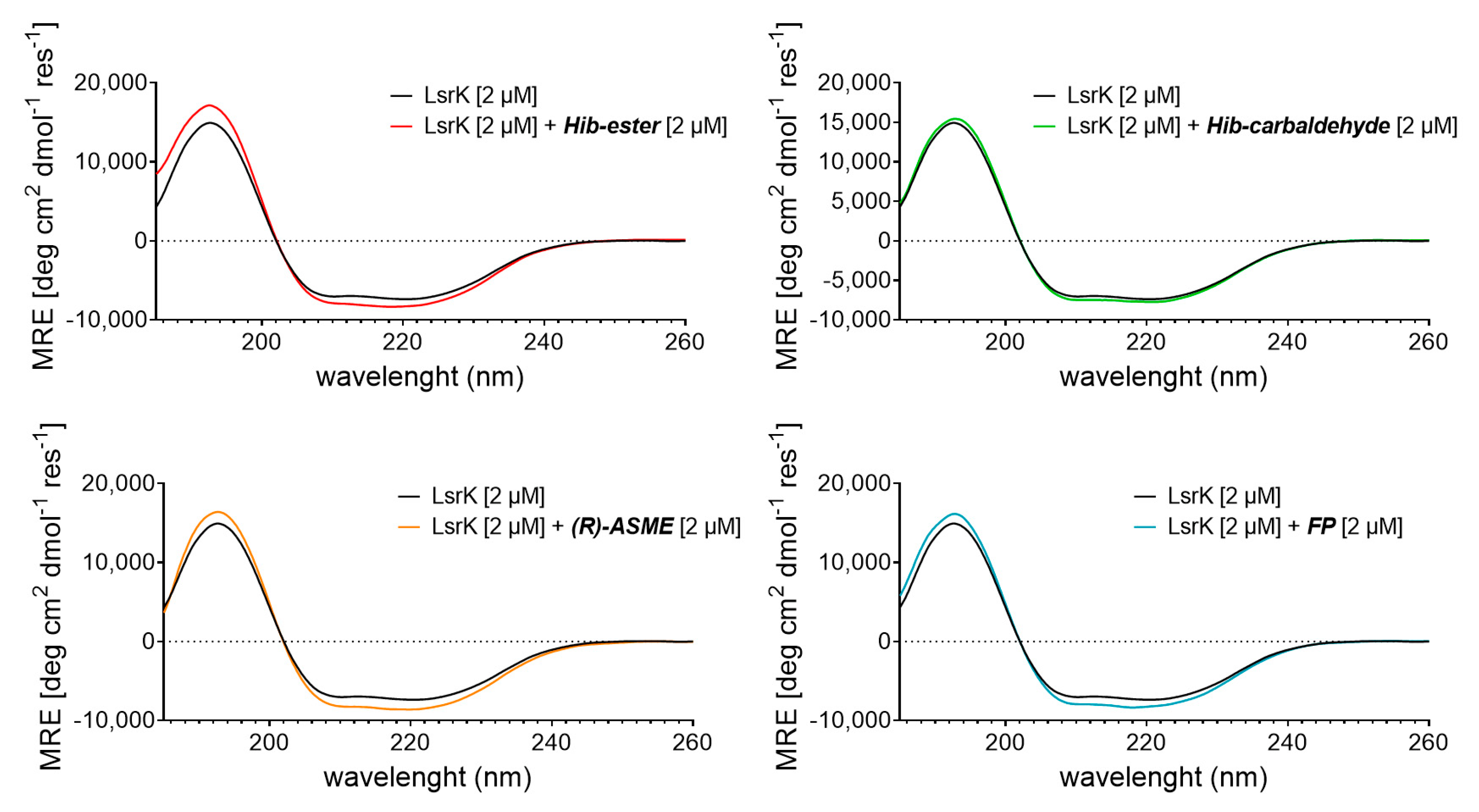
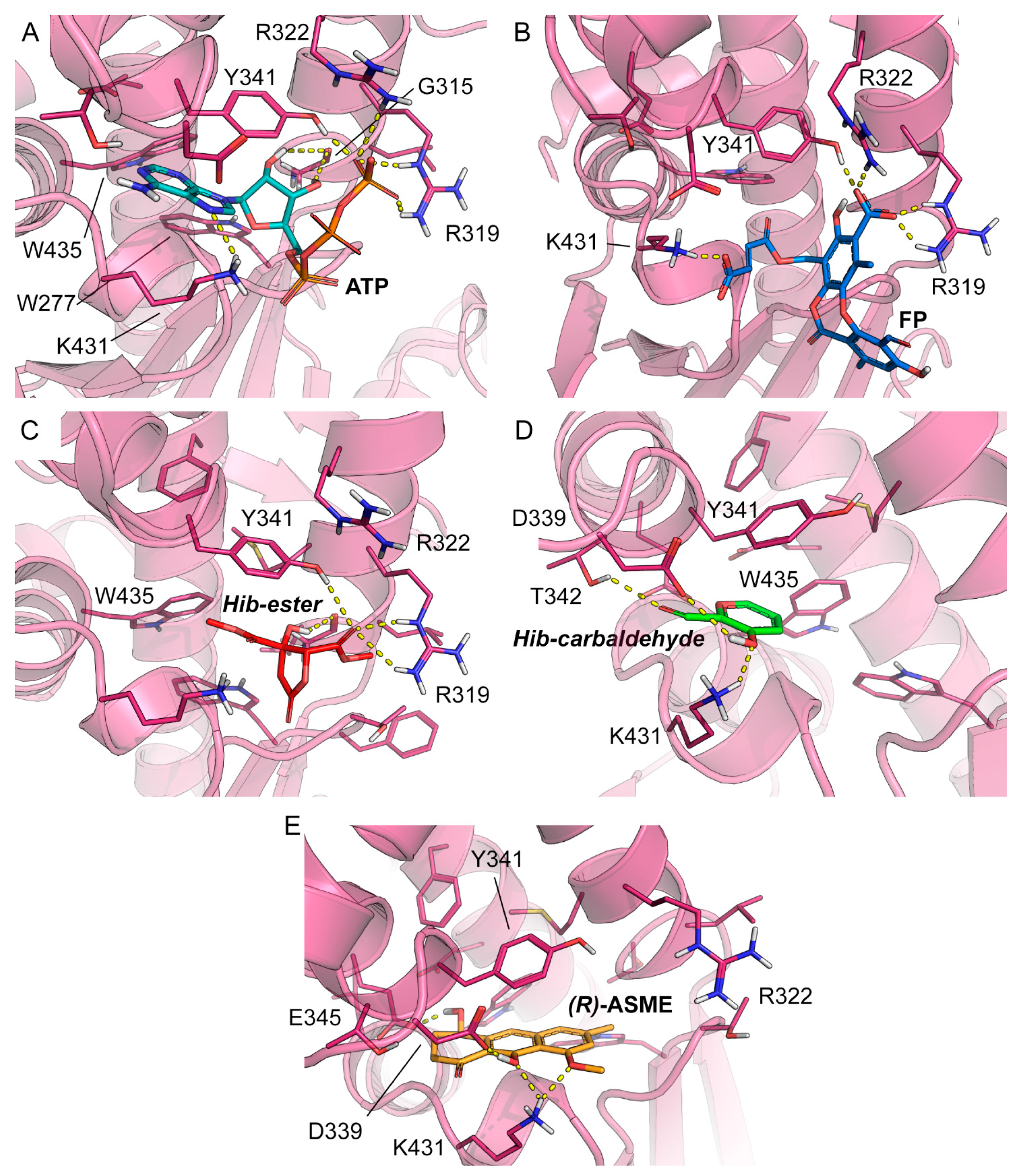
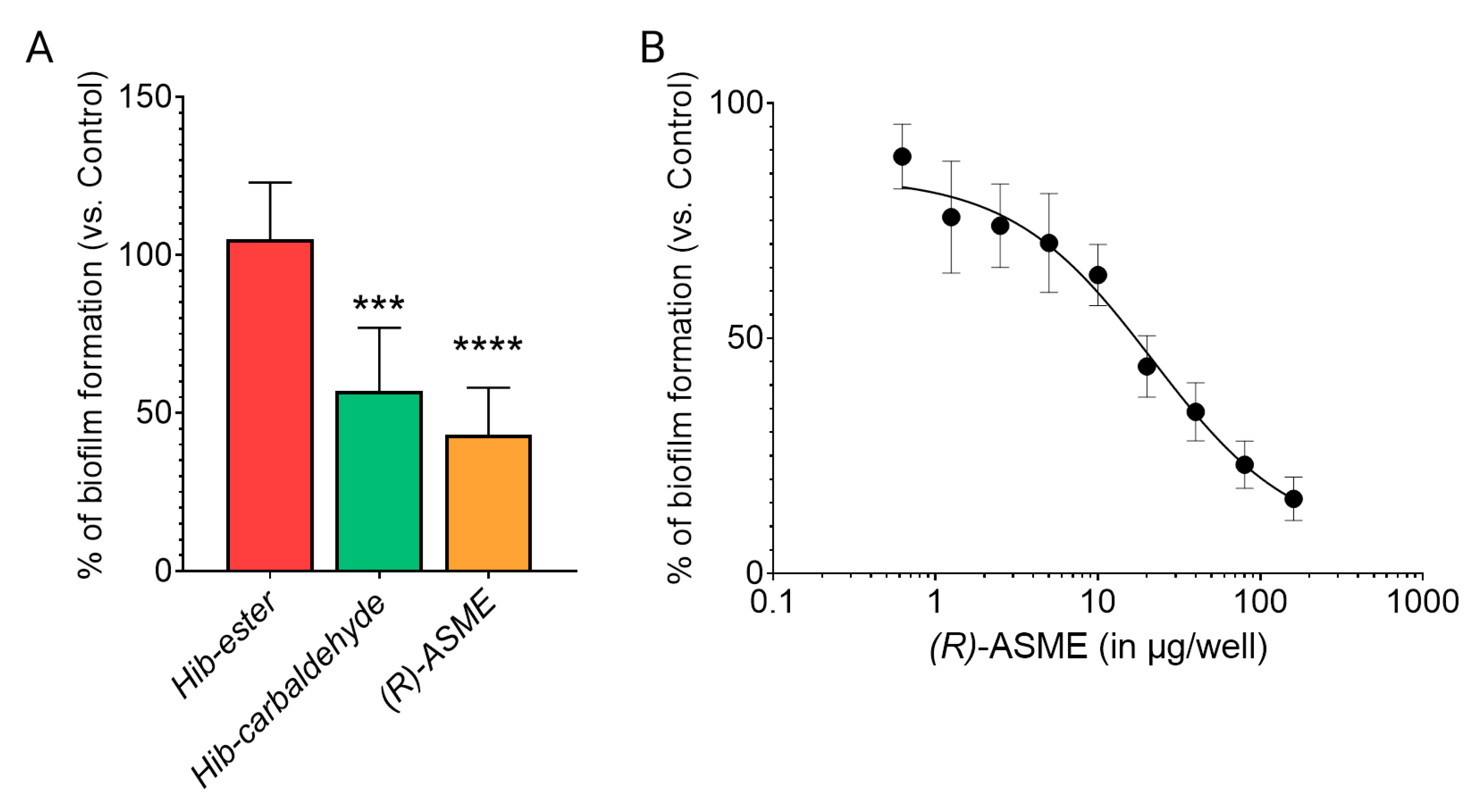
2. Results and Discussion
3. Materials and Methods
3.1. Isolation and Purification of Hib-Ester and Hib-Carbaldehyde
3.2. Isolation and Purification of (R)-ASME
3.3. Isolation and Purification of Fumarprotocetraric Acid
3.4. Expression and Purification of LsrK
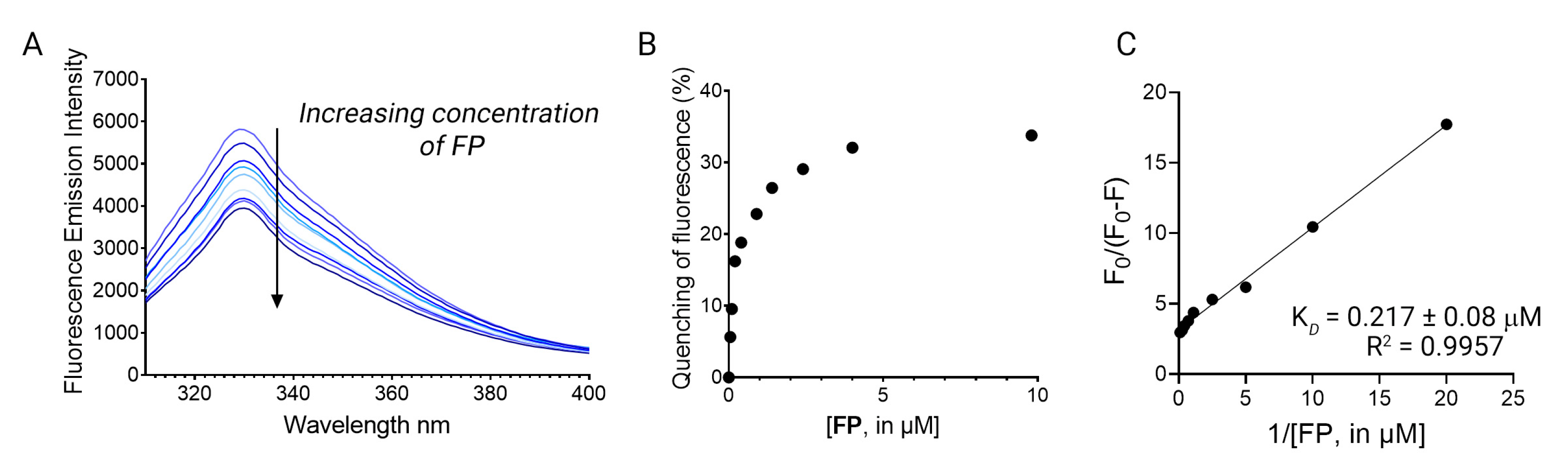
3.5. Fluorescence Binding Studies
3.6. Circular Dichroism
3.7. Docking
3.8. Biofilm Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Bank. Drug-Resistant Infections: A Threat to Our Economic Future (Final Report); The World Bank: Washington, DC, USA, 2017; pp. 1–132. [Google Scholar]
- World Health Organization. Global Antimicrobial Resistance Surveillance System (GLASS) Report; WHO: Geneva, Switzerland, 2017; ISBN 978-92-4-151344-9. [Google Scholar]
- Irfan, M.; Almotiri, A.; AlZeyadi, Z.A. Antimicrobial Resistance and Its Drivers—A Review. Antibiotics 2022, 11, 1362. [Google Scholar] [CrossRef]
- McEwen, S.A.; Collignon, P.J. Antimicrobial Resistance: A One Health Perspective. Microbiol. Spectr. 2018, 6, 521–547. [Google Scholar] [CrossRef] [Green Version]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 481–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, J.; Rafiq, Q.A.; Ratcliffe, E. Antimicrobial resistance mechanisms and potential synthetic treatments. Futur. Sci. OA 2018, 4, FSO290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idrees, M.; Sawant, S.; Karodia, N.; Rahman, A. Staphylococcus aureus Biofilm: Morphology, Genetics, Pathogenesis and Treatment Strategies. Int. J. Environ. Res. Public Health 2021, 18, 7602. [Google Scholar] [CrossRef]
- Miller, S.T.; Xavier, K.B.; Campagna, S.R.; Taga, M.E.; Semmelhack, M.F.; Bassler, B.L.; Hughson, F.M. Salmonella typhimurium Recognizes a Chemically Distinct Form of the Bacterial Quorum-Sensing Signal AI-2. Mol. Cell 2004, 15, 677–687. [Google Scholar] [CrossRef]
- Xavier, K.B.; Bassler, B.L. Interference with AI-2-mediated bacterial cell-cell communication. Nature 2005, 437, 750–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Attila, C.; Wang, L.; Wood, T.K.; Valdes, J.J.; Bentley, W.E. Quorum Sensing in Escherichia coli Is Signaled by AI-2/LsrR: Effects on Small RNA and Biofilm Architecture. J. Bacteriol. 2007, 189, 6011–6020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.-H.; Eo, Y.; Grishaev, A.; Guo, M.; Smith, J.A.I.; Sintim, H.O.; Kim, E.-H.; Cheong, H.-K.; Bentley, W.E.; Ryu, K.-S. Crystal Structures of the LsrR Proteins Complexed with Phospho-AI-2 and Two Signal-Interrupting Analogues Reveal Distinct Mechanisms for Ligand Recognition. J. Am. Chem. Soc. 2013, 135, 15526–15535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linciano, P.; Cavalloro, V.; Martino, E.; Kirchmair, J.; Listro, R.; Rossi, D.; Collina, S. Tackling Antimicrobial Resistance with Small Molecules Targeting LsrK: Challenges and Opportunities. J. Med. Chem. 2020, 63, 15243–15257. [Google Scholar] [CrossRef]
- Xavier, K.B.; Bassler, B.L. Regulation of uptake and processing of the quorum-sensing autoinducer AI-2 in Escherichia coli. J. Bacteriol. 2005, 187, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, V.; Fernandes, R.; Tsao, C.-Y.; Bentley, W.E. Cross Species Quorum Quenching Using a Native AI-2 Processing Enzyme. ACS Chem. Biol. 2010, 5, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Medarametla, P.; Kronenberger, T.; Laitinen, T.; Poso, A. Structural Characterization of LsrK as a Quorum Sensing Target and a Comparison between X-ray and Homology Models. J. Chem. Inf. Model. 2021, 61, 1346–1353. [Google Scholar] [CrossRef]
- Stotani, S.; Gatta, V.; Medarametla, P.; Padmanaban, M.; Karawajczyk, A.; Giordanetto, F.; Tammela, P.; Laitinen, T.; Poso, A.; Tzalis, D.; et al. DPD-Inspired Discovery of Novel LsrK Kinase Inhibitors: An Opportunity to Fight Antimicrobial Resistance. J. Med. Chem. 2019, 62, 2720–2737. [Google Scholar] [CrossRef] [Green Version]
- Gatta, V.; Ilina, P.; Porter, A.; McElroy, S.; Tammela, P. Targeting quorum sensing: High-throughput screening to identify novel lsrk inhibitors. Int. J. Mol. Sci. 2019, 20, 3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.H.; Hauk, P.; Cho, K.; Eo, Y.; Ma, X.; Stephens, K.; Cha, S.; Jeong, M.; Suh, J.Y.; Sintim, H.O.; et al. Evidence of link between quorum sensing and sugar metabolism in Escherichia coli revealed via cocrystal structures of LsrK and HPr. Sci. Adv. 2018, 4, eaar7063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghisaidoobe, A.; Chung, S. Intrinsic Tryptophan Fluorescence in the Detection and Analysis of Proteins: A Focus on Förster Resonance Energy Transfer Techniques. Int. J. Mol. Sci. 2014, 15, 22518–22538. [Google Scholar] [CrossRef]
- Yammine, A.; Gao, J.; Kwan, A. Tryptophan Fluorescence Quenching Assays for Measuring Protein-ligand Binding Affinities: Principles and a Practical Guide. BIO-PROTOCOL 2019, 9, e3253. [Google Scholar] [CrossRef]
- Kosanić, M.; Ranković, B. Antioxidant and Antimicrobial Properties of Some Lichens and Their Constituents. J. Med. Food 2011, 14, 1624–1630. [Google Scholar] [CrossRef]
- Htun, T. A Negative Deviation from Stern–Volmer Equation in Fluorescence Quenching. J. Fluoresc. 2004, 14, 217–222. [Google Scholar] [CrossRef]
- Lehrer, S.S. Solute perturbation of protein fluorescence. The quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry 1971, 10, 3254–3263. [Google Scholar] [CrossRef] [PubMed]
- Gehlen, M.H. The centenary of the Stern-Volmer equation of fluorescence quenching: From the single line plot to the SV quenching map. J. Photochem. Photobiol. C Photochem. Rev. 2020, 42, 100338. [Google Scholar] [CrossRef]
- Mátyus, L.; Szöllősi, J.; Jenei, A. Steady-state fluorescence quenching applications for studying protein structure and dynamics. J. Photochem. Photobiol. B Biol. 2006, 83, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Quenching of Fluorescence. In Principles of Fluorescence Spectroscopy; Springer: Boston, MA, USA, 2006; pp. 277–330. [Google Scholar]
- Samworth, C.M.; Esposti, M.D.; Lenaz, G. Quenching of the intrinsic tryptophan fluorescence of mitochondrial ubiquinol-cytochrome-c reductase by the binding of ubiquinone. Eur. J. Biochem. 1988, 171, 81–86. [Google Scholar] [CrossRef]
- Pietrocola, G.; Valtulina, V.; Rindi, S.; Jost, B.H.; Speziale, P. Functional and structural properties of CbpA, a collagen-binding protein from Arcanobacterium pyogenes. Microbiology 2007, 153, 3380–3389. [Google Scholar] [CrossRef] [Green Version]
- Geethanjali, H.S.; Nagaraja, D.; Melavanki, R.M.; Kusanur, R.A. Fluorescence quenching of boronic acid derivatives by aniline in alcohols—A Negative deviation from Stern–Volmer equation. J. Lumin. 2015, 167, 216–221. [Google Scholar] [CrossRef]
- Alam, M.F.; Varshney, S.; Khan, M.A.; Laskar, A.A.; Younus, H. In vitro DNA binding studies of therapeutic and prophylactic drug citral. Int. J. Biol. Macromol. 2018, 113, 300–308. [Google Scholar] [CrossRef]
- Cvijetić, I.N.; Petrović, D.D.; Verbić, T.Ž.; Juranić, I.O.; Drakulić, B.J. Human Serum Albumin Binding of 2-[(Carboxymethyl)sulfanyl]-4-oxo-4-(4-tert-butylphenyl)butanoic Acid and its Mono-Me Ester. ADMET DMPK 2014, 2, 126–142. [Google Scholar] [CrossRef]
- Phopin, K.; Ruankham, W.; Prachayasittikul, S.; Prachayasittikul, V.; Tantimongcolwat, T. Insight into the Molecular Interaction of Cloxyquin (5-chloro-8-hydroxyquinoline) with Bovine Serum Albumin: Biophysical Analysis and Computational Simulation. Int. J. Mol. Sci. 2019, 21, 249. [Google Scholar] [CrossRef] [Green Version]
- Chaves, O.; Amorim, A.; Castro, L.; Sant’Anna, C.; de Oliveira, M.; Cesarin-Sobrinho, D.; Netto-Ferreira, J.; Ferreira, A. Fluorescence and Docking Studies of the Interaction between Human Serum Albumin and Pheophytin. Molecules 2015, 20, 19526–19539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, D.; Cingolani, M.; Rampazzo, E.; Prodi, L.; Zaccheroni, N. Static quenching upon adduct formation: A treatment without shortcuts and approximations. Chem. Soc. Rev. 2021, 50, 8414–8427. [Google Scholar] [CrossRef]
- Malacrida, A.; Cavalloro, V.; Martino, E.; Costa, G.; Ambrosio, F.A.; Alcaro, S.; Rigolio, R.; Cassetti, A.; Miloso, M.; Collina, S. Anti-Multiple Myeloma Potential of Secondary Metabolites from Hibiscus sabdariffa—Part 2. Molecules 2021, 26, 6596. [Google Scholar] [CrossRef]
- Pimentel-Moral, S.; Borrás-Linares, I.; Lozano-Sánchez, J.; Arráez-Román, D.; Martínez-Férez, A.; Segura-Carretero, A. Microwave-assisted extraction for Hibiscus sabdariffa bioactive compounds. J. Pharm. Biomed. Anal. 2018, 156, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Malacrida, A.; Cavalloro, V.; Martino, E.; Cassetti, A.; Nicolini, G.; Rigolio, R.; Cavaletti, G.; Mannucci, B.; Vasile, F.; Di Giacomo, M.; et al. Anti-multiple myeloma potential of secondary metabolites from hibiscus sabdariffa. Molecules 2019, 24, 2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Ahmed, K.; Gaggeri, R.; Della Volpe, S.; Maggi, L.; Mazzeo, G.; Longhi, G.; Abbate, S.; Corana, F.; Martino, E.; et al. (R)-(−)-Aloesaponol III 8-Methyl Ether from Eremurus persicus: A Novel Compound against Leishmaniosis. Molecules 2017, 22, 519. [Google Scholar] [CrossRef] [Green Version]
- Collina, S.; Azzolina, O.; Vercesi, D.; Sbacchi, M.; Scheideler, M.A.; Barbieri, A.; Lanza, E.; Ghislandi, V. Synthesis and antinociceptive activity of pyrrolidinylnaphthalenes. Bioorg. Med. Chem. 2000, 8, 1925–1930. [Google Scholar] [CrossRef] [PubMed]
- Ghislandi, V.; Collina, S.; Azzolina, O.; Barbieri, A.; Lanza, E.; Tadini, C. Preparation and configuration of racemic and optically active analgesic cycloaminoalkylnaphthalenes. Chirality 1999, 11, 21–28. [Google Scholar] [CrossRef]
- Della Volpe, S.; Listro, R.; Parafioriti, M.; Di Giacomo, M.; Rossi, D.; Ambrosio, F.A.; Costa, G.; Alcaro, S.; Ortuso, F.; Hirsch, A.K.H.; et al. BOPC1 Enantiomers Preparation and HuR Interaction Study. From Molecular Modeling to a Curious DEEP-STD NMR Application. ACS Med. Chem. Lett. 2020, 11, 883–888. [Google Scholar] [CrossRef]
- Della Volpe, S.; Nasti, R.; Queirolo, M.; Unver, M.Y.; Jumde, V.K.; Dömling, A.; Vasile, F.; Potenza, D.; Ambrosio, F.A.; Costa, G.; et al. Novel Compounds Targeting the RNA-Binding Protein HuR. Structure-Based Design, Synthesis, and Interaction Studies. ACS Med. Chem. Lett. 2019, 10, 615–620. [Google Scholar] [CrossRef]
- Nasti, R.; Rossi, D.; Amadio, M.; Pascale, A.; Unver, M.Y.; Hirsch, A.K.H.; Collina, S. Compounds Interfering with Embryonic Lethal Abnormal Vision (ELAV) Protein–RNA Complexes: An Avenue for Discovering New Drugs. J. Med. Chem. 2017, 60, 8257–8267. [Google Scholar] [CrossRef]
- Rui, M.; Rossi, D.; Marra, A.; Paolillo, M.; Schinelli, S.; Curti, D.; Tesei, A.; Cortesi, M.; Zamagni, A.; Laurini, E.; et al. Synthesis and biological evaluation of new aryl-alkyl(alkenyl)-4-benzylpiperidines, novel Sigma Receptor (SR) modulators, as potential anticancer-agents. Eur. J. Med. Chem. 2016, 124, 649–665. [Google Scholar] [CrossRef] [Green Version]
- Rogers, D.M.; Jasim, S.B.; Dyer, N.T.; Auvray, F.; Réfrégiers, M.; Hirst, J.D. Electronic Circular Dichroism Spectroscopy of Proteins. Chem 2019, 5, 2751–2774. [Google Scholar] [CrossRef]
- Rodger, A.; Marrington, R.; Roper, D.; Windsor, S. Circular Dichroism Spectroscopy for the Study of Protein-Ligand Interactions. In Protein-Ligand Interactions; Humana Press: Totowa, NJ, USA, 2005; pp. 343–363. [Google Scholar]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Schrödinger Release 2021-3: Maestro 2021; Schrödinger LLC: New York, NY, USA, 2021. [Google Scholar]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Residues Involved in the Binding | GlideScore [kcal mol−1] |
|---|---|---|
| ATP | G315 b,d, R319 a,c, R322 a,c, Y341 a,c, K431 a,c, Y341 b,c | n/a |
| FP | G315 b,d, R319 a,c, R322 a,c, Y341 a,c, Y341 c | −6.490 |
| Hib-ester | G315 b,d, R319 a,c, Y341 b,c | −4.984 |
| Hib-carbaldehyde | T342 a,c, K341 a,c, Y341 b,c | −6.169 |
| (R)-ASME | D339 a,c, K431 a,c, E345 a,c, Y341 b,c | −8.184 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Listro, R.; Milli, G.; Pellegrini, A.; Motta, C.; Cavalloro, V.; Martino, E.; Kirchmair, J.; Pietrocola, G.; Rossi, D.; Linciano, P.; et al. Structural Insights into the Ligand–LsrK Kinase Binding Mode: A Step Forward in the Discovery of Novel Antimicrobial Agents. Molecules 2023, 28, 2542. https://doi.org/10.3390/molecules28062542
Listro R, Milli G, Pellegrini A, Motta C, Cavalloro V, Martino E, Kirchmair J, Pietrocola G, Rossi D, Linciano P, et al. Structural Insights into the Ligand–LsrK Kinase Binding Mode: A Step Forward in the Discovery of Novel Antimicrobial Agents. Molecules. 2023; 28(6):2542. https://doi.org/10.3390/molecules28062542
Chicago/Turabian StyleListro, Roberta, Giorgio Milli, Angelica Pellegrini, Chiara Motta, Valeria Cavalloro, Emanuela Martino, Johannes Kirchmair, Giampiero Pietrocola, Daniela Rossi, Pasquale Linciano, and et al. 2023. "Structural Insights into the Ligand–LsrK Kinase Binding Mode: A Step Forward in the Discovery of Novel Antimicrobial Agents" Molecules 28, no. 6: 2542. https://doi.org/10.3390/molecules28062542